
Pathogen and Host Response Dynamics in a Mouse Model of Borrelia hermsii Relapsing Fever

Abstract
:1. Introduction
1.1. Early Animal Experiments
1.2. The North American Species B. hermsii
1.3. Overview
2. Materials and Methods
2.1. Animals
2.2. Bacterial Strains and Culture Conditions
2.3. Mouse Infections
2.4. Antibody Agglutination Assays
2.5. Growth Inhibition Assay
2.6. Indirect Immunofluorescence Assay (IFA)
2.7. Enzyme-Linked Immunosorbent (ELISA) Assay for IgM Antibodies
2.8. Assays for Cytokines, Chemokines, and Other Serum Components
2.9. Iron Assay
2.10. In Vitro Blood Cell Aggregation Assay
2.11. Nucleic Acid Extraction and cDNA Synthesis
2.12. Quantitative PCR (qPCR)
2.13. Growth Rate Determinations
2.14. Statistics
3. Results and Discussion
3.1. Growth of B. hermsii in the Blood
3.1.1. Background
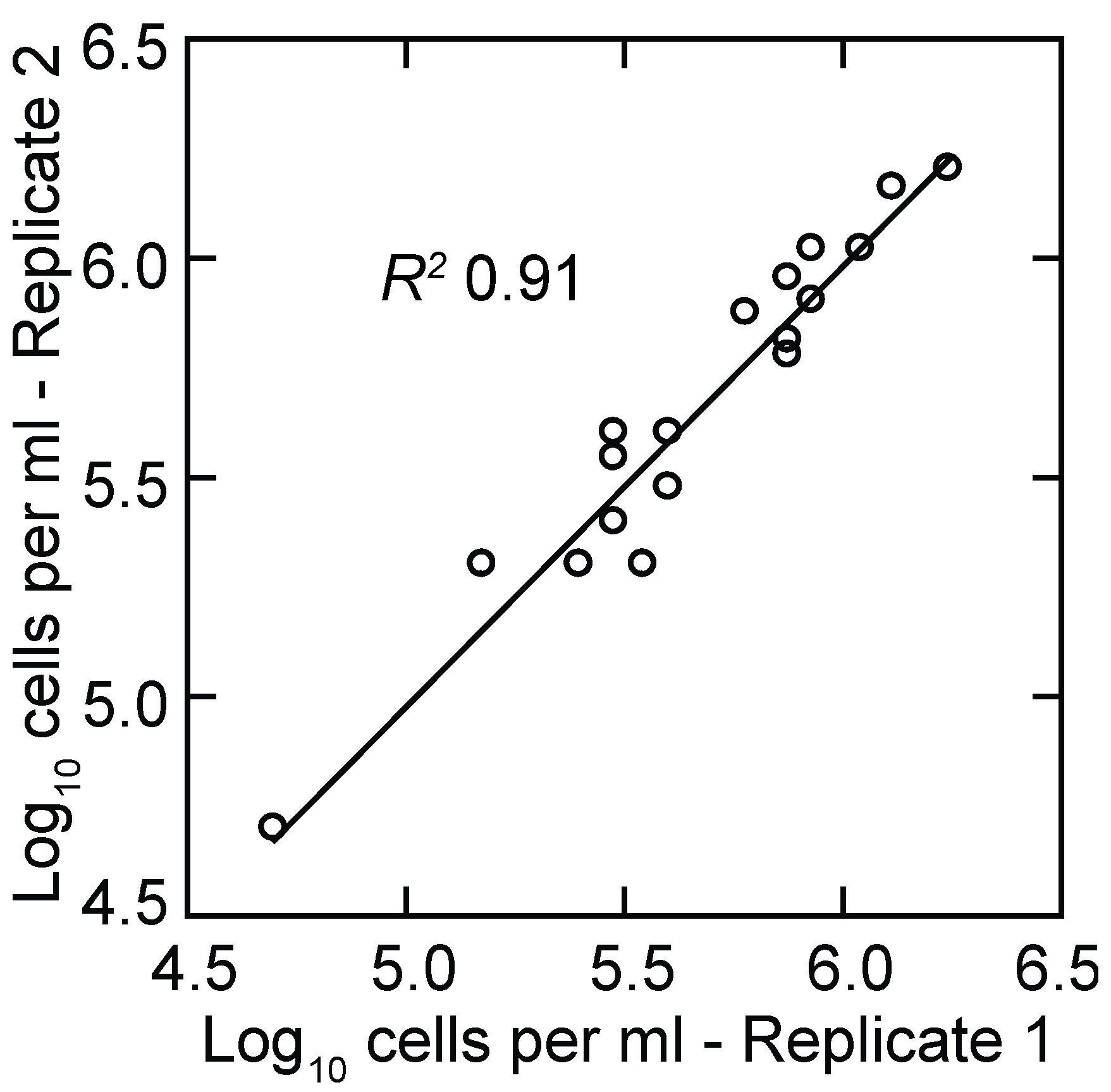
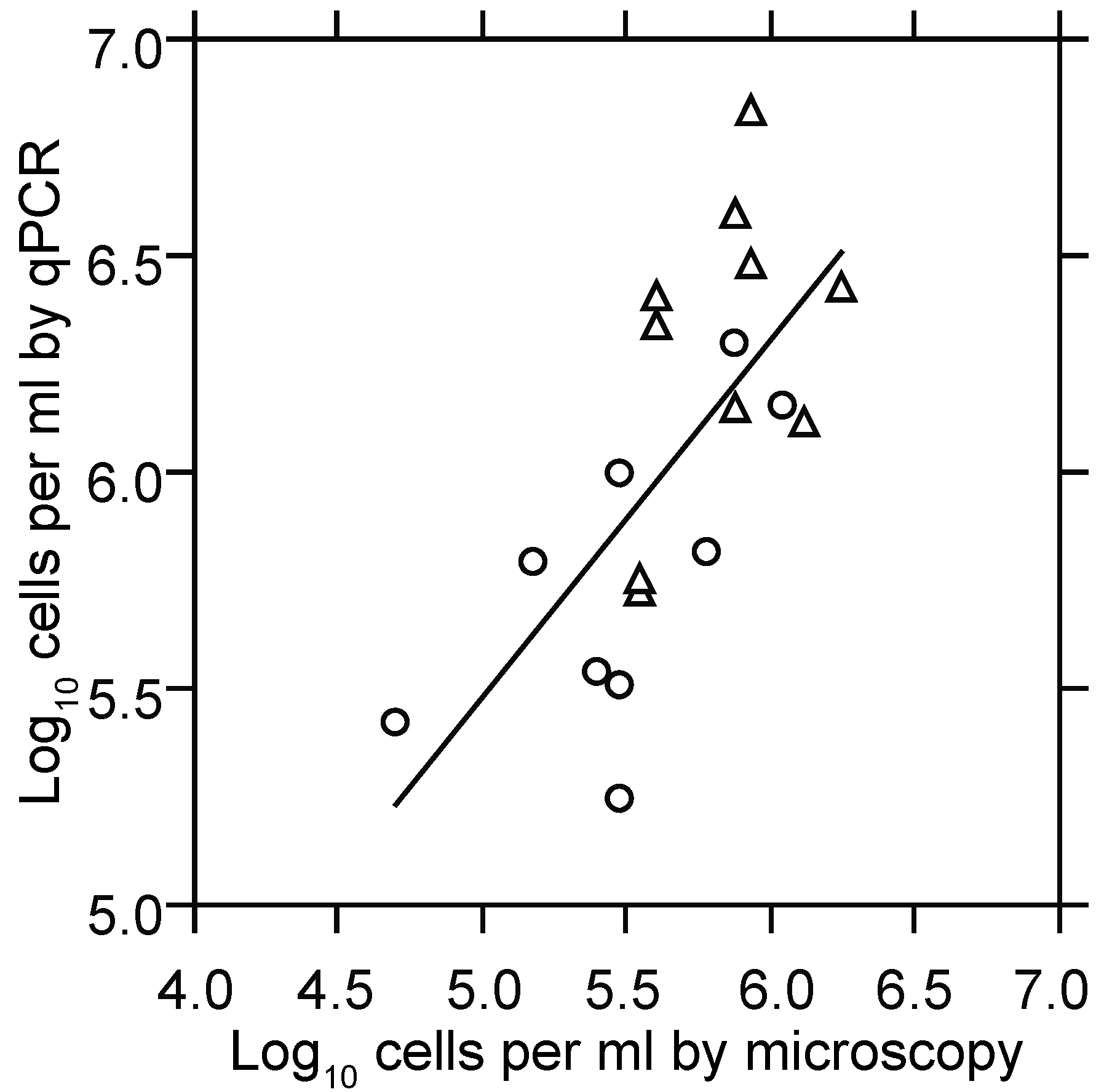
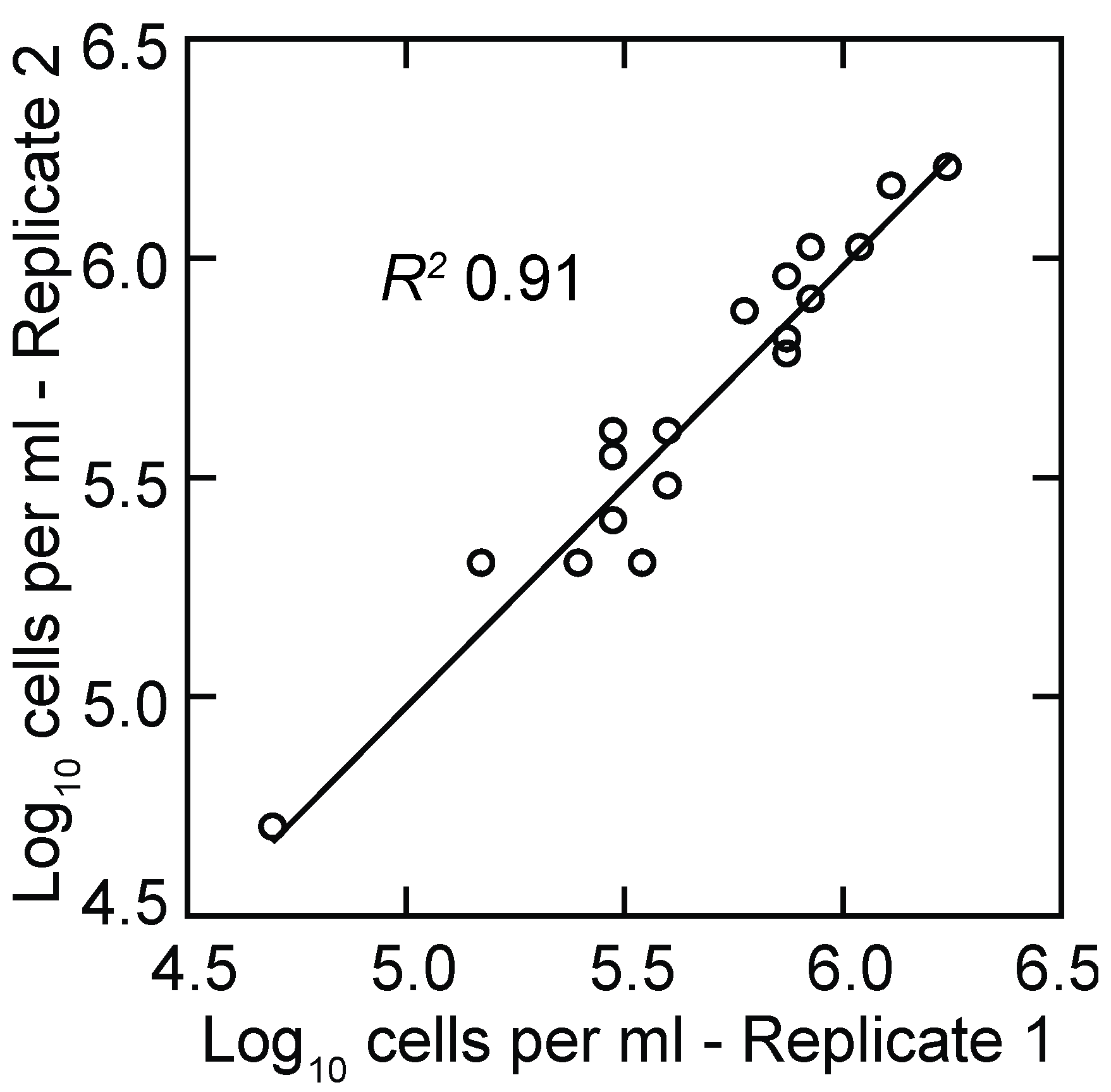
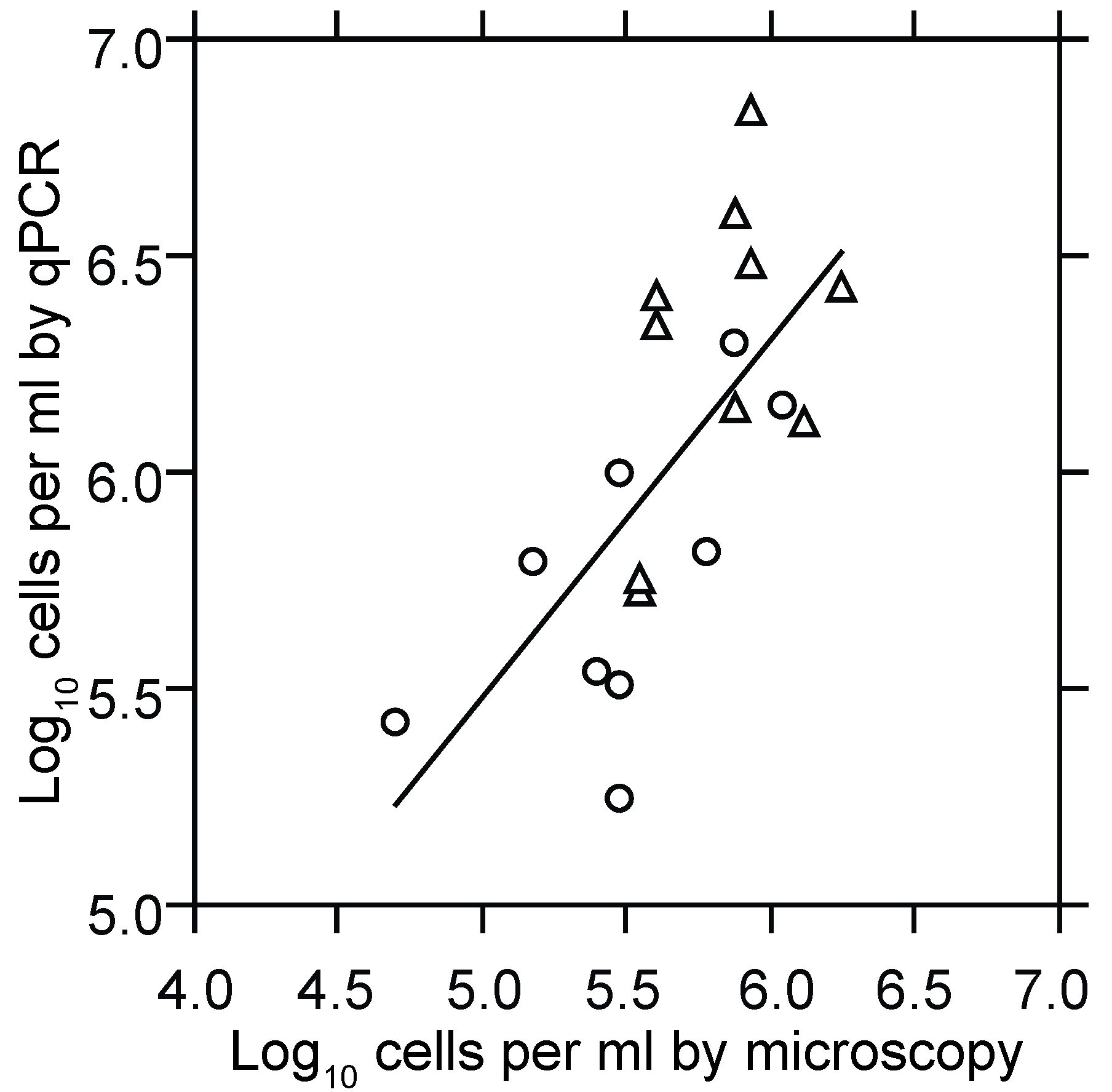
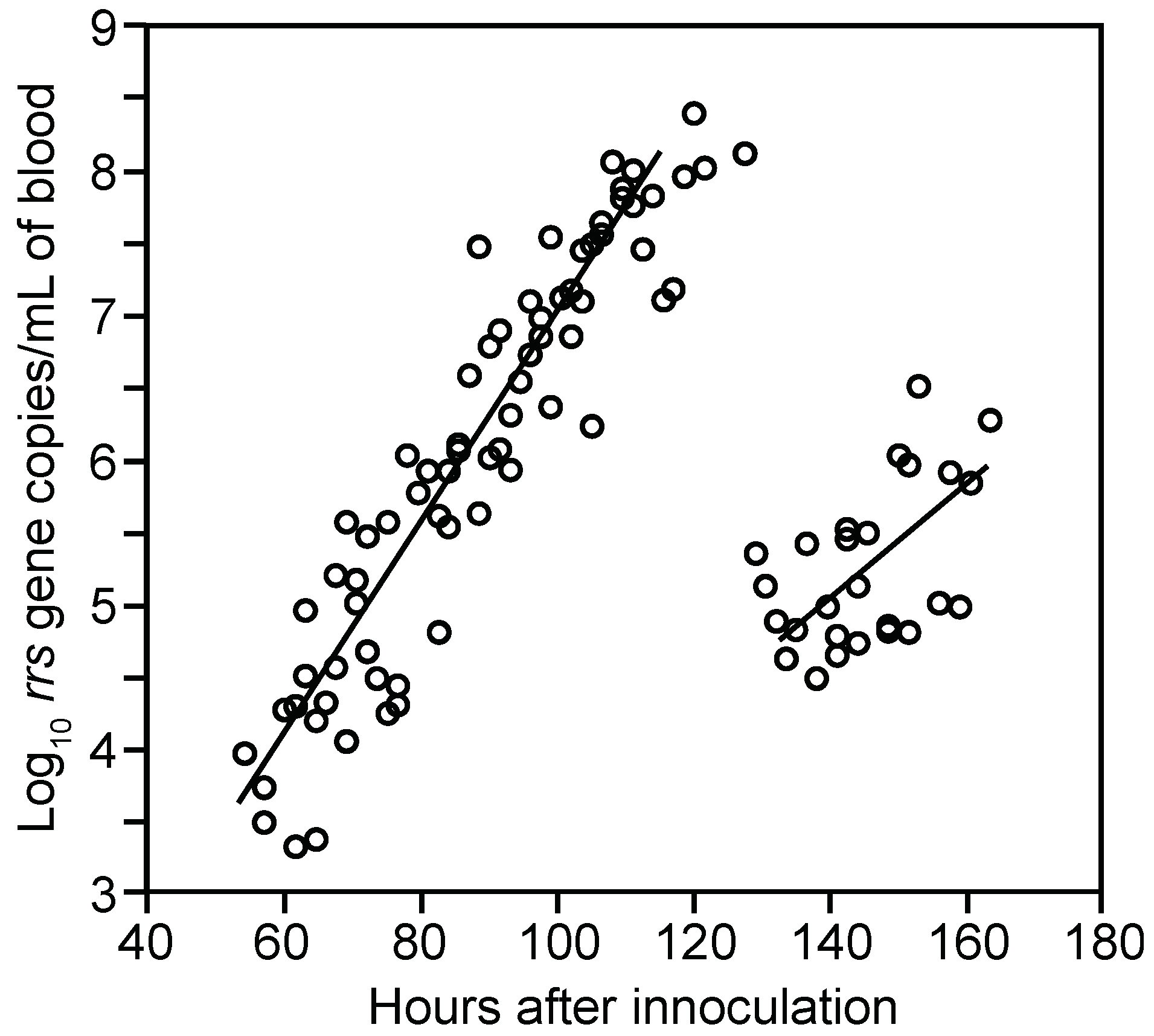
3.1.2. PCR Quantitation of Bacteria in the Blood
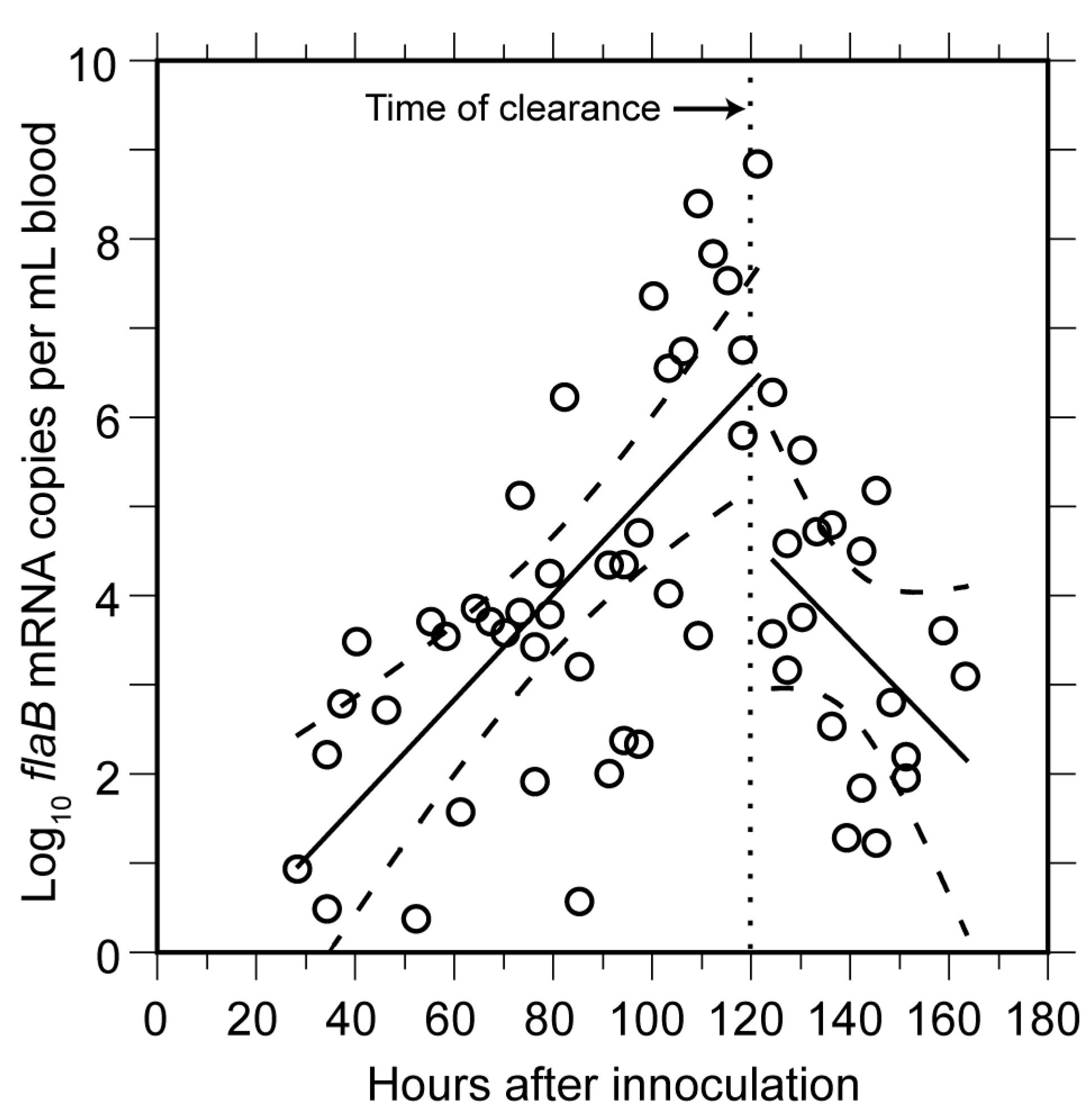
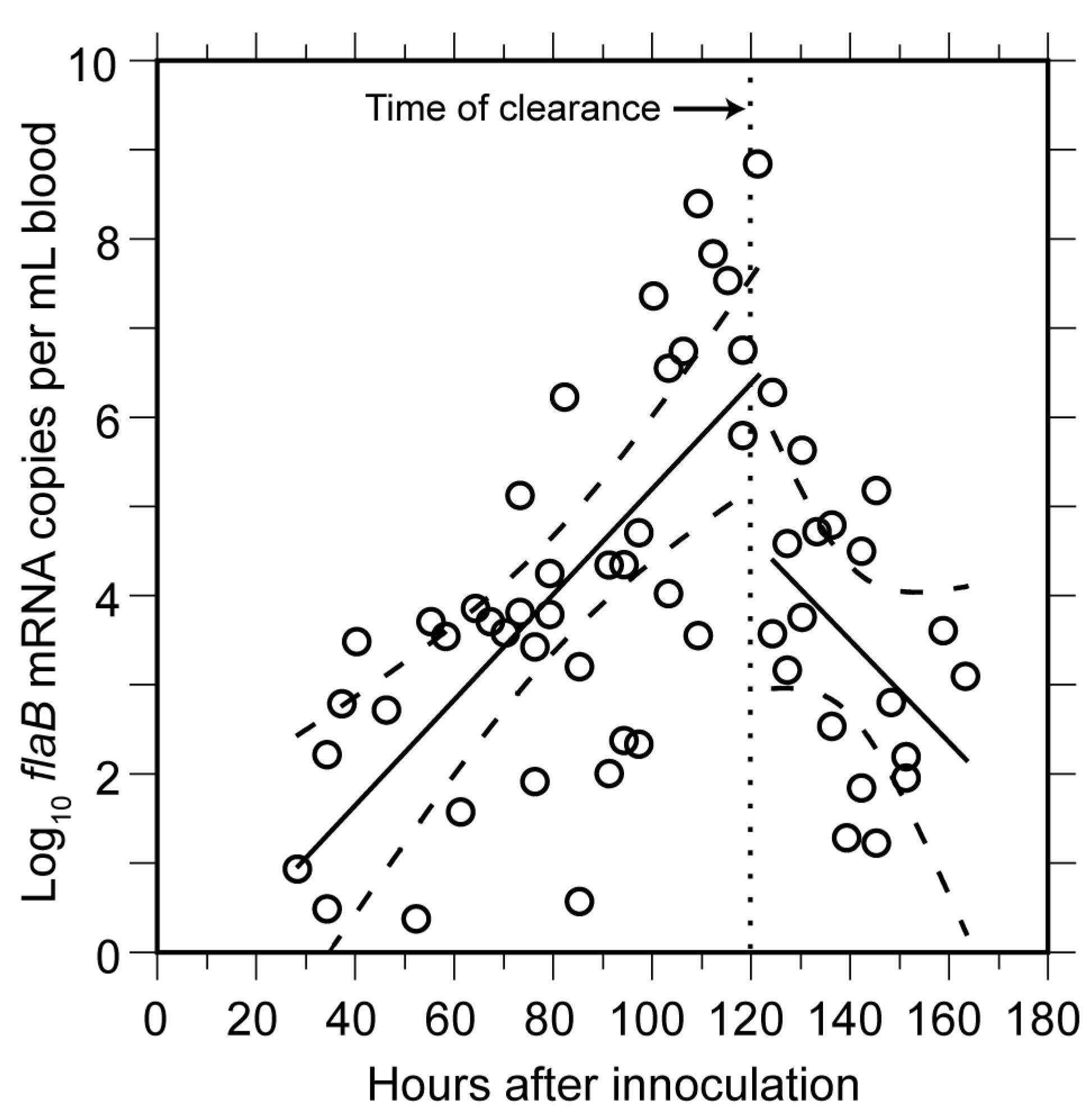
3.1.3. Clearance of Bacteria from Blood
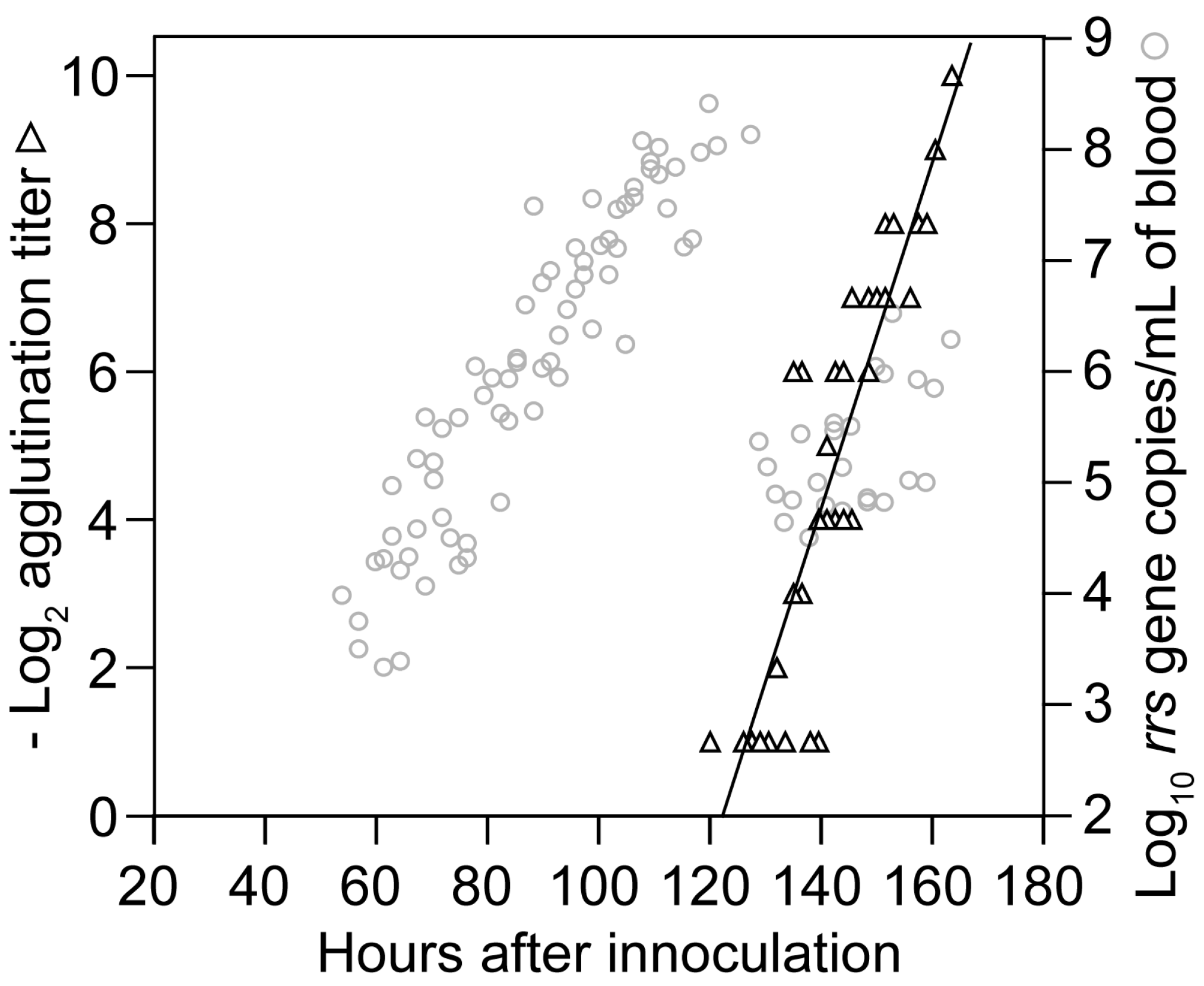
3.1.4. Relapse of Bacteria in the Blood
3.1.5. Comments
3.2. Innate Host Responses to B. hermsii Infection
3.2.1. Background
3.2.2. Analysis of Proteins in Plasma during Experimental Infection
3.2.3. Comments
3.3. Antibody Responses to B. hermsii Infection
3.3.1. Background
3.3.2. Agglutination Assays
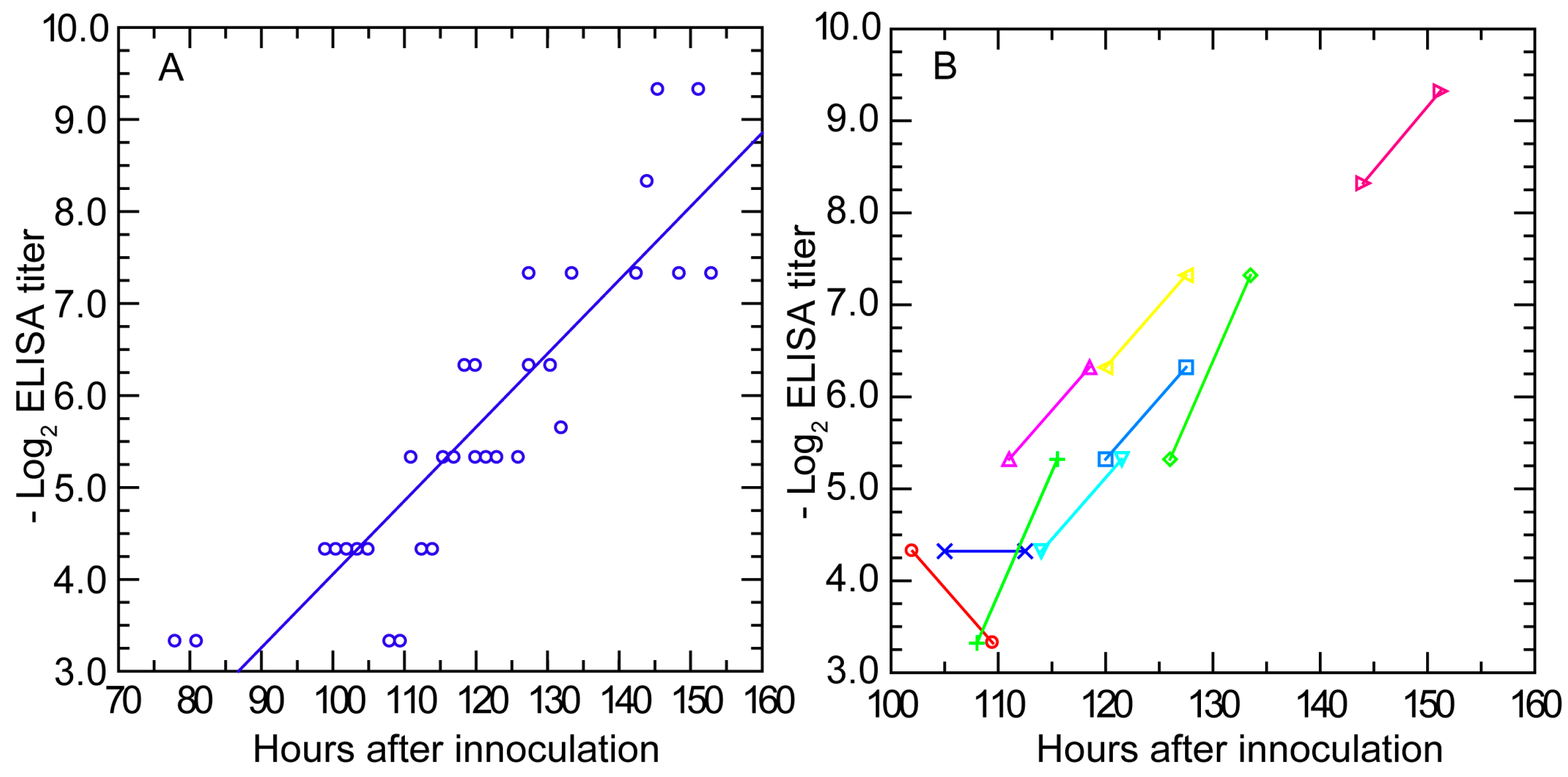
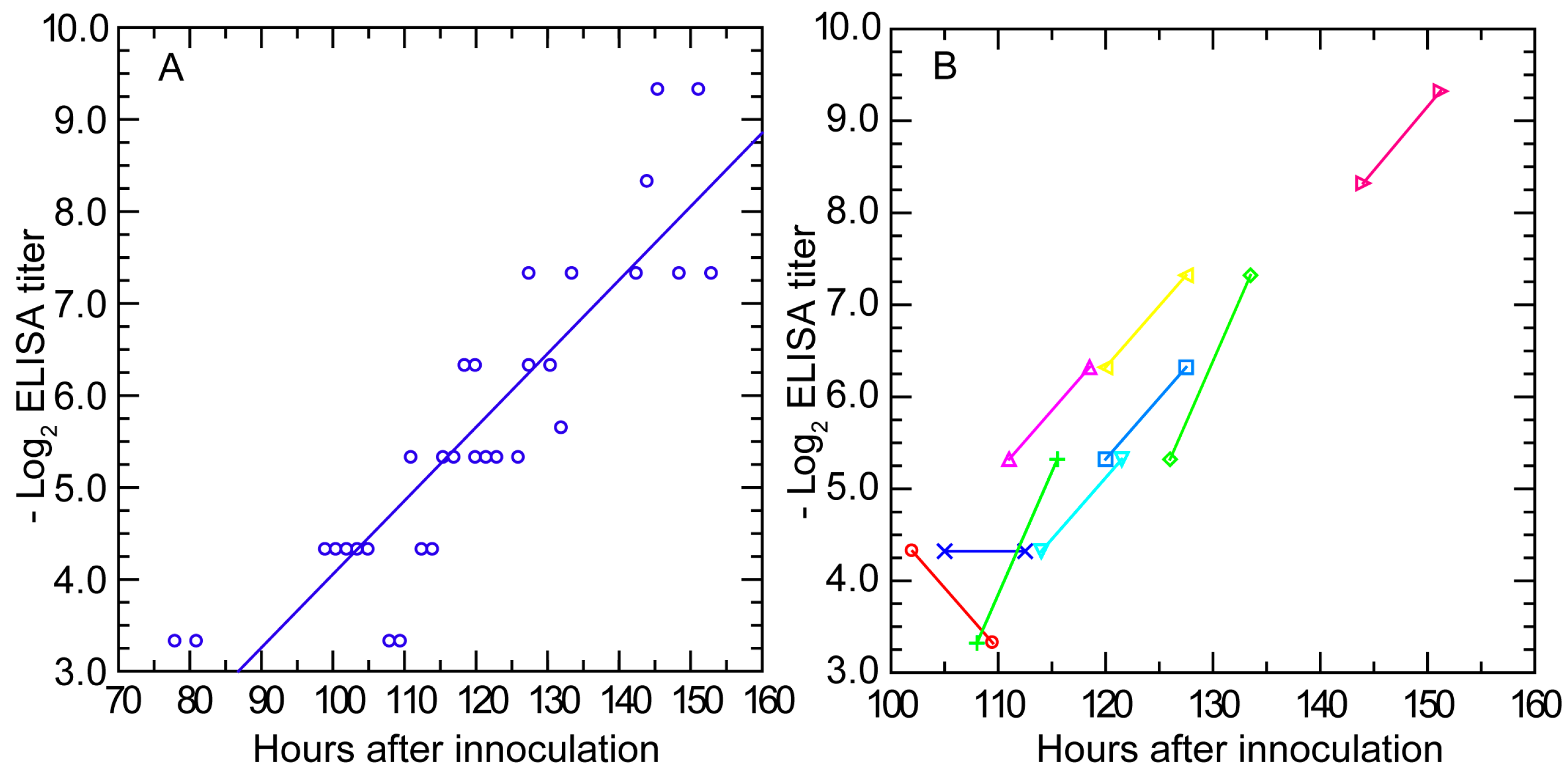
3.3.3. Antibody Response Detected by ELISA and IFA
3.3.4. Antibody Response in Nude Mice
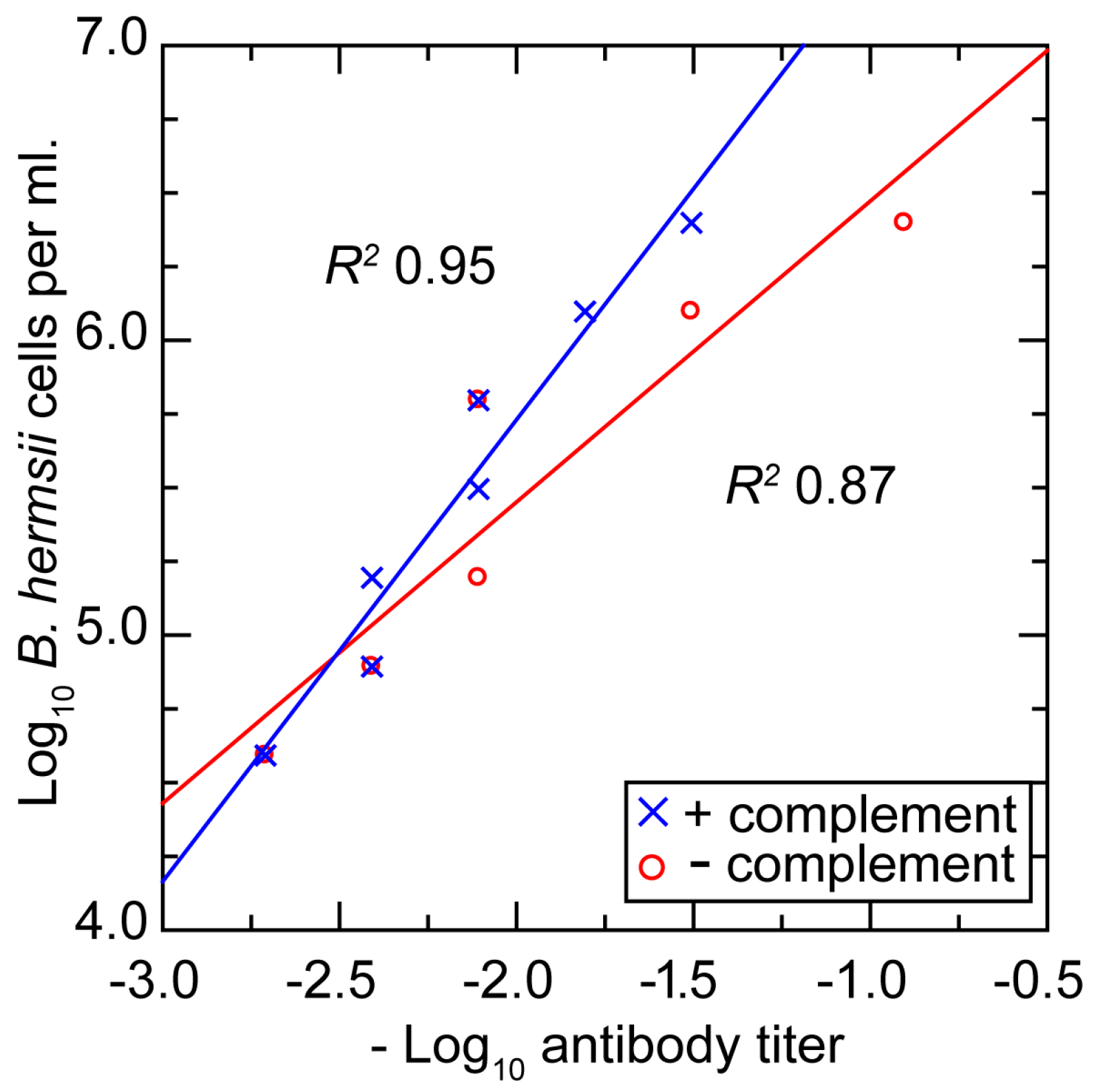
3.3.5. In Vitro Neutralization with a Monoclonal Antibody
3.3.6. Comments
3.4. Anemia during Infection
3.4.1. Background
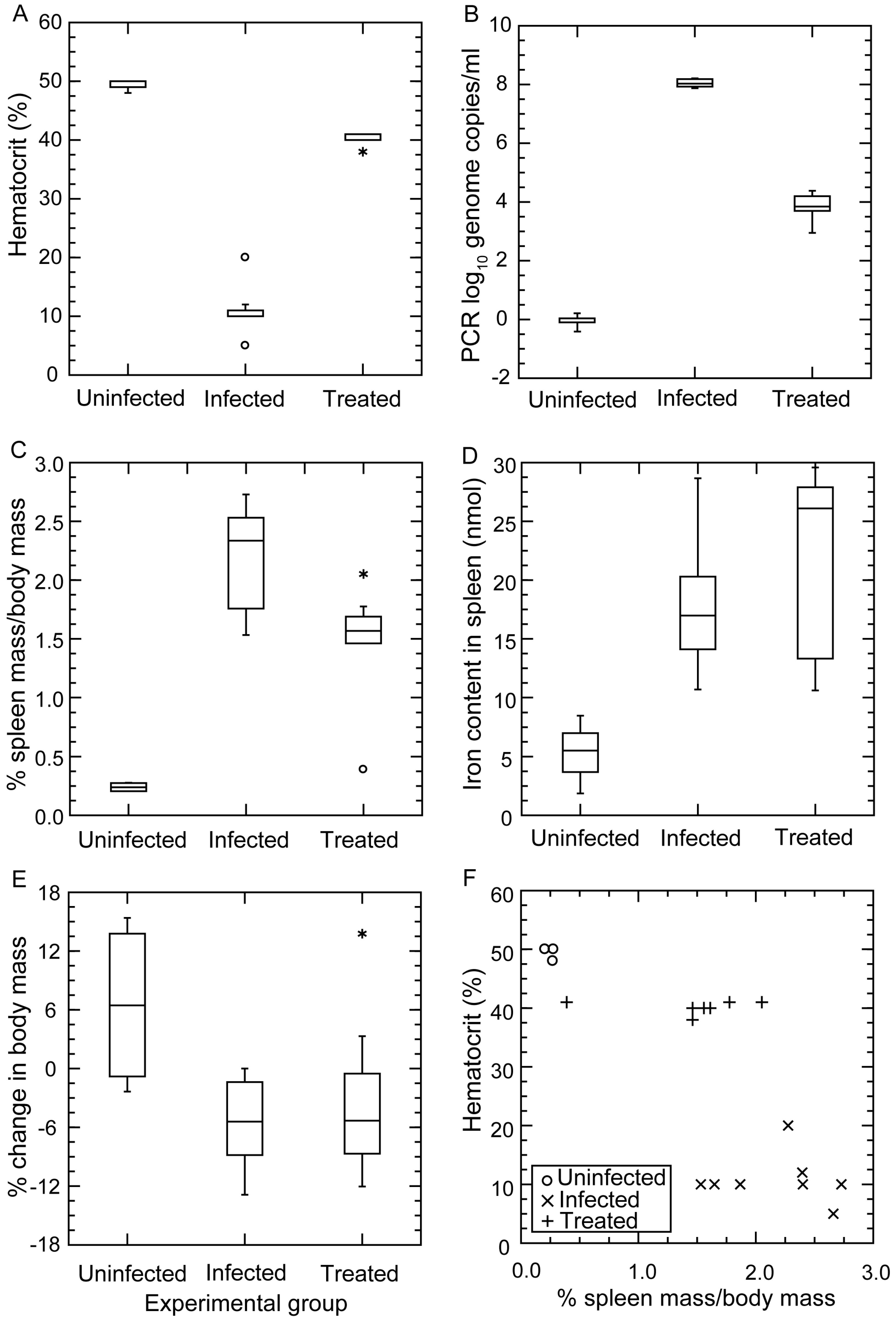
3.4.2. Changes in Hematocrit during Infection and after Antibiotic Treatment
3.4.3. Histopathology
3.4.4. Comments
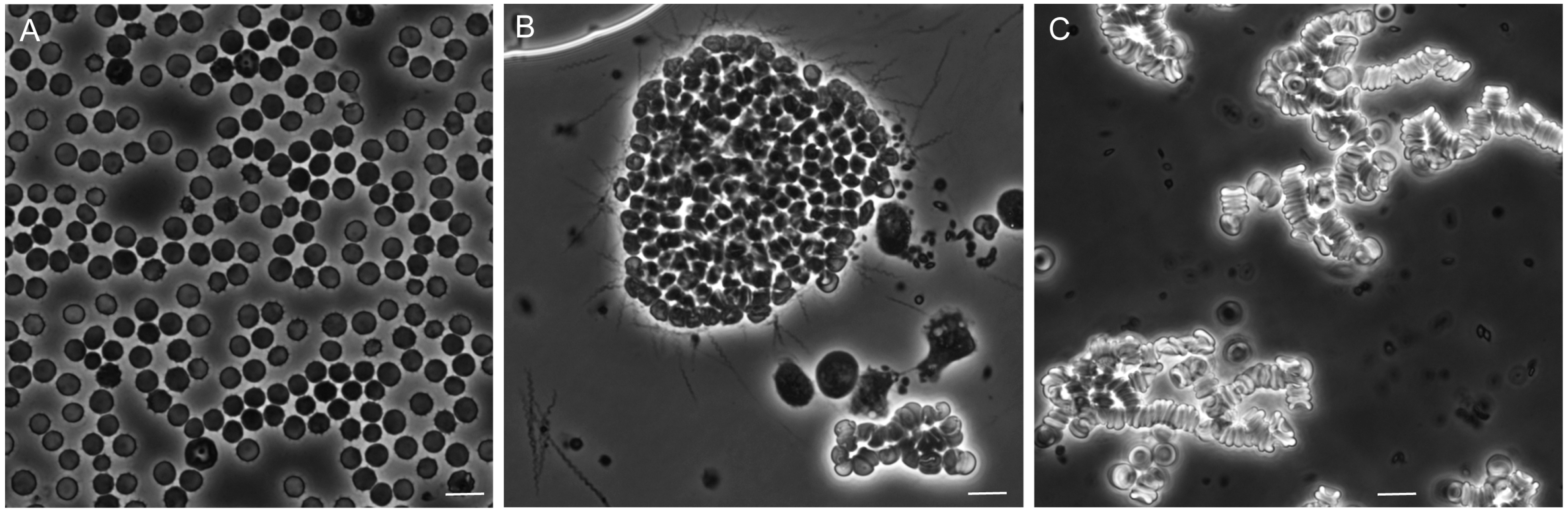
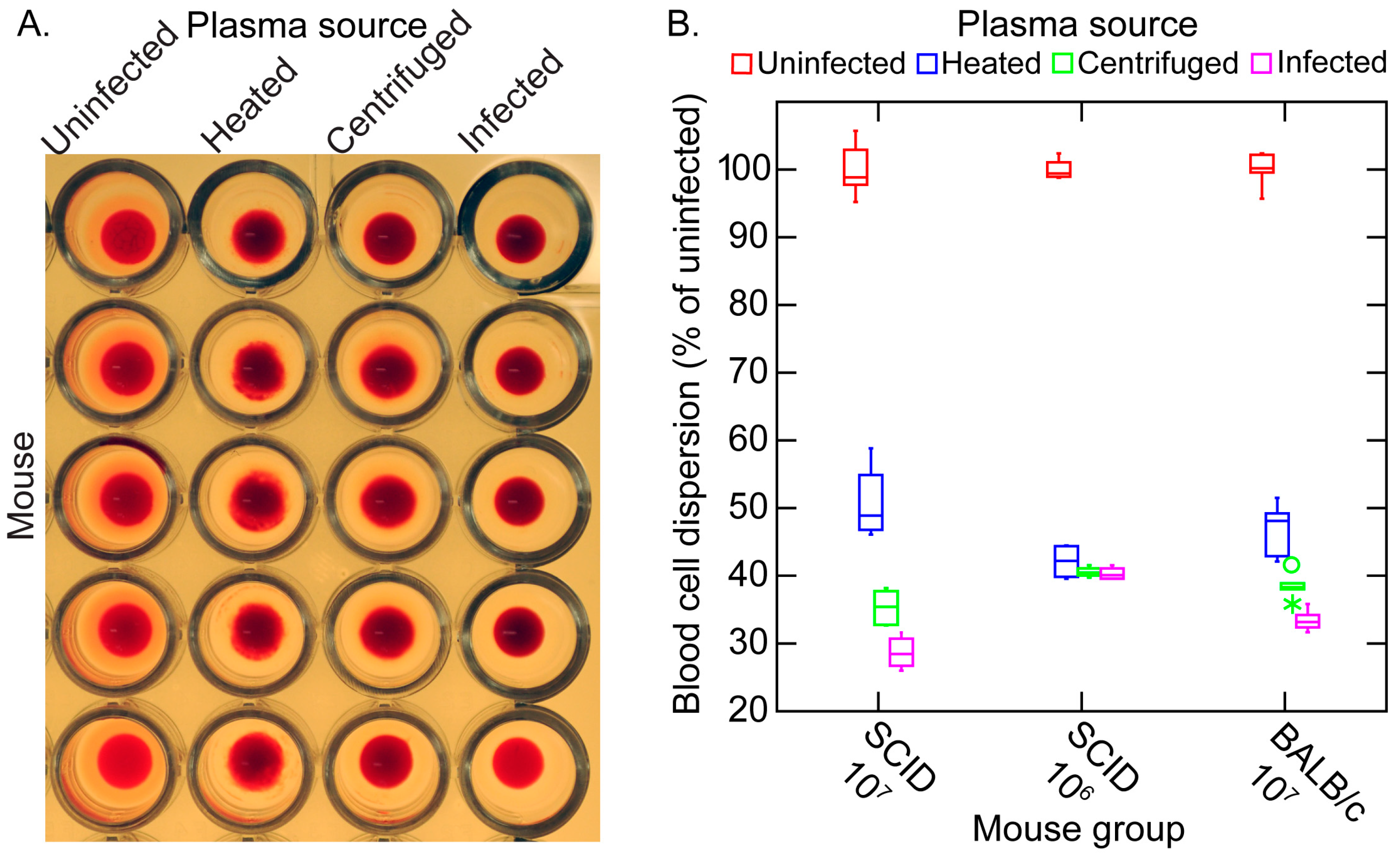
3.5. Erythrocyte Aggregation during Infection
3.5.1. Background
3.5.2. In Vitro Aggregation Experiment
3.5.3. Comments
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| J-H | Jarisch-Herxheimer |
| IFA | indirect immunofluorescence assay |
| PBS | phosphate-buffered saline |
| qPCR | quantitative polymerase chain reaction |
| SCID | severe combined immunodeficient |
| TLR | Toll-like receptor |
References
- Obermeier, O. Vorkommen feinster, eine eigenbewegung zeigender fäden im Blute von recurrenskranken, vorläufige mittheilung. Centrablatt Med. Wiss. 1873, 10, 145–147. (In German) [Google Scholar]
- Fitz, R.H. Report on pathology and pathological anatomy. Boston Med. Surg. J. 1873, 88, 393–397. [Google Scholar] [CrossRef]
- Cutler, S.J.; Abdissa, A.; Trape, J.F. New concepts for the old challenge of African relapsing fever borreliosis. Clin. Microbiol. Infect. 2009, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Motschutkoffsky, D. Experimentelle studien iiber die impfbarkeit typhoser fieber. Zentralbl. Med. Wissensch 1876, 14, 193–197. (In German) [Google Scholar]
- Manson, P. Tropical Diseases, a Manual of the Diseases of the Warm Climate; Cassell and Co.: London, UK, 1898. [Google Scholar]
- Christy, C. Ornithodoros moubata and tick fever in man. Br. Med. J. 1903, 2, 652–653. [Google Scholar]
- Ross, P.H.; Milne, A.D. Tick fever. Br. Med. J. 1904, 2, 1453–1454. [Google Scholar] [CrossRef] [PubMed]
- Dutton, J.E.; Todd, J.L. The nature of tick fever in the eastern part of the Congo Free State. Br. Med. J. 1905, 2, 1259–1260. [Google Scholar]
- Davis, G.E.; Burgdorfer, W. On the susceptibility of the guinea pig to the relapsing fever spirochete Borrelia duttonii. Bull. Soc. Pathol. Exot. Filiales 1954, 47, 498–501. [Google Scholar] [PubMed]
- Koch, R. Vorläufige mitteilungen über die ergebnisse einer forschungsreise nach Ostafrika. Dtsch. Med. Wochenschr. 1905, 31, 1865–1869. (In German) [Google Scholar] [CrossRef]
- Carlisle, R.J. Two cases of relapsing fever; with notes on the occurrence of this disease throughout the world at the present day. J. Infect. Dis. 1906, 3, 233–265. [Google Scholar] [CrossRef]
- Breinl, A. On the specific anture of the spirochaeta of the African tick fever. Lancet 1906, 167, 1690–1691. [Google Scholar] [CrossRef]
- Norris, C.; Pappenheimer, A.M.; Flournoy, T. Study of a spirochete obtained from a case of relapsing fever in man, with notes on morphology, animal reactions, and attempts at cultivation. J. Infect. Dis. 1906, 3, 266–290. [Google Scholar]
- Novy, F.G.; Knapp, R.E. Studies on Spirillum obermeieri and related organisms. J. Infect. Dis. 1906, 3, 291–293. [Google Scholar] [CrossRef]
- Kemp, H.; Moursund, W.; Wright, H. Relapsing fever in Texas. III. Some notes on the biological characteristics of the causative organism. Am. J. Trop. Med. 1934, 14, 163–169. [Google Scholar]
- Baltazard, M.; Bahmanyar, M.; Chamsa, M. Sur l’usage du cobaye pour la differenciation des spirochetes recurrents. Bull. Soc. Pathol. Exot. 1954, 47, 864–877. (In French) [Google Scholar]
- Brumpt, E. Etude de la fievre recurrente sporadique des etats-unis, transmise dans la nature par Ornithodoros turicata. C. R. Soc. Biol. 1933, 113, 1366–1369. (In French) [Google Scholar]
- Davis, G.E. The spirochetes. Ann. Rev. Microbiol. 1948, 2, 305–334. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, O. Borrelia, human relapsing fever, and parasite-vector-host relationships. Bacteriol. Rev. 1965, 29, 46–74. [Google Scholar] [PubMed]
- Southern, P.M.; Sanford, J.P. Relapsing fever: A clinical and microbiological review. Medicine 1969, 48, 129–150. [Google Scholar] [CrossRef]
- Felsenfeld, O. Borrelia. Strains, Vectors, Human and Animal Borreliosis; Warren H. Greene, Inc.: St. Louis, MO, USA, 1971; p. 180. [Google Scholar]
- Goubau, P.F. Relapsing fevers. A review. Ann. Soc. Belg. Med. Trop. 1984, 64, 335–364. [Google Scholar] [PubMed]
- Barbour, A.G.; Hayes, S.F. Biology of Borrelia species. Microbiol. Rev. 1986, 50, 381–400. [Google Scholar] [PubMed]
- Barbour, A.G. Immunobiology of relapsing fever. Contrib. Microbiol. Immunol. 1987, 8, 125–137. [Google Scholar] [PubMed]
- Cadavid, D.; Barbour, A.G. Neuroborreliosis during relapsing fever: Review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin. Infect. Dis. 1998, 26, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Barthold, S.W.; Cadavid, D.; Phillip, M.T. Animal models of borreliosis. In Borrelia: Molecular Biology, Host Interaction, and Pathogenesis; Radolf, J.D., Samuels, D.S., Eds.; Caister Academic Press: Norfolk, UK, 2010; pp. 359–412. [Google Scholar]
- Wheeler, C.; Herms, W.; Meyer, K. A new tick vector of relapsing fever in California. Proc. Soc. Exp. Biol. Med. 1935, 32, 1290–1292. [Google Scholar] [CrossRef]
- Wheeler, C.M. Relapsing fever in California. Attempts to transmit spirochaetes of california relapsing fever to human subjects by means of the bite of the vector Ornithodoros hermsi. Am. J. Trop. Med. 1938, 18, 641–657. [Google Scholar]
- Barbour, A. Relapsing fever. In Tick-Borne Diseases of Humans; Dennis, D.T., Goodman, J.L., Sonenshine, D.E., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 220–236. [Google Scholar]
- Briggs, L.H. Relapsing fever in California. JAMA 1922, 79, 941–944. [Google Scholar] [CrossRef]
- Boyer, K.M.; Munford, R.S.; Maupin, G.O.; Pattison, C.P.; Fox, M.D.; Barnes, A.M.; Jones, W.L.; Maynard, J.E. Tick-borne relapsing fever: An interstate outbreak originating at Grand Canyon National Park. Am. J. Epidemiol. 1977, 105, 469–479. [Google Scholar] [PubMed]
- Fritz, C.L.; Payne, J.R.; Schwan, T.G. Serologic evidence for Borrelia hermsii infection in rodents on federally owned recreational areas in california. Vector Borne Zoonotic Dis. 2013, 13, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.C.; Teglas, M.B. Relapsing fever group Borrelia in Southern California rodents. J. Med. Entomol. 2014, 51, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Schwan, T.G.; Raffel, S.J.; Schrumpf, M.E.; Schrumpf, M.E.; Webster, L.S.; Marques, A.R.; Spano, R.; Rood, M.; Burns, J.; Hu, R. Tick-borne relapsing fever and Borrelia hermsii, Los Angeles County, California, USA. Emerg. Infect. Dis. 2009, 15, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G. Phylogeny of a relapsing fever Borrelia species transmitted by the hard tick Ixodes scapularis. Infect. Genet. Evol. 2014, 27, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Elbir, H.; Abi-Rached, L.; Pontarotti, P.; Yoosuf, N.; Drancourt, M. African relapsing fever borreliae genomospecies revealed by comparative genomics. Front. Public Health 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Audic, S.; Robert, C.; Nguyen, T.T.; Blanc, G.; Cutler, S.J.; Wincker, P.; Couloux, A.; Claverie, J.M.; Raoult, D.; et al. The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet. 2008, 4, e1000185. [Google Scholar] [CrossRef] [PubMed]
- McNeil, E.; Hinshaw, W.R.; Kissling, R.E. A study of Borrelia anserina infection (spirochetosis) in turkeys. J. Bacteriol. 1949, 57, 191–206. [Google Scholar] [PubMed]
- Davis, G.E. The relapsing fevers: Tick-spirochete specificity studies. Exp. Parasitol. 1952, 1, 406–410. [Google Scholar] [CrossRef]
- Burgdorfer, W.; Mavros, A.J. Susceptibility of various species of rodents to the relapsing fever spirochete, Borrelia hermsii. Infect. Immun. 1970, 2, 256–259. [Google Scholar] [PubMed]
- Beck, M. California field and laboratory studies on relapsing fever. J. Infect. Dis. 1937, 60, 64–80. [Google Scholar] [CrossRef]
- Coffey, E.M.; Eveland, W.C. Experimental relapsing fever initiated by Borrelia hermsii. II. Sequential appearance of major serotypes in the rat. J. Infect. Dis. 1967, 117, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.M.; Eveland, W.C. Experimental relapsing fever initiated by Borrelia hermsii. I. Identification of major serotypes by immunofluorescence. J. Infect. Dis. 1967, 117, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R. Cultivation of Borrelia hermsii. Science 1971, 173, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Stoenner, H.G. Biology of Borrelia hermsii in Kelly’s medium. Appl. Microbiol. 1974, 28, 540–543. [Google Scholar] [PubMed]
- Thompson, R.S.; Burgdorfer, W.; Russell, R.; Francis, B.J. Outbreak of tick-borne relapsing fever in Spokane County, Washington. JAMA 1969, 210, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Stoenner, H.G.; Dodd, T.; Larsen, C. Antigenic variation of Borrelia hermsii. J. Exp. Med. 1982, 156, 1297–1311. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Tessier, S.L.; Stoenner, H.G. Variable major proteins of Borrelia hermsii. J. Exp. Med. 1982, 156, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Schwan, T.G.; Hinnebusch, B.J. Bloodstream- versus tick-associated variants of a relapsing fever bacterium. Science 1998, 280, 1938–1940. [Google Scholar] [CrossRef] [PubMed]
- Magoun, L.; Zuckert, W.R.; Robbins, D.; Parveen, N.; Alugupalli, K.R.; Schwan, T.G.; Barbour, A.G.; Leong, J.M. Variable Small Protein (Vsp)-dependent and Vsp-independent pathways for glycosaminoglycan recognition by relapsing fever spirochaetes. Mol. Microbiol. 2000, 36, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.J.; Barbour, A.G.; Restrepo, B.I.; Schwan, T.G. Population structure of the relapsing fever spirochete Borrelia hermsii as indicated by polymorphism of two multigene families that encode immunogenic outer surface lipoproteins. Infect. Immun. 1998, 66, 432–440. [Google Scholar] [PubMed]
- Barbour, A.G. The chromosome and plasmids of the tick-borne relapsing fever agent Borrelia hermsii. Genome Announc. 2016, 4, e00528-16. [Google Scholar] [CrossRef] [PubMed]
- Alugupalli, K.R.; Michelson, A.D.; Joris, I.; Schwan, T.G.; Hodivala-Dilke, K.; Hynes, R.O.; Leong, J.M. Spirochete-platelet attachment and thrombocytopenia in murine relapsing fever borreliosis. Blood 2003, 102, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Breitschwerdt, E.B.; Nicholson, W.L.; Kiehl, A.R.; Steers, C.; Meuten, D.J.; Levine, J.F. Natural infections with Borrelia spirochetes in two dogs from Florida. J. Clin. Microbiol. 1994, 32, 352–357. [Google Scholar] [PubMed]
- Whitney, M.S.; Schwan, T.G.; Sultemeier, K.B.; McDonald, P.S.; Brillhart, M.N. Spirochetemia caused by Borrelia turicatae infection in 3 dogs in Texas. Vet. Clin. Pathol. 2007, 36, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.L.; Raffel, S.J.; Fischer, R.J.; Bellinghausen, M.; Stevenson, C.; Schwan, T.G. First isolation of the relapsing fever spirochete, Borrelia hermsii, from a domestic dog. Ticks Tick Borne Dis. 2014, 5, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Baneth, G.; Nachum-Biala, Y.; Halperin, T.; Hershko, Y.; Kleinerman, G.; Anug, Y.; Abdeen, Z.; Lavy, E.; Aroch, I.; Straubinger, R.K. Borrelia persica infection in dogs and cats: Clinical manifestations, clinicopathological findings and genetic characterization. Parasites Vectors 2016. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Kitten, T.; Carter, C.J.; Infante, D.; Barbour, A.G. Subtelomeric expression regions of Borrelia hermsii linear plasmids are highly polymorphic. Mol. Microbiol. 1992, 6, 3299–3311. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Restrepo, B.I.; Porcella, S.F.; Raffel, S.J.; Schwan, T.G.; Barbour, A.G. Antigenic variation by Borrelia hermsii occurs through recombination between extragenic repetitive elements on linear plasmids. Mol. Microbiol. 2006, 60, 1329–1343. [Google Scholar] [CrossRef] [PubMed]
- Marcsisin, R.A.; Lewis, E.R.; Barbour, A.G. Expression of the tick-associated Vtp protein of Borrelia hermsii in a murine model of relapsing fever. PLoS ONE 2016, 11, e0149889. [Google Scholar]
- Barbour, A.G. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 1984, 57, 521–525. [Google Scholar] [PubMed]
- Kazragis, R.J.; Dever, L.L.; Jorgensen, J.H.; Barbour, A.G. In vivo activities of ceftriaxone and vancomycin against Borrelia spp. In the mouse brain and other sites. Antimicrob. Agents Chemother. 1996, 40, 2632–2636. [Google Scholar] [PubMed]
- Sadziene, A.; Thompson, P.A.; Barbour, A.G. In vitro inhibition of Borrelia burgdorferi growth by antibodies. J. Infect. Dis. 1993, 167, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Tessier, S.L.; Hayes, S.F. Variation in a major surface protein of Lyme disease spirochetes. Infect. Immun. 1984, 45, 94–100. [Google Scholar] [PubMed]
- Tsao, J.I.; Wootton, J.T.; Bunikis, J.; Luna, M.G.; Fish, D.; Barbour, A.G. An ecological approach to preventing human infection: Vaccinating wild mouse reservoirs intervenes in the Lyme disease cycle. Proc. Natl. Acad. Sci. USA 2004, 101, 18159–18164. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Bunikis, J.; Travinsky, B.; Hoen, A.G.; Diuk-Wasser, M.A.; Fish, D.; Tsao, J.I. Niche partitioning of Borrelia burgdorferi and Borrelia miyamotoi in the same tick vector and mammalian reservoir species. Am. J. Trop. Med. Hyg. 2009, 81, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. Tm4: A free, open-source system for microarray data management and analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [PubMed]
- Russell, H. Human and experimental relapsing fever, Accra, Gold Coast, 1929–1930. West Afr. Med. J. 1931, 4, 59–66. [Google Scholar]
- Eidmann, E.; Lippelt, H.; Poespodihardjo, J. Quantitative Untersuchungen über die Vermehrung von Borrelia erratici in der weißen Maus. Z. Tropenmed. Parasitol. 1959, 10, 339. (In German) [Google Scholar] [PubMed]
- Pennington, P.M.; Allred, C.D.; West, C.S.; Alvarez, R.; Barbour, A.G. Arthritis severity and spirochete burden are determined by serotype in the Borrelia turicatae-mouse model of Lyme disease. Infect. Immun. 1997, 65, 285–292. [Google Scholar] [PubMed]
- Cadavid, D.; Thomas, D.D.; Crawley, R.; Barbour, A.G. Variability of a bacterial surface protein and disease expression in a possible mouse model of systemic Lyme borreliosis. J. Exp. Med. 1994, 179, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Benoit, V.M.; Petrich, A.; Alugupalli, K.R.; Marty-Roix, R.; Moter, A.; Leong, J.M.; Boyartchuk, V.L. Genetic control of the innate immune response to Borrelia hermsii influences the course of relapsing fever in inbred strains of mice. Infect. Immun. 2010, 78, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Sadziene, A.; Barbour, A.G.; Rosa, P.A.; Thomas, D.D. An OspB mutant of Borrelia burgdorferi has reduced invasiveness in vitro and reduced infectivity in vivo. Infect. Immun. 1993, 61, 3590–3596. [Google Scholar] [PubMed]
- Kitten, T.; Barbour, A.G. The relapsing fever agent Borrelia hermsii has multiple copies of its chromosome and linear plasmids. Genetics 1992, 132, 311–324. [Google Scholar] [PubMed]
- Barbour, A.G. Antigenic variation of a relapsing fever Borrelia species. Ann. Rev. Microbiol. 1990, 44, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Barbour, A.G. Cross-species hybridization of a Borrelia burgdorferi DNA array reveals infection- and culture-associated genes of the unsequenced genome of the relapsing fever agent Borrelia hermsii. Mol. Microbiol. 2004, 51, 729–748. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Carter, C.J.; Barbour, A.G. Activation of a vmp pseudogene in Borrelia hermsii: An alternate mechanism of antigenic variation during relapsing fever. Mol. Microbiol. 1994, 13, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Dai, Q.; Restrepo, B.I.; Stoenner, H.G.; Frank, S.A. Pathogen escape from host immunity by a genome program for antigenic variation. Proc. Natl. Acad. Sci. USA 2006, 103, 18290–18295. [Google Scholar] [CrossRef] [PubMed]
- Bolz, D.D.; Sundsbak, R.S.; Ma, Y.; Akira, S.; Weis, J.H.; Schwan, T.G.; Weis, J.J. Dual role of Myd88 in rapid clearance of relapsing fever Borrelia spp. Infect. Immun. 2006, 74, 6750–6760. [Google Scholar] [CrossRef] [PubMed]
- Alugupalli, K.R.; Akira, S.; Lien, E.; Leong, J.M. Myd88- and Bruton’s tyrosine kinase-mediated signals are essential for T cell-independent pathogen-specific IgM responses. J. Immunol. 2007, 178, 3740–3749. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Barbour, A.G. Antigen diversity in the bacterium B. hermsii through “somatic” mutations in rearranged vmp genes. Cell 1994, 78, 867–876. [Google Scholar] [CrossRef]
- Geigy, R.; Sarasin, G. Isolatstamme von Borrelia duttonii und immunisierungsverhalten gegenuber der weissen maus. Acta Trop. 1958, 15, 254–258. (In German) [Google Scholar] [PubMed]
- Schuhardt, V.T.; Wilkerson, M. Relapse phenomena in rats infected with single spirochetes (Borrelia recurrentis var. turicatae). J. Bacteriol. 1951, 62, 215–219. [Google Scholar] [PubMed]
- Cunningham, J.; Fraser, A.G.L. Further observations on Indian relapsing fever. III. Persistence of spirochaetes in the blood and organs of infected animals. Indian J. Med. Res. 1937, 24, 581–592. [Google Scholar]
- Aslam, N.; Turner, C.M. The relationship of variable antigen expression and population growth rates in trypanosoma brucei. Parasitol. Res. 1992, 78, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, A. Clinical pathology of the Jarisch-Herxheimer reaction. J. Infect. Dis. 1976, 133, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Negussie, Y.; Remick, D.G.; DeForge, L.E.; Kunkel, S.L.; Eynon, A.; Griffin, G.E. Detection of plasma tumor necrosis factor, interleukins 6, and 8 during the Jarisch-Herxheimer reaction of relapsing fever. J. Exp. Med. 1992, 175, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Galloway, R.E.; Levin, J.; Butler, T.; Naff, G.B.; Goldsmith, G.H.; Saito, H.; Awoke, S.; Wallace, C.K. Activation of protein mediators of inflammation and evidence for endotoxemia in Borrelia recurrentis infection. Am. J. Med. 1977, 63, 933–938. [Google Scholar] [CrossRef]
- Takayama, K.; Rothenberg, R.J.; Barbour, A.G. Absence of lipopolysaccharide in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 1987, 55, 2311–2313. [Google Scholar] [PubMed]
- Young, E.J.; Weingarten, N.M.; Baughn, R.E.; Duncan, W.C. Studies on the pathogenesis of the Jarisch-Herxheimer reaction: Development of an animal model and evidence against a role for classical endotoxin. J. Infect. Dis. 1982, 146, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Vidal, V.; Scragg, I.G.; Cutler, S.J.; Rockett, K.A.; Fekade, D.; Warrell, D.A.; Wright, D.J.; Kwiatkowski, D. Variable major lipoprotein is a principal TNF-inducing factor of louse-borne relapsing fever. Nat. Med. 1998, 4, 1416–1420. [Google Scholar] [CrossRef] [PubMed]
- Wooten, R.M.; Ma, Y.; Yoder, R.A.; Brown, J.P.; Weis, J.H.; Zachary, J.F.; Kirschning, C.J.; Weis, J.J. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol. 2002, 168, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Londoño, D.; Marques, A.; Hornung, R.L.; Cadavid, D. Relapsing fever borreliosis in IL-10 deficient mice. Infect. Immun. 2008, 76, 5508–5513. [Google Scholar] [CrossRef] [PubMed]
- Londoño, D.; Marques, A.; Hornung, R.L.; Cadavid, D. IL-10 helps control pathogen load during high-level bacteremia. J. Immunol. 2008, 181, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Hirsch, C.M.; Ghalyanchi Langeroudi, A.; Meinardi, S.; Lewis, E.R.; Estabragh, A.S.; Blake, D.R. Elevated carbon monoxide in the exhaled breath of mice during a systemic bacterial infection. PLoS ONE 2013, 8, e69802. [Google Scholar] [CrossRef] [PubMed]
- Langeroudi, A.G.; Hirsch, C.M.; Estabragh, A.S.; Meinardi, S.; Blake, D.R.; Barbour, A.G. Elevated carbon monoxide to carbon dioxide ratio in the exhaled breath of mice treated with a single dose of lipopolysaccharide. Open Forum Infect. Dis. 2014, 1, ofu085. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Londono, D.; Bai, Y.; Cabral, E.S.; Quandt, J.; Hornung, R.; Martin, R.; Marques, A.; Cadavid, D. High production of CXCL13 in blood and brain during persistent infection with the relapsing fever spirochete Borrelia turicatae. J. Neuropathol. Exp. Neurol. 2007, 66, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Gabritchewsky, G.N. Les bases de la sérothérapie de la fievre récurrente. Ann. Inst. Pasteur 1896, 10, 629–659. (In French) [Google Scholar]
- Barbour, A.G.; Bundoc, V. In vitro and in vivo neutralization of the relapsing fever agent Borrelia hermsii with serotype-specific immunoglobulin M antibodies. Infect. Immun. 2001, 69, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Newman, K., Jr.; Johnson, R.C. In vivo evidence that an intact lytic complement pathway is not essential for successful removal of circulating Borrelia turicatae from mouse blood. Infect. Immun. 1981, 31, 465–469. [Google Scholar] [PubMed]
- Newman, K., Jr.; Johnson, R.C. T-cell-independent elimination of Borrelia turicatae. Infect. Immun. 1984, 45, 572–576. [Google Scholar] [PubMed]
- Arimitsu, Y.; Akama, K. Characterization of protective antibodies produced in mice infected with Borrelia duttonii. Jpn. J. Med. Sci. Biol. 1973, 26, 229–237. [Google Scholar] [CrossRef]
- Sadziene, A.; Jonsson, M.; Bergström, S.; Bright, R.K.; Kennedy, R.C.; Barbour, A.G. A bactericidal antibody to Borrelia burgdorferi is directed against a variable region of the OspB protein. Infect. Immun. 1994, 62, 2037–2045. [Google Scholar] [PubMed]
- Connolly, S.E.; Benach, J.L. Cutting edge: The spirochetemia of murine relapsing fever is cleared by complement-independent bactericidal antibodies. J. Immunol. 2001, 167, 3029–3032. [Google Scholar] [CrossRef] [PubMed]
- Luke, C.J.; Marshall, M.A.; Zahradnik, J.M.; Bybel, M.; Menefee, B.E.; Barbour, A.G. Growth-inhibiting antibody responses of humans vaccinated with recombinant outer surface protein a or infected with Borrelia burgdorferi or both. J. Infect. Dis. 2000, 181, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Sadziene, A.; Thompson, P.A.; Barbour, A.G. A flagella-less mutant of Borrelia burgdorferi as a live attenuated vaccine in the murine model of Lyme disease. J. Infect. Dis. 1996, 173, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.V.; Anders, R.F.; Knowles, G. Differential effect of immunoglobulin on the in vitro growth of several isolates of Plasmodium falciparum. Infect. Immun. 1983, 39, 1228–1235. [Google Scholar] [PubMed]
- Barbour, A.G.; Tessier, S.L.; Todd, W.J. Lyme disease spirochetes and ixodid tick spirochetes share a common surface antigenic determinant defined by a monoclonal antibody. Infect. Immun. 1983, 41, 795–804. [Google Scholar] [PubMed]
- Chung, H.; Chang, F. Relapsing fever; clinical and statistical study of 337 cases. Chin. Med. J. 1939, 55, 6–33. [Google Scholar]
- Burman, N.; Shamaei-Tousi, A.; Bergström, S. The spirochete Borrelia crocidurae causes erythrocyte rosetting during relapsing fever. Infect. Immun. 1998, 66, 815–819. [Google Scholar] [PubMed]
- Meleney, H.E. Relapse phenomena of Spironema recurrentis. J. Exp. Med. 1928, 48, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Baltazard, M. Identification des spirochètes récurrents. Individualité de l'espèce Spirochaeta recurrentis. Bull. Soc. Pathol. Exot. 1947, 40, 77–81. (In French) [Google Scholar]
- Alugupalli, K.R.; Gerstein, R.M.; Chen, J.; Szomolanyi-Tsuda, E.; Woodland, R.T.; Leong, J.M. The resolution of relapsing fever borreliosis requires IgM and is concurrent with expansion of B1b lymphocytes. J. Immunol. 2003, 170, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Schwegmann, A.; Atkinson, R.; Claes, F.; Drennan, M.; de Baetselier, P.; Brombacher, F. The role of B-cells and IgM antibodies in parasitemia, anemia, and VSG switching in Trypanosoma brucei-infected mice. PLoS Pathog. 2008, 4, e1000122. [Google Scholar] [CrossRef] [PubMed]
- Amole, B.O.; Clarkson, A.B., Jr.; Shear, H.L. Pathogenesis of anemia in Trypanosoma brucei-infected mice. Infect. Immun. 1982, 36, 1060–1068. [Google Scholar] [PubMed]
- Naessens, J.; Kitani, H.; Nakamura, Y.; Yagi, Y.; Sekikawa, K.; Iraqi, F. TNF-alpha mediates the development of anaemia in a murine Trypanosoma brucei rhodesiense infection, but not the anaemia associated with a murine Trypanosoma congolense infection. Clin. Exp. Immunol. 2005, 139, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Cnops, J.; de Trez, C.; Stijlemans, B.; Keirsse, J.; Kauffmann, F.; Barkhuizen, M.; Keeton, R.; Boon, L.; Brombacher, F.; Magez, S. NK-, NKT- and CD8-derived IFNgamma drives myeloid cell activation and erythrophagocytosis, resulting in trypanosomosis-associated acute anemia. PLoS Pathog. 2015, 11, e1004964. [Google Scholar] [CrossRef] [PubMed]
- Cook, A.R. Relapsing fever in Uganda. J. Trop. Med. 1904, 7, 24–26. [Google Scholar]
- Shamaei-Tousi, A.; Martin, P.; Bergh, A.; Burman, N.; Brannstrom, T.; Bergström, S. Erythrocyte-aggregating relapsing fever spirochete Borrelia crocidurae induces formation of microemboli. J. Infect. Dis. 1999, 180, 1929–1938. [Google Scholar] [CrossRef]
- Guyard, C.; Chester, E.M.; Raffel, S.J.; Schrumpf, M.E.; Policastro, P.F.; Porcella, S.F.; Leong, J.M.; Schwan, T.G. Relapsing fever spirochetes contain chromosomal genes with unique direct tandemly repeated sequences. Infect. Immun. 2005, 73, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Brust, M.; Aouane, O.; Thiebaud, M.; Flormann, D.; Verdier, C.; Kaestner, L.; Laschke, M.W.; Selmi, H.; Benyoussef, A.; Podgorski, T.; et al. The plasma protein fibrinogen stabilizes clusters of red blood cells in microcapillary flows. Sci. Rep. 2014, 4, 4348. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Todd, W.J.; Stoenner, H.G. Action of penicillin on borrelia hermsii. Antimicrob. Agents Chemother. 1982, 21, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Barthold, S.W.; Beck, D.S.; Hansen, G.M.; Terwilliger, G.A.; Moody, K.D. Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis. 1990, 162, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Barthold, S.W. Infectivity of Borrelia burgdorferi relative to route of inoculation and genotype in laboratory mice. J. Infect. Dis. 1991, 163, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Weis, J.J.; McCracken, B.A.; Ma, Y.; Fairbairn, D.; Roper, R.J.; Morrison, T.B.; Weis, J.H.; Zachary, J.F.; Doerge, R.W.; Teuscher, C. Identification of quantitative trait loci governing arthritis severity and humoral responses in the murine model of Lyme disease. J. Immunol. 1999, 162, 948–956. [Google Scholar]
- Lopez, J.E.; McCoy, B.N.; Krajacich, B.J.; Schwan, T.G. Acquisition and subsequent transmission of Borrelia hermsii by the soft tick Ornithodoros hermsi. J. Med. Entomol. 2011, 48, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.E.; Wilder, H.K.; Hargrove, R.; Brooks, C.P.; Peterson, K.E.; Beare, P.A.; Sturdevant, D.E.; Nagarajan, V.; Raffel, S.J.; Schwan, T.G. Development of genetic system to inactivate a Borrelia turicatae surface protein selectively produced within the salivary glands of the arthropod vector. PLoS Negl. Trop. Dis. 2013, 7, e2514. [Google Scholar] [CrossRef] [PubMed]
- Policastro, P.F.; Raffel, S.J.; Schwan, T.G. Cotransmission of divergent relapsing fever spirochetes by artificially infected Ornithodoros hermsi. Appl. Environ. Microbiol. 2011, 77, 8494–8499. [Google Scholar] [CrossRef] [PubMed]
- Aylor, D.L.; Valdar, W.; Foulds-Mathes, W.; Buus, R.J.; Verdugo, R.A.; Baric, R.S.; Ferris, M.T.; Frelinger, J.A.; Heise, M.; Frieman, M.B.; et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011, 21, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Shusterman, A.; Salyma, Y.; Nashef, A.; Soller, M.; Wilensky, A.; Mott, R.; Weiss, E.I.; Houri-Haddad, Y.; Iraqi, F.A. Genotype is an important determinant factor of host susceptibility to periodontitis in the Collaborative Cross and inbred mouse populations. BMC Genet. 2013, 14. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Difference in Means (Lower, Upper 95% Confidence Limits) | ||||
|---|---|---|---|---|---|
| Unit a | Peak vs. Uninfected | Clearance vs. Uninfected | Clearance vs. Peak | Change b | |
| β-2 Microglobulin | µg | +0.1 (−0.2, +0.3) | +0.4 (+0.2, +0.6) c | +0.3 (+0.1, +0.5) | C |
| Clusterin | µg | +62 (+19, +106) | +131 (+91, +171) | +69 (+7, +130) | P,C |
| C-reactive protein | µg | +3.8 (+2.5, +5.0) | +3.6 (+2.3, +4.8) | 0.2 (−2.0, +1.6) | P,C |
| EGF d | pg | −3.6 (−8.0, +0.7) | −6.5 (−10.8, −2.3) | −2.9 (−8.0, +2.2) | (P,C) |
| Endothelin-1 | pg | +5.5 (−9.6, +20.5) | +11.2 (+3.3, +19.2) | +5.8 (−10.9, +22.4) | C |
| Fibrinogen | µg | +3421 (+1710, +5131) | +3703 (+2135, +5269) | +282 (−1643, +2207) | P,C |
| Growth hormone | ng | −15.8 (−28.2, −3.4) | −14.3 (−26.5, −2.0) | +1.5 (−3.2, +6.2) | (P,C) |
| Haptoglobin | µg | +88 (+70, +105) | +112 (+103, +121) | +25 (+1.7, +48) | P,C |
| IP-10 (CXCL10) e | pg | +151 (+108, +195) | +29 (+13, +45) | −122 (−173, −71) | P |
| Interferon-γ | pg | +6.0 (-0.5, +12.6) | +1.3 (−1.1, +3.6) | −4.8 (−13.0, +3.5) | |
| IL-1α | pg | −26 (−62, +9) | −28 (−64, +7.9) | 1.4 (−4.2, +1.4) | |
| IL-1β | pg | +120 (−670, +910) | −900 (−1810, +10) | −1020 (−1690, −350) | (C) |
| IL-2 | pg | +7.0 (−8.6, +23) | +0.4 (−14.4, +15.2) | −6.7 (−27.9, +14.5) | |
| IL-6 | pg | +14.4 (+6.0, +22.9) | +1.3 (−0.2, +2.7) | −13.2 (−23.4, −3.0) | P |
| IL-7 | pg | +60 (+40, +70) | +10 (0, +30) | −50 (−70, −20) | P |
| IL-10 | pg | +378 (+237, +520) | −38 (−103, +28) | −416 (−581, −251) | P |
| IL-11 | pg | +64 (+2.0, +126) | 6.5 (−25.0, +12.1) | 71 (−143, +2.0) | P |
| IL-18 | pg | +520 (+300, +740) | −50 (−520, +430) | −570 (−1130, −10) | P |
| Insulin | µIU | −0.7 (−1.1, −0.2) | −1.1 (−1.5, −0.7) | −0.5 (−1.0, +0.1) | (P,C) |
| KC/GROα (CXCL1) | pg | +680 (+460, +890) | +40 (+40, +120) | −640 (−900, −370) | P |
| Lymphotactin (XCL1) | pg | +144 (+101, +188) | +41 (+15, +68) | −103 (−154, −51) | P |
| MCP-1 (CCL2) | pg | +700 (+563, +837) | +65 (+5.9, +124) | −635 (−796, −474) | P |
| MCP-3 (CCL7) | pg | +550 (+396, +703) | +78 (−45, +201) | −472 (−635, −310) | P |
| MCP-5 (CCL12) | pg | +145 (+190, +100) | +46 (+90, +2.8) | −99 (−44, −153) | P |
| M-CSF | pg | +1180 (+780, +1590) | +180 (−220, +580) | −1000 (−1460, −540) | P |
| MDC (CCL22) | pg | +29 (−44, +102) | −97 (−166, −28) | −126 (−207, −45) | (C) |
| MIP-1α (CCL3) | pg | +90 (+20, +200) | +120 (+30, +220) | +30 (−50, +110) | P,C |
| MIP-1β (CCL4) | pg | +216 (+180, +253) | +15 (−13, +44) | −201 (−240, −163) | P |
| MIP-1γ (CCL9) | pg | +7420 (+4980, +9860) | +7230 (+4540, +10,010) | −180 (−4100, +3730) | P,C |
| MIP-2 (CXL1) | pg | +11.3 (+5.7, +16.9) | +2.3 (+0.7, +5.3) | +9.0 (+15.8, +2.2) | P,C |
| MIP-3β (CCL19) | pg | +210 (+120, +310) | +80 (−40, +190) | −140 (−240, −40) | P |
| MMP-9 | ng | +37 (+14, +60) | +12 (−1.3, +25) | −25 (−54, +4.1) | P |
| Myeloperoxidase | ng | +93 (+62, +123) | +118 (+102, +135) | +25 (−14, +65) | P,C |
| Oncostatin M | pg | +100 (+70, +150) | −10 (−20, +10) | −120 (−160, −70) | P |
| Osteopontin | pg | +22.9 (+8.8, +37.0) | +81 (+52, +109) | +58 (+23, +93) | P,C |
| Serum Amyloid P | µg | +15.4 (+10.1, +20.6) | +14.0 (+9.5, +18.5) | −1.4 (−8.5, +5.8) | P,C |
| SGOT | µg | +5.6 (−4.9, +16.1) | +13.7 (+2.8, +24.6) | +8.1 (−8.2, +24.4) | C |
| Stem Cell Factor | pg | +68 (+39, +97) | −6.3 (−32, +19.5) | −74 (−115, −34) | P |
| TIMP-1 | pg | +940 (+480, +1,400) | +980 (+630, +1,330) | +50 (−460, +560) | P,C |
| TNF-α | pg | +20 (0, +40) | 0 (−10, 0) | −20 (−50, 0) | P |
| VCAM-1 | ng | +405 (+257, +552) | +540 (+395, +685) | +135 (−67, +337) | P,C |
| VEGF | pg | +49 (+8.5, +90) | +6.6 (−44, +57) | −43 (−88, +2.9) | P |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crowder, C.D.; Ghalyanchi Langeroudi, A.; Shojaee Estabragh, A.; Lewis, E.R.G.; Marcsisin, R.A.; Barbour, A.G. Pathogen and Host Response Dynamics in a Mouse Model of Borrelia hermsii Relapsing Fever. Vet. Sci. 2016, 3, 19. https://doi.org/10.3390/vetsci3030019
Crowder CD, Ghalyanchi Langeroudi A, Shojaee Estabragh A, Lewis ERG, Marcsisin RA, Barbour AG. Pathogen and Host Response Dynamics in a Mouse Model of Borrelia hermsii Relapsing Fever. Veterinary Sciences. 2016; 3(3):19. https://doi.org/10.3390/vetsci3030019
Chicago/Turabian StyleCrowder, Christopher D., Arash Ghalyanchi Langeroudi, Azadeh Shojaee Estabragh, Eric R. G. Lewis, Renee A. Marcsisin, and Alan G. Barbour. 2016. "Pathogen and Host Response Dynamics in a Mouse Model of Borrelia hermsii Relapsing Fever" Veterinary Sciences 3, no. 3: 19. https://doi.org/10.3390/vetsci3030019
APA StyleCrowder, C. D., Ghalyanchi Langeroudi, A., Shojaee Estabragh, A., Lewis, E. R. G., Marcsisin, R. A., & Barbour, A. G. (2016). Pathogen and Host Response Dynamics in a Mouse Model of Borrelia hermsii Relapsing Fever. Veterinary Sciences, 3(3), 19. https://doi.org/10.3390/vetsci3030019