cGMP Signaling and Vascular Smooth Muscle Cell Plasticity


Abstract
:1. Introduction
2. Role of VSMCs in Physiology and Pathophysiology
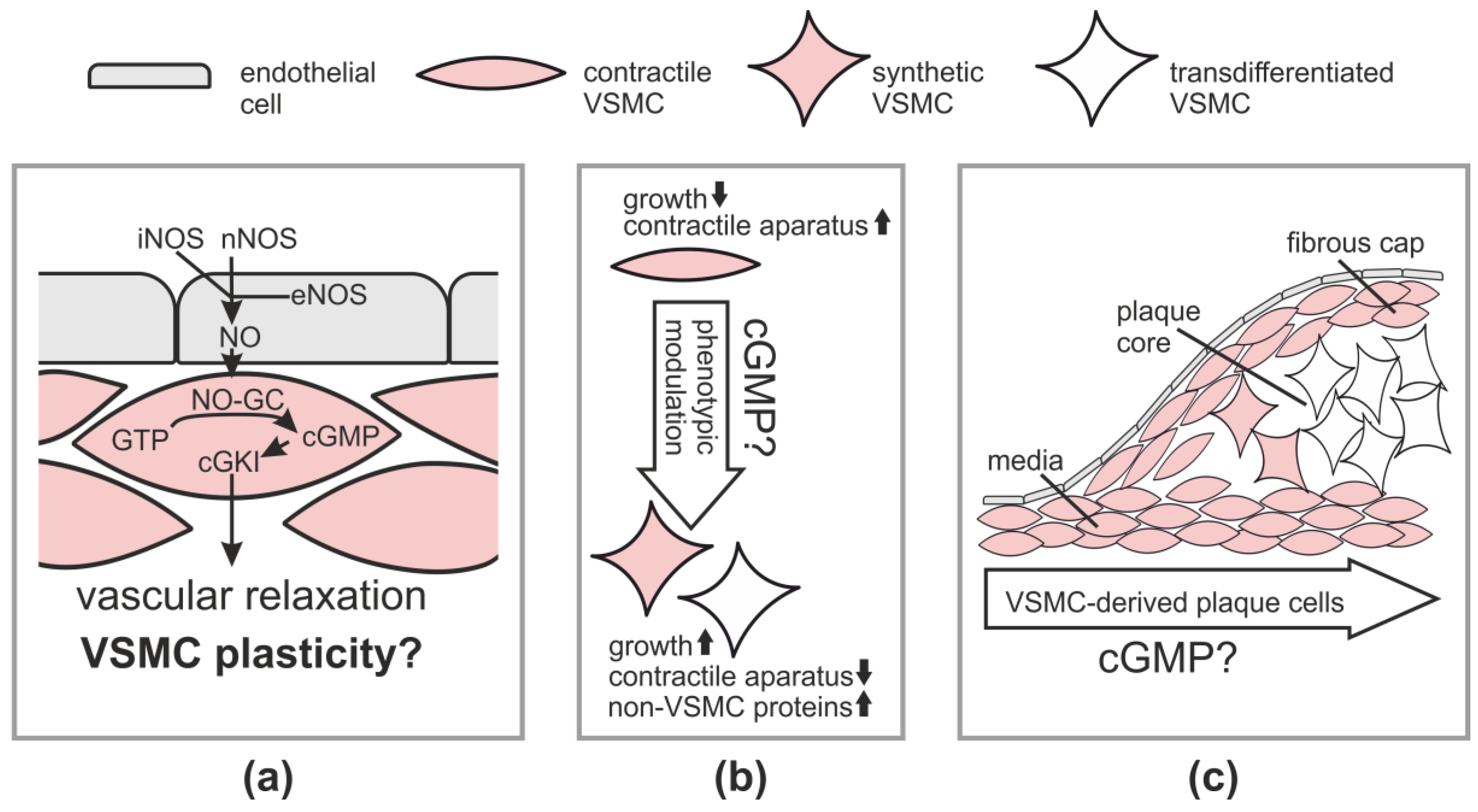
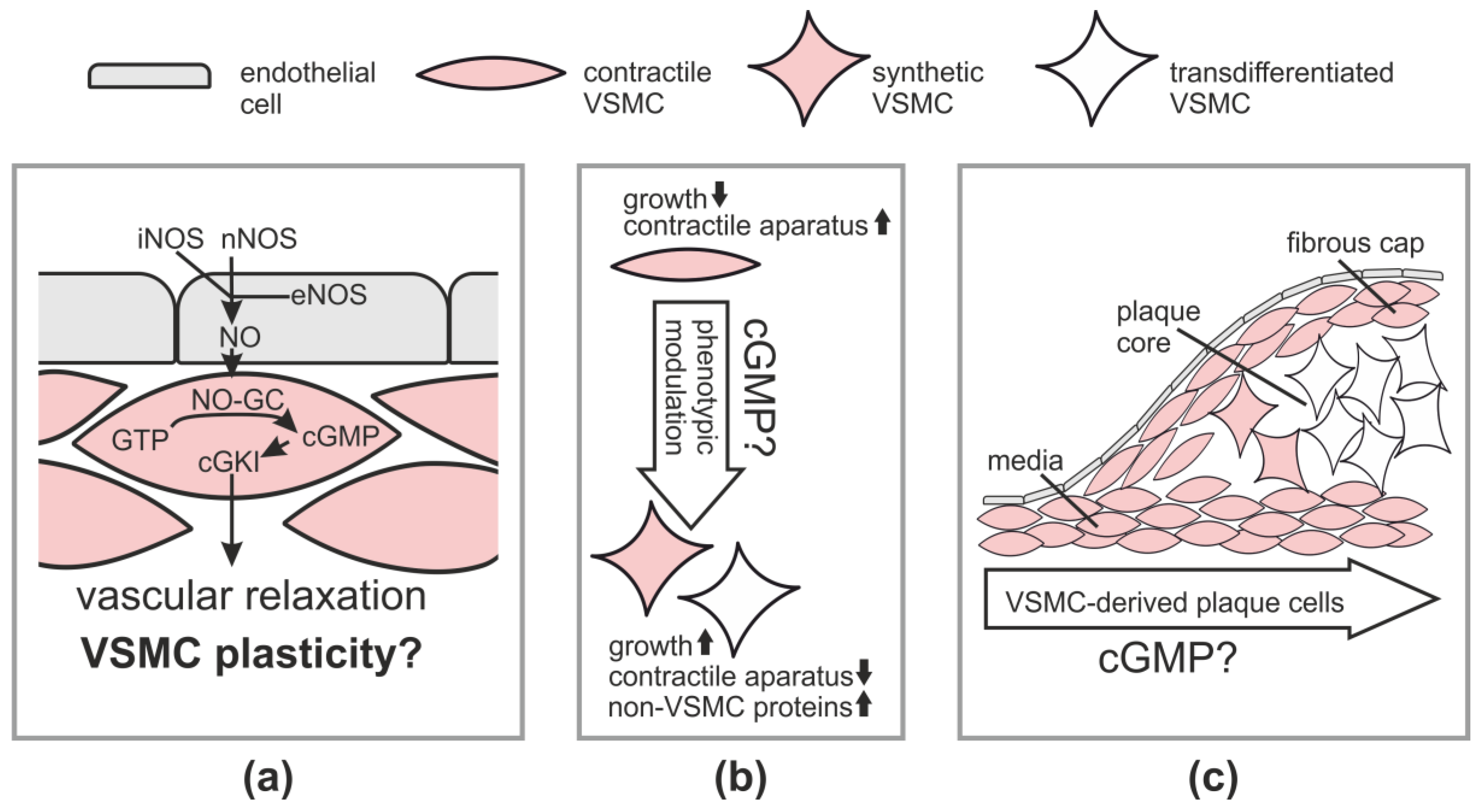
2.1. Vasodilation via the NO-cGMP-cGKI Pathway
2.2. VSMCs in Vascular Diseases
2.2.1. Atherosclerosis
2.2.2. Restenosis
3. cGMP and VSMC Plasticity in Cell Culture
4. cGMP and Vascular Diseases
4.1. Role of cGMP in Atherosclerosis
4.2. Role of cGMP in Restenosis
4.3. Role of cGMP in Angiogenesis
5. Limitations and Future Directions
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.; Feil, R. Meeting report: cGMP matters. Sci. Sign. 2008, 1, pe12. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Koesling, D. The function of NO-sensitive guanylyl cyclase: What we can learn from genetic mouse models. Nitric Oxide 2009, 21, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular physiology of membrane guanylyl cyclase receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 111–136. [Google Scholar]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, T.; Wei, H.; Miano, J.M.; Abe, J.; Berk, B.C.; Yan, C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ. Res. 2003, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Surks, H.K. cGMP-dependent protein kinase i and smooth muscle relaxation: A tale of two isoforms. Circ. Res. 2007, 101, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Bernhard, D.; Lukowski, R.; Weinmeister, P.; Worner, R.; Wegener, J.W.; Valtcheva, N.; Feil, S.; Schlossmann, J.; Hofmann, F.; et al. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ. Res. 2007, 101, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Feil, S.; Hofmann, F. A heretical view on the role of NO and cGMP in vascular proliferative diseases. Trends Mol. Med. 2005, 11, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.; Schmidt, H.H.H.W. cGMP in the vasculature. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 447–467. [Google Scholar]
- Lincoln, T.M.; Wu, X.; Sellak, H.; Dey, N.; Choi, C.S. Regulation of vascular smooth muscle cell phenotype by cyclic GMP and cyclic GMP-dependent protein kinase. Front. Biosci. 2006, 11, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.P.; Somlyo, A.V. Signal transduction and regulation in smooth muscle. Nature 1994, 372, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Buys, E.S.; Sips, P.; Vermeersch, P.; Raher, M.J.; Rogge, E.; Ichinose, F.; Dewerchin, M.; Bloch, K.D.; Janssens, S.; Brouckaert, P. Gender-specific hypertension and responsiveness to nitric oxide in sGCα1 knockout mice. Cardiovasc. Res. 2008, 79, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.; Konig, P.; Wirth, A.; Offermanns, S.; Koesling, D.; Friebe, A. Smooth muscle-specific deletion of nitric oxide-sensitive guanylyl cyclase is sufficient to induce hypertension in mice. Circulation 2010, 121, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Investig. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Thoonen, R.; Cauwels, A.; Decaluwe, K.; Geschka, S.; Tainsh, R.E.; Delanghe, J.; Hochepied, T.; De Cauwer, L.; Rogge, E.; Voet, S.; et al. Cardiovascular and pharmacological implications of haem-deficient NO-unresponsive soluble guanylate cyclase knock-in mice. Nat. Commun. 2015, 6, 8482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, M.; Feil, R.; Siegl, D.; Feil, S.; Hofmann, F.; Pohl, U.; de Wit, C. cGMP-dependent protein kinase mediates NO- but not acetylcholine-induced dilations in resistance vessels in vivo. Hypertension 2004, 44, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszodi, A.; Andersson, K.E.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Shah, P.K.; Rajavashisth, T.B. Cellular origins of atherosclerosis: Towards ontogenetic endgame? FASEB J. 2003, 17, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Manabe, I.; Nagai, R. Lineage of bone marrow-derived cells in atherosclerosis. Circ. Res. 2013, 112, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth muscle cell plasticity: Fact or fiction? Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Wang, A.; Wang, D.; Li, S. Smooth muscle cells: To be or not to be? Response to Nguyen et al. Circ. Res. 2013, 112, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Hofmann, F.; Feil, R. SM22α modulates vascular smooth muscle cell phenotype during atherogenesis. Circ. Res. 2004, 94, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Wolfsgruber, W.; Feil, S.; Brummer, S.; Kuppinger, O.; Hofmann, F.; Feil, R. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, E.R.; Pugach, I.M.; Orekhov, A.N. Subendothelial smooth muscle cells of human aorta express macrophage antigen in situ and in vitro. Atherosclerosis 1997, 135, 19–27. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Albarran-Juarez, J.; Kaur, H.; Grimm, M.; Offermanns, S.; Wettschureck, N. Lineage tracing of cells involved in atherosclerosis. Atherosclerosis 2016, 251, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Fuchtbauer, E.M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Do vascular smooth muscle cells differentiate to macrophages in atherosclerotic lesions? Circ. Res. 2014, 115, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Totary-Jain, H.; Marks, A.R. Vascular smooth muscle cell proliferation in restenosis. Circ. Cardiovasc. Interv. 2011, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Chamley-Campbell, J.; Campbell, G.R.; Ross, R. The smooth muscle cell in culture. Physiol. Rev. 1979, 59, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Worth, N.F.; Rolfe, B.E.; Song, J.; Campbell, G.R. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil. Cytoskel. 2001, 49, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Segura-Puimedon, M.; Mergia, E.; Al-Hasani, J.; Aherrahrou, R.; Stoelting, S.; Kremer, F.; Freyer, J.; Koesling, D.; Erdmann, J.; Schunkert, H.; et al. Proatherosclerotic effect of the alpha1-subunit of soluble guanylyl cyclase by promoting smooth muscle phenotypic switching. Am. J. Pathol. 2016, 186, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, R.; Weinmeister, P.; Bernhard, D.; Feil, S.; Gotthardt, M.; Herz, J.; Massberg, S.; Zernecke, A.; Weber, C.; Hofmann, F.; et al. Role of smooth muscle cGMP/cGKI signaling in murine vascular restenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.B.; Foley, K.F.; Lincoln, T.M.; Dostmann, W.R. Inhibition of cGMP-dependent protein kinase reverses phenotypic modulation of vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 2005, 45, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Butt, E. cGMP-dependent protein kinase modulators. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 409–421. [Google Scholar]
- Gambaryan, S.; Geiger, J.; Schwarz, U.R.; Butt, E.; Begonja, A.; Obergfell, A.; Walter, U. Potent inhibition of human platelets by cGMP analogs independent of cGMP-dependent protein kinase. Blood 2004, 103, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.J.; Senis, Y.A.; Auger, J.M.; Feil, R.; Hofmann, F.; Salmon, G.; Peterson, J.T.; Burslem, F.; Watson, S.P. Gpib-dependent platelet activation is dependent on src kinases but not MAP kinase or cGMP-dependent kinase. Blood 2004, 103, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, M.; Glazova, M.; Gambaryan, S.; Vollkommer, T.; Butt, E.; Bader, B.; Heermeier, K.; Lincoln, T.M.; Walter, U.; Palmetshofer, A. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 2000, 275, 33536–33541. [Google Scholar] [CrossRef] [PubMed]
- Valtcheva, N.; Nestorov, P.; Beck, A.; Russwurm, M.; Hillenbrand, M.; Weinmeister, P.; Feil, R. The commonly used cGMP-dependent protein kinase type I (cGKI) inhibitor Rp-8-Br-PET-cGMPS can activate cGKI in vitro and in intact cells. J. Biol. Chem. 2009, 284, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S.; Butt, E.; Kobsar, A.; Geiger, J.; Rukoyatkina, N.; Parnova, R.; Nikolaev, V.O.; Walter, U. The oligopeptide DT-2 is a specific PKG I inhibitor only in vitro, not in living cells. Br. J. Pharmacol. 2012, 167, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Tulis, D.A.; Bohl Masters, K.S.; Lipke, E.A.; Schiesser, R.L.; Evans, A.J.; Peyton, K.J.; Durante, W.; West, J.L.; Schafer, A.I. YC-1-mediated vascular protection through inhibition of smooth muscle cell proliferation and platelet function. Biochem. Biophys. Res. Commun. 2002, 291, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.; Stasch, J.P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Pacher, P.; Evgenov, O.V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Griebenow, N.; Hahn, M.G.; Hartung, I.; Mais, F.J.; Mittendorf, J.; Schafer, M.; Schirok, H.; Stasch, J.P.; Stoll, F.; et al. The chemistry and biology of soluble guanylate cyclase stimulators and activators. Angew. Chem. Int. Ed. Engl. 2013, 52, 9442–9462. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.D.; Schlutsmeyer, S.M.; Bloch, D.B.; de la Monte, S.M.; Roberts, J.D., Jr.; Filippov, G.; Janssens, S.P.; Rosenzweig, A.; Bloch, K.D. Adenovirus-mediated gene transfer of cGMP-dependent protein kinase increases the sensitivity of cultured vascular smooth muscle cells to the antiproliferative and pro-apoptotic effects of nitric oxide/cGMP. J. Biol. Chem. 1998, 273, 34263–34271. [Google Scholar] [CrossRef] [PubMed]
- Boerth, N.J.; Dey, N.B.; Cornwell, T.L.; Lincoln, T.M. Cyclic GMP-dependent protein kinase regulates vascular smooth muscle cell phenotype. J. Vasc. Res. 1997, 34, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Sellak, H.; Brown, F.M.; Lincoln, T.M. cGMP-dependent protein kinase and the regulation of vascular smooth muscle cell gene expression: Possible involvement of ELK-1 sumoylation. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1660–H1670. [Google Scholar] [CrossRef] [PubMed]
- Hassid, A.; Arabshahi, H.; Bourcier, T.; Dhaunsi, G.S.; Matthews, C. Nitric oxide selectively amplifies FGF-2-induced mitogenesis in primary rat aortic smooth muscle cells. Am. J. Physiol. 1994, 267, H1040–H1048. [Google Scholar] [CrossRef] [PubMed]
- Komalavilas, P.; Shah, P.K.; Jo, H.; Lincoln, T.M. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 34301–34309. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Buga, G.M.; Wei, L.H.; Bauer, P.M.; Wu, G.; del Soldato, P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic guanosine monophosphate-dependent protein kinase I promotes adhesion of primary vascular smooth muscle cells. Mol. Biol. Cell 2008, 19, 4434–4441. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Rolli-Derkinderen, M.; Sauzeau, V.; Boyer, L.; Lemichez, E.; Baron, C.; Henrion, D.; Loirand, G.; Pacaud, P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 2005, 96, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Sandner, P.; Schmidtko, A. Meeting report of the 8(th) international conference on cGMP “cGMP: Generators, effectors, and therapeutic implications” at Bamberg, Germany, from June 23 to 25, 2017. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Kraehling, J.R.; Sessa, W.C. Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ. Res. 2017, 120, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Kemp-Harper, B. cGMP signalling: From bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Oettrich, J.M.; Dao, V.T.; Frijhoff, J.; Kleikers, P.; Casas, A.I.; Hobbs, A.J.; Schmidt, H.H. Clinical relevance of cyclic GMP modulators: A translational success story of network pharmacology. Clin. Pharmacol. Ther. 2016, 99, 360–362. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Leineweber, K.; Moosmang, S.; Paulson, D. Genetics of NO deficiency. Am. J. Cardiol. 2017, 120, S80–S88. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B.; Munroe, P.B.; Rice, K.M.; Bochud, M.; Johnson, A.D.; Chasman, D.I.; Smith, A.V.; Tobin, M.D.; Verwoert, G.C.; Hwang, S.J.; et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emdin, C.A.; Khera, A.V.; Klarin, D.; Natarajan, P.; Zekavat, S.M.; Nomura, A.; Haas, M.; Aragam, K.; Ardissino, D.; Wilson, J.G.; et al. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 2018, 137, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Stark, K.; Esslinger, U.B.; Rumpf, P.M.; Koesling, D.; de Wit, C.; Kaiser, F.J.; Braunholz, D.; Medack, A.; Fischer, M.; et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 2013, 504, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Wobst, J.; Wolf, B.; Eckhold, J.; Vilne, B.; Hollstein, R.; von Ameln, S.; Dang, T.A.; Sager, H.B.; Moritz Rumpf, P.; et al. Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 2017, 136, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Maass, P.G.; Aydin, A.; Luft, F.C.; Schachterle, C.; Weise, A.; Stricker, S.; Lindschau, C.; Vaegler, M.; Qadri, F.; Toka, H.R.; et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat. Genet. 2015, 47, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.C.; Regalado, E.; Casteel, D.E.; Santos-Cortez, R.L.; Gong, L.; Kim, J.J.; Dyack, S.; Horne, S.G.; Chang, G.; Jondeau, G.; et al. Recurrent gain-of-function mutation in prkg1 causes thoracic aortic aneurysms and acute aortic dissections. Am. J. Hum. Genet. 2013, 93, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Herve, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Friebe, A.; Mergia, E.; Dangel, O.; Lange, A.; Koesling, D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide-sensitive guanylyl cyclase. Proc. Natl. Acad. Sci. USA 2007, 104, 7699–7704. [Google Scholar] [CrossRef] [PubMed]
- Feil, R. Conditional somatic mutagenesis in the mouse using site-specific recombinases. In Conditional Mutagenesis: An Approach to Disease Models; Feil, R., Metzger, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–28. [Google Scholar]
- Wegener, J.W.; Nawrath, H.; Wolfsgruber, W.; Kuhbandner, S.; Werner, C.; Hofmann, F.; Feil, R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res. 2002, 90, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kuhlencordt, P.J.; Astern, J.; Gyurko, R.; Huang, P.L. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation 2001, 104, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.W.; Reddick, R.L.; Jennette, J.C.; Shesely, E.G.; Smithies, O.; Maeda, N. Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)Apoe(−/−) mice are ameliorated by enalapril treatment. J. Clin. Investig. 2000, 105, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.H.; Huang, P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein e/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Moroi, M.; Zhang, L.; Yasuda, T.; Virmani, R.; Gold, H.K.; Fishman, M.C.; Huang, P.L. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J. Clin. Investig. 1998, 101, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Yogo, K.; Shimokawa, H.; Funakoshi, H.; Kandabashi, T.; Miyata, K.; Okamoto, S.; Egashira, K.; Huang, P.; Akaike, T.; Takeshita, A. Different vasculoprotective roles of NO synthase isoforms in vascular lesion formation in mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, e96–e100. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L., 2nd; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, H1600–H1608. [Google Scholar] [CrossRef] [PubMed]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Symes, J.F.; Fishman, M.C.; et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Investig. 1998, 101, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Hotten, S.; Schodel, J.; Rutzel, S.; Hu, K.; Widder, J.; Marx, A.; Huang, P.L.; Ertl, G. Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Tsutsui, M.; Shimokawa, H.; Horiuchi, M.; Tanimoto, A.; Suda, O.; Tasaki, H.; Huang, P.L.; Sasaguri, Y.; Yanagihara, N.; et al. Vasculoprotective roles of neuronal nitric oxide synthase. FASEB J. 2002, 16, 1994–1996. [Google Scholar] [CrossRef] [PubMed]
- Detmers, P.A.; Hernandez, M.; Mudgett, J.; Hassing, H.; Burton, C.; Mundt, S.; Chun, S.; Fletcher, D.; Card, D.J.; Lisnock, J.; et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein e-deficient mice. J. Immunol. 2000, 165, 3430–3435. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Chen, J.; Han, F.; Astern, J.; Huang, P.L. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein e-knockout mice. Circulation 2001, 103, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Dimayuga, P.; Zhu, J.; Nilsson, J.; Kaul, S.; Shah, P.K.; Cercek, B. Decreased neointimal thickening after arterial wall injury in inducible nitric oxide synthase knockout mice. Circ. Res. 1999, 85, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Sennlaub, F.; Courtois, Y.; Goureau, O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J. Clin. Investig. 2001, 107, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, P.; Buys, E.; Sips, P.; Pokreisz, P.; Marsboom, G.; Gillijns, H.; Pellens, M.; Dewerchin, M.; Bloch, K.D.; Brouckaert, P.; et al. Gender-specific modulation of the response to arterial injury by soluble guanylate cyclase alpha1. Open Cardiovasc. Med. J. 2009, 3, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bettaga, N.; Jager, R.; Dunnes, S.; Groneberg, D.; Friebe, A. Cell-specific impact of nitric oxide-dependent guanylyl cyclase on arteriogenesis and angiogenesis in mice. Angiogenesis 2015, 18, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Aicher, A.; Heeschen, C.; Feil, S.; Hofmann, F.; Mendelsohn, M.E.; Feil, R.; Dimmeler, S. cGMP-dependent protein kinase I is crucial for angiogenesis and postnatal vasculogenesis. PLoS ONE 2009, 4, e4879. [Google Scholar] [CrossRef] [PubMed]
- Yamahara, K.; Itoh, H.; Chun, T.H.; Ogawa, Y.; Yamashita, J.; Sawada, N.; Fukunaga, Y.; Sone, M.; Yurugi-Kobayashi, T.; Miyashita, K.; et al. Significance and therapeutic potential of the natriuretic peptides/cGMP/cGMP-dependent protein kinase pathway in vascular regeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 3404–3409. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.Y.; Chen, C.Y.; Zhao, J.F.; Su, K.H.; Lee, H.T.; Lin, S.J.; Shyue, S.K.; Hsiao, S.H.; Lee, T.S. Activation of soluble guanylyl cyclase prevents foam cell formation and atherosclerosis. Acta Physiol. 2014, 210, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Fukumura, D.; Jain, R.K. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol. Med. 2004, 10, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Sinnaeve, P.; Chiche, J.D.; Gillijns, H.; Van Pelt, N.; Wirthlin, D.; Van De Werf, F.; Collen, D.; Bloch, K.D.; Janssens, S. Overexpression of a constitutively active protein kinase g mutant reduces neointima formation and in-stent restenosis. Circulation 2002, 105, 2911–2916. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Chang, W.C.; Chang, G.Y.; Kuo, S.C.; Teng, C.M. The inhibitory mechanism of YC-1, a benzyl indazole, on smooth muscle cell proliferation: An in vitro and in vivo study. J. Pharmacol. Sci. 2004, 94, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Tulis, D.A.; Durante, W.; Peyton, K.J.; Chapman, G.B.; Evans, A.J.; Schafer, A.I. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem. Biophys. Res. Commun. 2000, 279, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, K.; Tarcea, V.; Pali, S.; Barnucz, E.; Gwanmesia, P.N.; Korkmaz, S.; Radovits, T.; Loganathan, S.; Merkely, B.; Karck, M.; et al. Cinaciguat prevents neointima formation after arterial injury by decreasing vascular smooth muscle cell migration and proliferation. Int. J. Cardiol. 2013, 167, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Hoenicka, M.; Schmid, C. Cardiovascular effects of modulators of soluble guanylyl cyclase activity. Cardiovasc. Hematol. Agents Med. Chem. 2008, 6, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; McLean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of p-selectin expression and leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Namba, T.; Koike, H.; Murakami, K.; Aoki, M.; Makino, H.; Hashiya, N.; Ogihara, T.; Kaneda, Y.; Kohno, M.; Morishita, R. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation 2003, 108, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, A.; Smith, R.D.; Khitha, J.; Arora, N.; Veerareddy, S.; Langston, W.; Chidlow, J.H., Jr.; Barlow, S.C.; Teng, X.; Patel, R.P.; et al. Sildenafil promotes ischemia-induced angiogenesis through a PKG-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Fomin, N.; Krawutschke, C.; Russwurm, M.; Feil, R. Visualization of cGMP with cGi biosensors. Methods Mol. Biol. 2013, 1020, 89–120. [Google Scholar] [PubMed]
- Nikolaev, V.O.; Lohse, M.J. Novel techniques for real-time monitoring of cGMP in living cells. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 229–243. [Google Scholar]
- Russwurm, M.; Mullershausen, F.; Friebe, A.; Jager, R.; Russwurm, C.; Koesling, D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: A systematic approach. Biochem. J. 2007, 407, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Wen, L.; Hillenbrand, M.; Vachaviolos, A.; Feil, S.; Ott, T.; Han, X.; Fukumura, D.; Jain, R.K.; Russwurm, M.; et al. Transgenic mice for cGMP imaging. Circ. Res. 2013, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Mohrle, D.; Reimann, K.; Wolter, S.; Wolters, M.; Varakina, K.; Mergia, E.; Eichert, N.; Geisler, H.S.; Sandner, P.; Ruth, P.; et al. NO-sensitive guanylate cyclase isoforms NO-GC1 and NO-GC2 contribute to noise-induced inner hair cell synaptopathy. Mol. Pharmacol. 2017, 92, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Shuhaibar, L.C.; Egbert, J.R.; Norris, R.P.; Lampe, P.D.; Nikolaev, V.O.; Thunemann, M.; Wen, L.; Feil, R.; Jaffe, L.A. Intercellular signaling via cyclic GMP diffusion through gap junctions restarts meiosis in mouse ovarian follicles. Proc. Natl. Acad. Sci. USA 2015, 112, 5527–5532. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Schmidt, K.; de Wit, C.; Han, X.; Jain, R.K.; Fukumura, D.; Feil, R. Correlative intravital imaging of cGMP signals and vasodilation in mice. Front. Physiol. 2014, 5, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.; Peters, S.; Frank, K.; Wen, L.; Feil, R.; Rathjen, F.G. Dorsal root ganglion axon bifurcation tolerates increased cyclic GMP levels: The role of phosphodiesterase 2A and scavenger receptor Npr3. Eur. J. Neurosci. 2016, 44, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Schorg, B.F.; Feil, S.; Lin, Y.; Voelkl, J.; Golla, M.; Vachaviolos, A.; Kohlhofer, U.; Quintanilla-Martinez, L.; Olbrich, M.; et al. Cre/lox-assisted non-invasive in vivo tracking of specific cell populations by positron emission tomography. Nat. Commun. 2017, 8, 444. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Smolenski, A.; Lohmann, S.M.; Kukreja, R.C. Cyclic GMP-dependent protein kinase ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J. Biol. Chem. 2006, 281, 38644–38652. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Muraski, J.; Chen, Y.; Tsujita, Y.; Wall, J.; Glembotski, C.C.; Schaefer, E.; Beckerle, M.; Sussman, M.A. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J. Clin. Investig. 2005, 115, 2716–2730. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, B.; Feil, R.; Hofmann, F.; Willenbockel, C.; Drexler, H.; Smolenski, A.; Lohmann, S.M.; Wollert, K.C. cGMP-dependent protein kinase type I inhibits TAB1-p38 mitogen-activated protein kinase apoptosis signaling in cardiac myocytes. J. Biol. Chem. 2006, 281, 32831–32840. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Volker, K.; Gabetaner, B.; Bayer, B.; Abebetaer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase i. Eur. Heart J. 2013, 34, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Kobsar, A.; Heeg, S.; Krohne, K.; Opitz, A.; Walter, U.; Bock, M.; Gambaryan, S.; Eigenthaler, M. Cyclic nucleotide-regulated proliferation and differentiation vary in human hematopoietic progenitor cells derived from healthy persons, tumor patients, and chronic myelocytic leukemia patients. Stem Cells Dev. 2008, 17, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; Fiscus, R.R. Essential roles of the nitric oxide (NO)/cGMP/protein kinase G type-Iα (PKG-Iα) signaling pathway and the atrial natriuretic peptide (ANP)/cGMP/PKG-Iα autocrine loop in promoting proliferation and cell survival of OP9 bone marrow stromal cells. J. Cell. Biochem. 2011, 112, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cGKI-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rukoyatkina, N.; Walter, U.; Friebe, A.; Gambaryan, S. Differentiation of cGMP-dependent and -independent nitric oxide effects on platelet apoptosis and reactive oxygen species production using platelets lacking soluble guanylyl cyclase. Thromb. Haemost. 2011, 106, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, H.; Schwappacher, R.; Marathe, N.; Zhuang, S.; Casteel, D.E.; Haas, B.; Chen, Y.; Pfeifer, A.; Kato, H.; Shattil, S.; et al. Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci. Signal. 2010, 3, ra91. [Google Scholar] [CrossRef] [PubMed]
- Jaumann, M.; Dettling, J.; Gubelt, M.; Zimmermann, U.; Gerling, A.; Paquet-Durand, F.; Feil, S.; Wolpert, S.; Franz, C.; Varakina, K.; et al. cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat. Med. 2012, 18, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Dhayade, S.; Kaesler, S.; Sinnberg, T.; Dobrowinski, H.; Peters, S.; Naumann, U.; Liu, H.; Hunger, R.E.; Thunemann, M.; Biedermann, T.; et al. Sildenafil potentiates a cGMP-dependent pathway to promote melanoma growth. Cell Rep. 2016, 14, 2599–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Qureshi, A.A.; Robinson, K.C.; Han, J. Sildenafil use and increased risk of incident melanoma in US men: A prospective cohort study. JAMA Intern. Med. 2014, 174, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Loeb, S.; Folkvaljon, Y.; Lambe, M.; Robinson, D.; Garmo, H.; Ingvar, C.; Stattin, P. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA 2015, 313, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Sanchez-Laorden, B.; Packer, L.; Hidalgo-Carcedo, C.; Hayward, R.; Viros, A.; Sahai, E.; Marais, R. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5a. Cancer Cell 2011, 19, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Feil, R. Viagra releases the brakes on melanoma growth. Mol. Cell. Oncol. 2017, 4, e1188874. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Mouse Model | Effect of Mutation on Vascular Remodeling | References |
|---|---|---|---|
| eNOS | Null mutation | Enhanced atherosclerosis on ApoE−/− 1 background | [92,93,94] |
| Enhanced neointima formation after vascular injury | [95,96,97] | ||
| Impaired angiogenesis | [98,99,100] | ||
| nNOS | Null mutation | Enhanced atherosclerosis on ApoE−/− background | [101] |
| Enhanced neointima formation after vascular injury | [102] | ||
| iNOS | Null mutation | Reduced atherosclerosis on ApoE−/− background | [103,104] |
| Reduced neointima formation after vascular injury | [105] | ||
| Reduced pathological neovascularization in the ischemic retina | [106] | ||
| NO-GCα1-subunit | Null mutation | Reduced atherosclerosis on ApoE−/− background | [50] |
| Reduced neointima formation after vascular injury in male mice | [107] | ||
| NO-GCβ1-subunit | Smooth muscle-specific knockout (tamoxifen-inducible) | Reduced arteriogenesis in hindlimb ischemia model | [108] |
| Null mutation | Reduced arteriogenesis in hindlimb ischemia model | [108] | |
| cGKI | Smooth muscle-specific knockout (tamoxifen-inducible) | Reduced atherosclerosis on ApoE−/− background | [35] |
| Smooth muscle-specific knockout | No effect on neointima formation after vascular injury | [51] | |
| Null mutation | Reduced angiogenesis | [109,110] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehners, M.; Dobrowinski, H.; Feil, S.; Feil, R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. J. Cardiovasc. Dev. Dis. 2018, 5, 20. https://doi.org/10.3390/jcdd5020020
Lehners M, Dobrowinski H, Feil S, Feil R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. Journal of Cardiovascular Development and Disease. 2018; 5(2):20. https://doi.org/10.3390/jcdd5020020
Chicago/Turabian StyleLehners, Moritz, Hyazinth Dobrowinski, Susanne Feil, and Robert Feil. 2018. "cGMP Signaling and Vascular Smooth Muscle Cell Plasticity" Journal of Cardiovascular Development and Disease 5, no. 2: 20. https://doi.org/10.3390/jcdd5020020