
Comparative Transcriptomic Profiling and Gene Expression for Myxomatous Mitral Valve Disease in the Dog and Human

{kind=link}
Abstract
1. Introduction
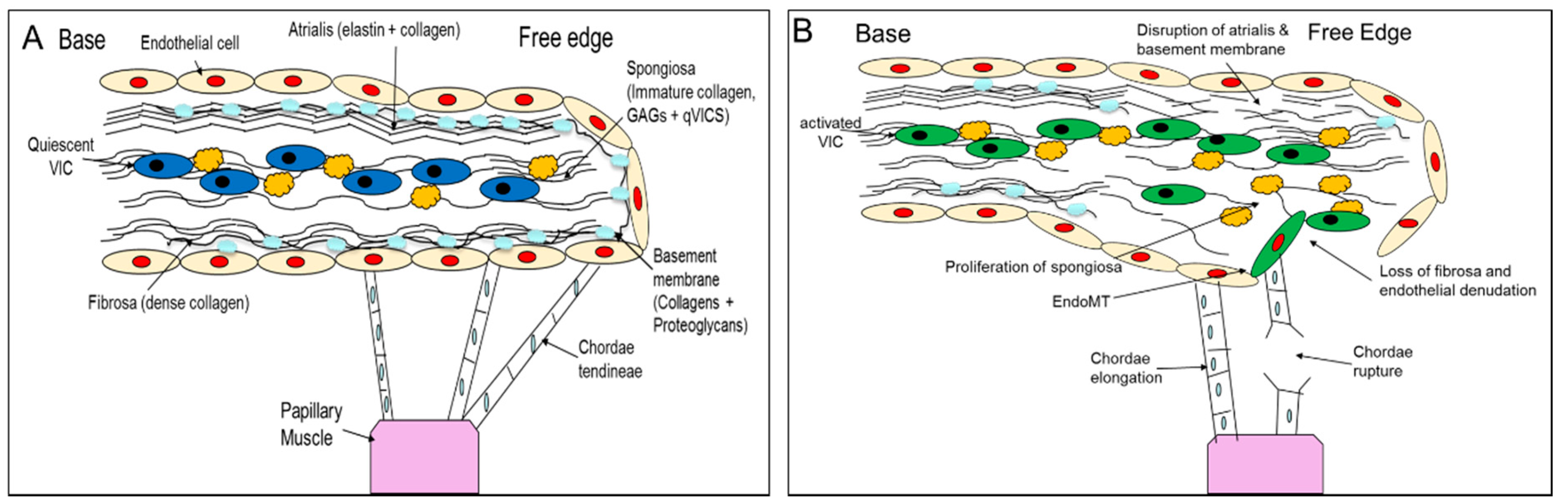
2. Comparative Pathology
3. Gene Changes in Canine and Human MMVD
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
Abbreviations
| MMVD | Myxomatous mitral valve disease |
| RT-PCR | Real time polymerase chain reaction |
| VEC | Valvular endothelial cell |
| LAMA | Laminin |
| NID | Nidogen |
| GAG | Glycosaminoglycan |
| VIC | Valvular interstitial cell |
| ECM | Extracellular matrix |
| FED | Fibroelastic deficiency |
| MVP | Mitral valve prolapse |
| FDR | False discovery rate |
| COL | Collagen |
| LUM | Lumican |
| VCAN | Versican |
| MMP | Matrix-metalloproteinase |
| TIMP | Tissue inhibitor of metalloproteinase |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| EndoMT | Endothelial-to-mesenchymal transition |
| α-SMA | Alpha-smooth muscle actin |
| CD31 | Platelet endothelial cell adhesion molecule 1 |
| CDH5 | Vascular endothelial cadherin |
| HAS2 | Hyaluronic synthase 2 |
| IHC | Immunohistochemistry |
| TGF-β | Transforming growth factor–β |
| SMAD | Suppressor of mothers against decapentaplegic |
| BMP | Bone morphogenetic proteins |
| COMP | Cartilage oligomeric matrix protein |
| ENG | Endoglin |
| TGFBI | TGF-β-induced |
| CTGF | Connective tissue growth factor |
| 5-HT | 5-hydroxytryptamine |
| TPH1 | Tryptophan hydroxylase 1 |
| AGT2 | Angiotensin 2 |
| AT1 | AGT2 type 1 receptor |
| ADAMTS | A Disintegrin And Metalloproteinase with Thrombospondin Motifs |
| SLRP | Small leucine-rich proteoglycans |
| CHAD | Chondroadherin |
| KERA | Keratocan |
| HAPLN1 | Hyaluronan and proteoglycan link protein 1 |
| TNF-α | Tumor necrosis factor α |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| LDLR | Lipoprotein receptor |
| HSP | Heat shock proteins |
| ROS | Reactive oxygen species |
| NOX | Nicotinamide adenine dinucleotide phosphate-oxidase |
| SOD | Superoxide dismutase |
| ncRNAs | Non-coding RNAs |
| miRs | MicroRNAs |
References
- Connell, P.S.; Han, R.I.; Grande-Allen, K.J. Differentiating the aging of the mitral valve from human and canine myxomatous degeneration. J. Vet. Cardiol. 2012, 14, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Greenhouse, D.G.; Murphy, A.; Mignatti, P.; Zavadil, J.; Galloway, A.C.; Balsam, L.B. Mitral valve prolapse is associated with altered extracellular matrix gene expression patterns. Gene 2016, 586, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Freed, L.A.; Levy, D.; Levine, R.A.; Larson, M.G.; Evans, J.C.; Fuller, D.L.; Lehman, B.; Benjamin, E.J. Prevalence and clinical outcome of mitral-valve prolapse. N. Engl. J. Med. 1999, 341, 1–7. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, J.L.; Prendergast, B.D.; Chambers, J.B.; Ray, S.G.; Bridgewater, B. Valvular heart disease: The next cardiac epidemic. Heart 2011, 97, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.D.; Haggstrom, J. Mitral valve prolapse in the dog: A model of mitral valve prolapse in man. Cardiovasc. Res. 2000, 47, 234–243. [Google Scholar] [CrossRef]
- Aupperle, H.; Disatian, S. Pathology, protein expression and signaling in myxomatous mitral valve degeneration: Comparison of dogs and humans. J. Vet. Cardiol. 2012, 14, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Hulin, A.; Deroanne, C.; Lambert, C.; Defraigne, J.O.; Nusgens, B.; Radermecker, M.; Colige, A. Emerging pathogenic mechanisms in human myxomatous mitral valve: Lessons from past and novel data. Cardiovasc. Pathol. 2013, 22, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Liu, M.M.; Culshaw, G.; Clinton, M.; Argyle, D.J.; Corcoran, B.M. Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: A microarray study. Vet. J. 2015, 204, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.A.; Chittur, S.V. Genomic expression patterns of mitral valve tissues from dogs with degenerative mitral valve disease. Am. J. Vet. Res. 2006, 67, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Thalji, N.M.; Hagler, M.A.; Zhang, H.; Casaclang-Verzosa, G.; Nair, A.A.; Suri, R.M.; Miller, J.D. Nonbiased Molecular Screening Identifies Novel Molecular Regulators of Fibrogenic and Proliferative Signaling in Myxomatous Mitral Valve Disease. Circ. Cardiovasc. Genet. 2015, 8, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Freeman, L.M.; Rush, J.E.; Huggins, G.S.; Kennedy, A.D.; Labuda, J.A.; Laflamme, D.P.; Hannah, S.S. Veterinary Medicine and Multi-Omics Research for Future Nutrition Targets: Metabolomics and Transcriptomics of the Common Degenerative Mitral Valve Disease in Dogs. OMICS 2015, 19, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Yutzey, K.E. Heart Valve Structure and Function in Development and Disease. Ann. Rev. Physiol. 2011, 73, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.A.; Hagege, A.A.; Judge, D.P.; Padala, M.; Dal-Bianco, J.P.; Aikawa, E.; Beaudoin, J.; Bischoff, J.; Bouatia-Naji, N.; Bruneval, P.; et al. Mitral valve disease—Morphology and mechanisms. Nat. Rev. Cardiol. 2015, 12, 689–710. [Google Scholar] [CrossRef] [PubMed]
- Disatian, S.; Ehrhart, E.J., 3rd; Zimmerman, S.; Orton, E.C. Interstitial cells from dogs with naturally occurring myxomatous mitral valve disease undergo phenotype transformation. J. Heart Valve Dis. 2008, 17, 402–411. [Google Scholar] [PubMed]
- Han, R.I.; Black, A.; Culshaw, G.J.; French, A.T.; Else, R.W.; Corcoran, B.M. Distribution of myofibroblasts, smooth muscle-like cells, macrophages, and mast cells in mitral valve leaflets of dogs with myxomatous mitral valve disease. Am. J. Vet. Res. 2008, 69, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, B.M.; Black, A.; Anderson, H.; McEwan, J.D.; French, A.; Smith, P.; Devine, C. Identification of surface morphologic changes in the mitral valve leaflets and chordae tendineae of dogs with myxomatous degeneration. Am. J. Vet. Res. 2004, 65, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, E.; Aikawa, M.; Stone, J.R.; Fukumoto, Y.; Libby, P.; Schoen, F.J. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 2001, 104, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Black, A.; French, A.T.; Dukes-McEwan, J.; Corcoran, B.M. Ultrastructural morphologic evaluation of the phenotype of valvular interstitial cells in dogs with myxomatous degeneration of the mitral valve. Am. J. Vet. Res. 2005, 66, 1408–1414. [Google Scholar] [CrossRef]
- Han, R.I.; Clark, C.H.; Black, A.; French, A.; Culshaw, G.J.; Kempson, S.A.; Corcoran, B.M. Morphological changes to endothelial and interstitial cells and to the extra-cellular matrix in canine myxomatous mitral valve disease (endocardiosis). Vet. J. 2013, 197, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.R. Pathology of myxomatous mitral valve disease in the dog. J. Vet. Cardiol. 2012, 14, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.C.; Vowels, T.J.; Ko, J.M.; Hebeler, R.F. Gross and Histological Features of Excised Portions of Posterior Mitral Leaflet in Patients Having Operative Repair of Mitral Valve Prolapse and Comments on the Concept of Missing (= Ruptured) Chordae Tendineae. J. Am. Coll. Cardiol. 2014, 63, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Han, R.I.; Black, A.; Culshaw, G.; French, A.T.; Corcoran, B.M. Structural and cellular changes in canine myxomatous mitral valve disease: An image analysis study. J. Heart Valve Dis. 2010, 19, 60–70. [Google Scholar]
- Hadian, M.; Corcoran, B.M.; Bradshaw, J.P. Molecular changes in fibrillar collagen in myxomatous mitral valve disease. Cardiovasc. Pathol. 2010, 19, e141–148. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grande-Allen, K.J.; Griffin, B.P.; Ratliff, N.B.; Cosgrove, D.M.; Vesely, I. Glycosaminoglycan myxomatous mitral profiles of leaflets and chordae parallel the severity of mechanical alterations. J. Am. Coll. Cardiol. 2003, 42, 271–277. [Google Scholar] [CrossRef]
- Gupta, V.; Barzilla, J.E.; Mendez, J.S.; Stephens, E.H.; Lee, E.L.; Collard, C.D.; Laucirica, R.; Weigel, P.H.; Grande-Allen, K.J. Abundance and location of proteoglycans and hyaluronan within normal and myxomatous mitral valves. Cardiovasc. Pathol. 2009, 18, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lester, W.M. Myxomatous mitral valve disease and related entities: The role of matrix in valvular heart disease. Cardiovasc. Pathol. 1995, 4, 257–264. [Google Scholar] [CrossRef]
- Davies, M.J.; Moore, B.P.; Braimbridge, M.V. The floppy mitral valve. Study of incidence, pathology, and complications in surgical, necropsy, and forensic material. Br. Heart J. 1978, 40, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.W. Chronic valvular disease (endocardiosis) in dogs. Adv. Vet. Sci. Comp. Med. 1977, 21, 75–106. [Google Scholar] [PubMed]
- McDonald, P.C.; Wilson, J.E.; McNeill, S.; Gao, M.; Spinelli, J.J.; Rosenberg, F.; Wiebe, H.; McManus, B.M. The challenge of defining normality for human mitral and aortic valves: Geometrical and compositional analysis. Cardiovasc. Pathol. 2002, 11, 193–209. [Google Scholar] [CrossRef]
- Matsumaru, I.; Eishi, K.; Hashizume, K.; Kawano, H.; Tsuneto, A.; Hayashi, T. Clinical and pathological features of degenerative mitral valve disease: Billowing mitral leaflet versus fibroelastic deficiency. Ann. Thorac. Cardiovasc. Surg. 2014, 20, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Anyanwu, A.C.; Adams, D.H. Etiologic classification of degenerative mitral valve disease: Barlow’s disease and fibroelastic deficiency. Semin. Thorac. Cardiovasc. Surg. 2007, 19, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Han, R.I.; Impoco, G.; Culshaw, G.; French, A.T.; Black, A.; Corcoran, B.M. Cell maceration scanning electron microscopy and computer-derived porosity measurements in assessment of connective tissue microstructure changes in the canine myxomatous mitral valve. Vet. J. 2013, 197, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Sainger, R.; Grau, J.B.; Branchetti, E.; Poggio, P.; Seefried, W.F.; Field, B.C.; Acker, M.A.; Gorman, R.C.; Gorman, J.H., 3rd; Hargrove, C.W., 3rd; et al. Human myxomatous mitral valve prolapse: Role of bone morphogenetic protein 4 in valvular interstitial cell activation. J. Cell. Physiol. 2012, 227, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Wang, C.H.; Riddle, J.M.; Sabbah, H.N.; Magilligan, D.J., Jr.; Hawkins, E.T. Scanning electron microscopy of operatively excised severely regurgitant floppy mitral valves. Am. J. Cardiol. 1989, 64, 392–394. [Google Scholar] [CrossRef]
- Mirzaie, M.; Meyer, T.; Schwarz, P.; Lotfi, S.; Rastan, A.; Schondube, F. Ultrastructural alterations in acquired aortic and mitral valve disease as revealed by scanning and transmission electron microscopical investigations. Ann. Thorac Cardiovasc Surg. 2002, 8, 24–30. [Google Scholar] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking About? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Liu, M.M.; Clinton, M.; Culshaw, G.; Argyle, D.J.; Corcoran, B.M. Developmental pathways and endothelial to mesenchymal transition in canine myxomatous mitral valve disease. Vet. J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dal-Bianco, J.P.; Aikawa, E.; Bischoff, J.; Guerrero, J.L.; Handschumacher, M.D.; Sullivan, S.; Johnson, B.; Titus, J.S.; Iwamoto, Y.; Wylie-Sears, J.; et al. Active Adaptation of the Tethered Mitral Valve Insights Into a Compensatory Mechanism for Functional Mitral Regurgitation. Circulation 2009, 120, 334. [Google Scholar] [CrossRef] [PubMed]
- Hagler, M.A.; Hadley, T.M.; Zhang, H.; Mehra, K.; Roos, C.M.; Schaff, H.V.; Suri, R.M.; Miller, J.D. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc. Res. 2013, 99, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Mow, T.; Pedersen, H.D. Increased endothelin-receptor density in myxomatous canine mitral valve leaflets. J. Cardiovasc. Pharmacol. 1999, 34, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.H.; Mortensen, K.; Martinussen, T.; Larsson, L.I.; Baandrup, U.; Pedersen, H.D. Increased NADPH-diaphorase activity in canine myxomatous mitral valve leaflets. J. Comp. Pathol. 2003, 129, 120–130. [Google Scholar] [CrossRef]
- Kogure, K. Pathology of chronic mitral valvular disease in the dog. Nihon Juigaku Zasshi. 1980, 42, 323–329, 335. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Liu, M.M.; Culshaw, G.; French, A.; Corcoran, B. Comparison of cellular changes in Cavalier King Charles spaniel and mixed breed dogs with myxomatous mitral valve disease. J. Vet. Cardiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hulin, A.; Deroanne, C.F.; Lambert, C.A.; Dumont, B.; Castronovo, V.; Defraigne, J.O.; Nusgens, B.V.; Radermecker, M.A.; Colige, A.C. Metallothionein-dependent up-regulation of TGF-2 participates in the remodelling of the myxomatous mitral valve. Cardiovascular Res. 2012, 93, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, H.; Marz, I.; Thielebein, J.; Schoon, H.A. Expression of transforming growth factor-beta1, -beta2 and -beta3 in normal and diseased canine mitral valves. J. Comp. Pathol. 2008, 139, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, H.; Thielebein, J.; Kiefer, B.; Marz, I.; Dinges, G.; Schoon, H.A.; Schubert, A. Expression of Genes Encoding Matrix Metalloproteinases (MMPs) and their Tissue Inhibitors (TIMPs) in Normal and Diseased Canine Mitral Valves. J. Comp. Pathol. 2009, 140, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Herpin, A.; Lelong, C.; Favrel, P. Transforming growth factor-beta-related proteins: An ancestral and widespread superfamily of cytokines in metazoans. Dev. Comp. Immunol. 2004, 28, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Horbelt, D.; Denkis, A.; Knaus, P. A portrait of Transforming Growth Factor beta superfamily signalling: Background matters. Int. J. Biochem. Cell. Biol. 2012, 44, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Pohlers, D.; Brenmoehl, J.; Loffler, I.; Muller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-beta and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Moesgaard, S.G.; Aupperle, H.; Rajamaki, M.M.; Falk, T.; Rasmussen, C.E.; Zois, N.E.; Olsen, L.H. Matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs) and transforming growth factor-beta (TGF-beta) in advanced canine myxomatous mitral valve disease. Res. Vet. Sci. 2014, 97, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Obayashi, K.; Miyagawa-Tomita, S.; Matsumoto, H.; Koyama, H.; Nakanishi, T.; Hirose, H. Effects of transforming growth factor-beta 3 and matrix metalloproteinase-3 on the pathogenesis of chronic mitral valvular disease in dogs. Am. J. Vet. Res. 2011, 72, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.S.; Kornreich, B.G.; Gould, R.A.; Moise, N.S.; Butcher, J.T. Interactions between TGFbeta1 and cyclic strain in modulation of myofibroblastic differentiation of canine mitral valve interstitial cells in 3D culture. J. Vet. Cardiol. 2012, 14, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cushing, M.C.; Liao, J.T.; Anseth, K.S. Activation of valvular interstitial cells is mediated by transforming growth factor-beta 1 interactions with matrix molecules. Matrix Biol. 2005, 24, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Disatian, S.; Orton, E.C. Autocrine serotonin and transforming growth factor beta 1 signaling mediates spontaneous myxomatous mitral valve disease. J. Heart Valve Dis. 2009, 18, 44–51. [Google Scholar] [PubMed]
- Connolly, J.M.; Bakay, M.A.; Fulmer, J.T.; Gorman, R.C.; Gorman, J.H., 3rd; Oyama, M.A.; Levy, R.J. Fenfluramine disrupts the mitral valve interstitial cell response to serotonin. Am. J. Pathol. 2009, 175, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Oyama, M.A.; Levy, R.J. Insights into Serotonin Signaling Mechanisms Associated with Canine Degenerative Mitral Valve Disease. J. Vet. Intern. Med. 2010, 24, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Therap. 2009, 123, 255–278. [Google Scholar]
- Atkins, C.E.; Keene, B.W.; Brown, W.A.; Coats, J.R.; Crawford, M.A.; DeFrancesco, T.C.; Edwards, N.J.; Fox, P.R.; Lehmkuhl, L.B.; Luethy, M.W.; et al. Results of the veterinary enalapril trial to prove reduction in onset of heart failure in dogs chronically treated with enalapril alone for compensated, naturally occurring mitral valve insufficiency. J. Am. Vet. Med. Assoc. 2007, 231, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Geirsson, A.; Singh, M.; Ali, R.; Abbas, H.; Li, W.; Sanchez, J.A.; Hashim, S.; Tellides, G. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation 2012, 126, S189–S197. [Google Scholar] [CrossRef] [PubMed]
- Jaffre, F.; Bonnin, P.; Callebert, J.; Debbabi, H.; Setola, V.; Doly, S.; Monassier, L.; Mettauer, B.; Blaxall, B.C.; Launay, J.M.; et al. Serotonin and angiotensin receptors in cardiac fibroblasts coregulate adrenergic-dependent cardiac hypertrophy. Circ. Res. 2009, 104, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Diaz, N.; Tandon, I.; Plate, R.; Martindale, C.; Balachandran, K. Elevated Serotonin Interacts with Angiotensin-II to Result in Altered Valve Interstitial Cell Contractility and Remodeling. Cardiovasc. Eng. Technol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, H.; Marz, I.; Thielebein, J.; Dinges, G.; Schoon, H.A. Histomorphological findings and expression of matrix metalloproteinases and their tissue specific inhibitors (TIMPs) in normal tricuspid valves and in chronic tricuspid valvular disease in dogs. Vet. J. 2010, 183, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Togashi, M.; Tamura, K.; Nitta, T.; Ishizaki, M.; Sugisaki, Y.; Fukuda, Y. Role of matrix metalloproteinases and their tissue inhibitor of metalloproteinases in myxomatous change of cardiac floppy valves. Pathol. Int. 2007, 57, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Aupperle, H.; Thielebein, J.; Kiefer, B.; Marz, I.; Dinges, G.; Schoon, H.A. An immunohistochemical study of the role of matrix metalloproteinases and their tissue inhibitors in chronic mitral valvular disease (valvular endocardiosis) in dogs. Vet. J. 2009, 180, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [PubMed]
- Dupuis, L.E.; McCulloch, D.R.; McGarity, J.D.; Bahan, A.; Wessels, A.; Weber, D.; Diminich, A.M.; Nelson, C.M.; Apte, S.S.; Kern, C.B. Altered versican cleavage in ADAMTS5 deficient mice; a novel etiology of myxomatous valve disease. Dev. Biol. 2011, 357, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.B.; Wessels, A.; McGarity, J.; Dixon, L.J.; Alston, E.; Argraves, W.S.; Geeting, D.; Nelson, C.M.; Menick, D.R.; Apte, S.S. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010, 29, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Grande-Allen, K.J.; Calabro, A.; Gupta, V.; Wight, T.N.; Hascall, V.C.; Vesely, I. Glycosaminoglycans and proteoglycans in normal mitral valve leaflets and chordae: Association with regions of tensile and compressive loading. Glycobiology 2004, 14, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.A.; Wang, L.W.; Bader, H.; Ho, J.C.; Majors, A.K.; Hollyfield, J.G.; Traboulsi, E.I.; Apte, S.S. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Hadian, M.; Corcoran, B.M.; Han, R.I.; Grossmann, J.G.; Bradshaw, J.P. Collagen organization in canine myxomatous mitral valve disease: An X-ray diffraction study. Biophys. J. 2007, 93, 2472–2476. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.T.; Lim, T.K.; Richards, A.M.; Kofidis, T.; Teoh, K.L.; Ling, L.H.; Chung, M.C. Unravelling the proteome of degenerative human mitral valves. Proteomics 2015, 15, 2934–2944. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, M.A.; Limet, R.; Lapiere, C.M.; Nusgens, B. Increased mRNA expression of decorin in the prolapsing posterior leaflet of the mitral valve. Interact. Cardiovasc. Thorac. Surg. 2003, 2, 389–394. [Google Scholar] [CrossRef]
- Chen, Y.T.; Wang, J.; Wee, A.S.; Yong, Q.W.; Tay, E.L.; Woo, C.C.; Sorokin, V.; Richards, A.M.; Ling, L.H. Differential MicroRNA Expression Profile in Myxomatous Mitral Valve Prolapse and Fibroelastic Deficiency Valves. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markby, G.R.; Summers, K.M.; MacRae, V.E.; Corcoran, B.M. Comparative Transcriptomic Profiling and Gene Expression for Myxomatous Mitral Valve Disease in the Dog and Human. Vet. Sci. 2017, 4, 34. https://doi.org/10.3390/vetsci4030034
Markby GR, Summers KM, MacRae VE, Corcoran BM. Comparative Transcriptomic Profiling and Gene Expression for Myxomatous Mitral Valve Disease in the Dog and Human. Veterinary Sciences. 2017; 4(3):34. https://doi.org/10.3390/vetsci4030034
Chicago/Turabian StyleMarkby, Greg R., Kim M. Summers, Vicky E. MacRae, and Brendan M. Corcoran. 2017. "Comparative Transcriptomic Profiling and Gene Expression for Myxomatous Mitral Valve Disease in the Dog and Human" Veterinary Sciences 4, no. 3: 34. https://doi.org/10.3390/vetsci4030034
APA StyleMarkby, G. R., Summers, K. M., MacRae, V. E., & Corcoran, B. M. (2017). Comparative Transcriptomic Profiling and Gene Expression for Myxomatous Mitral Valve Disease in the Dog and Human. Veterinary Sciences, 4(3), 34. https://doi.org/10.3390/vetsci4030034