Bioengineering of a Full-Thickness Skin Equivalent in a 96-Well Insert Format for Substance Permeation Studies and Organ-On-A-Chip Applications


Abstract
: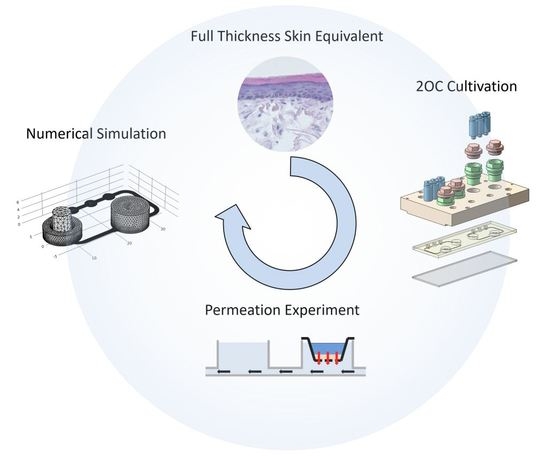
1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
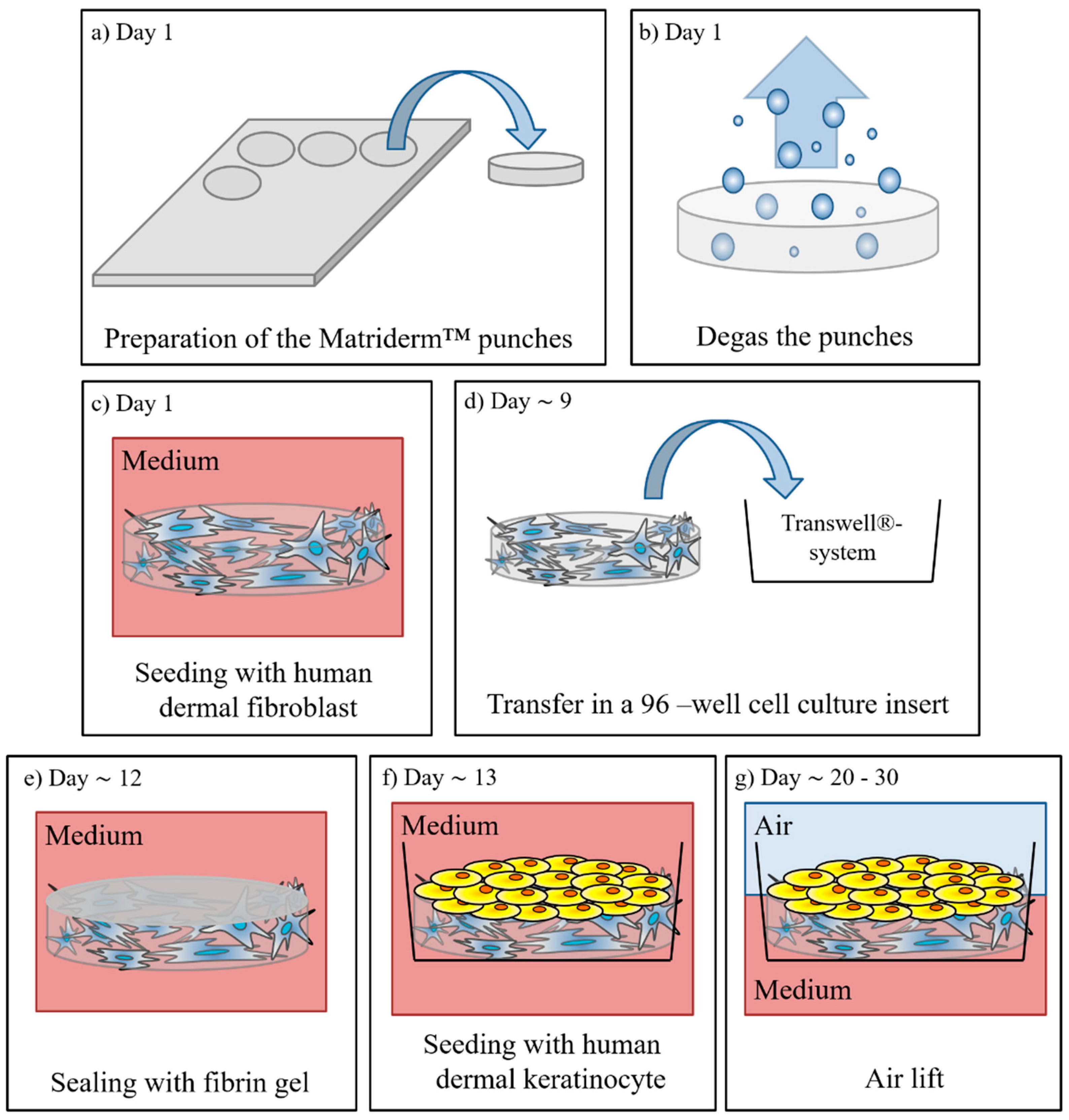
2.2. Manufacturing of Full Thickness Skin Equivalents
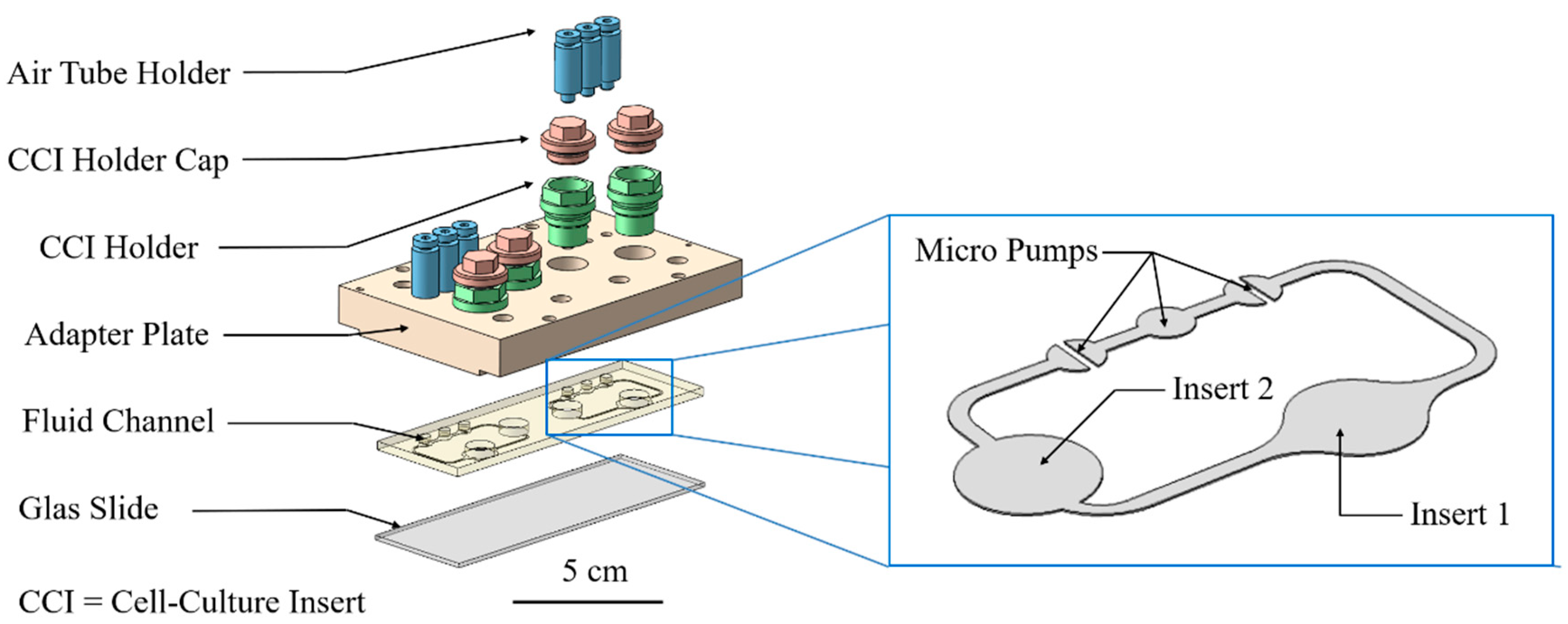
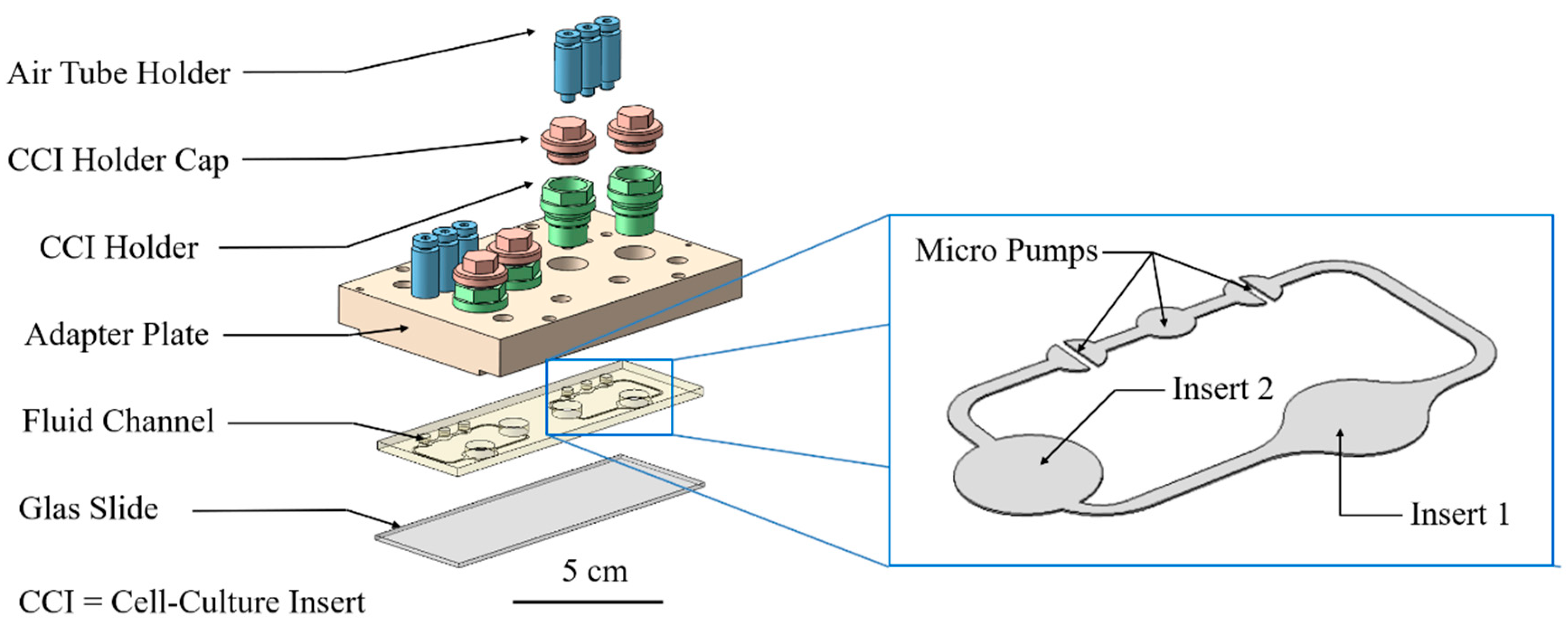
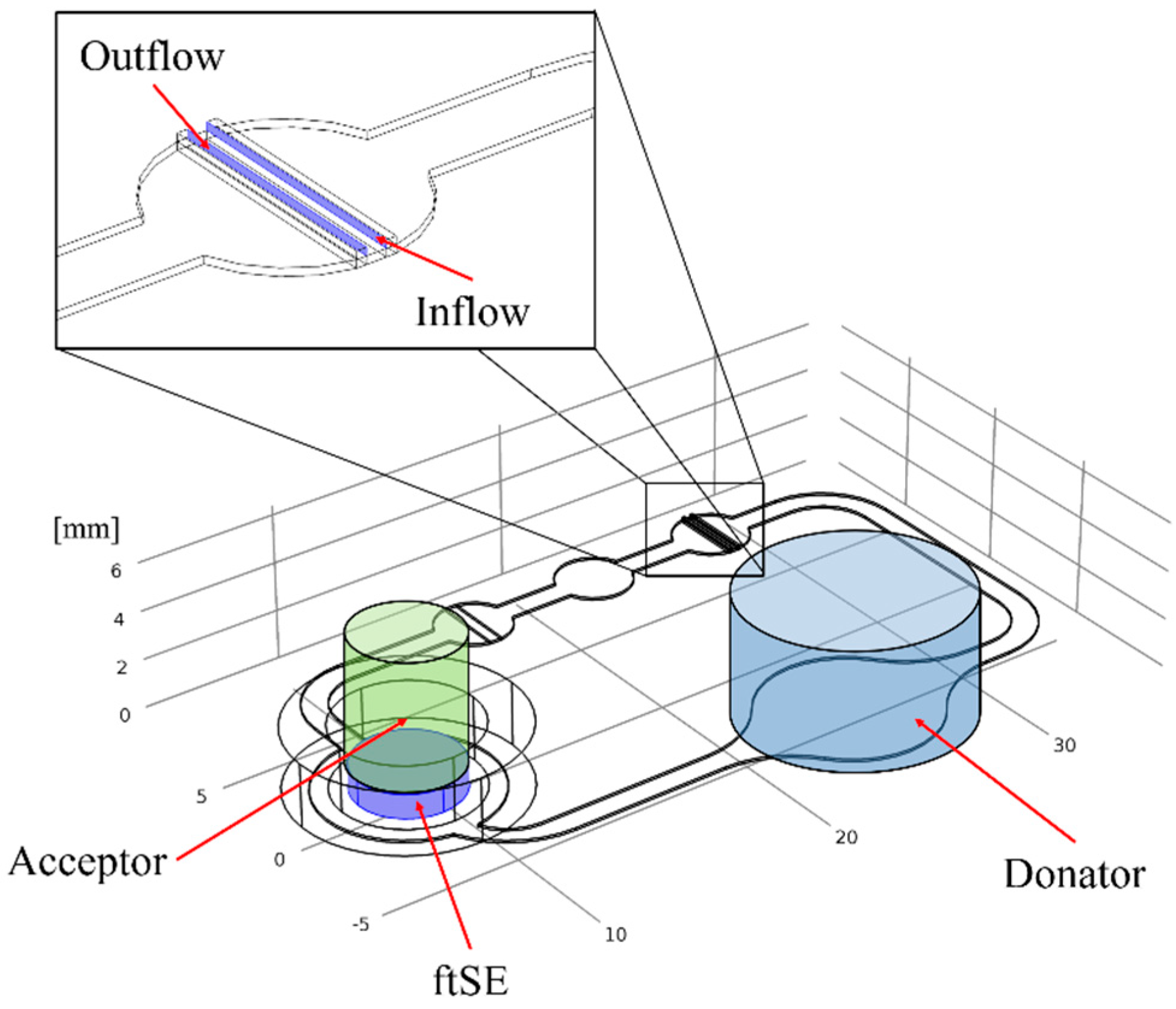
2.3. Cultivation in Two-Organ Chip
2.4. Staining
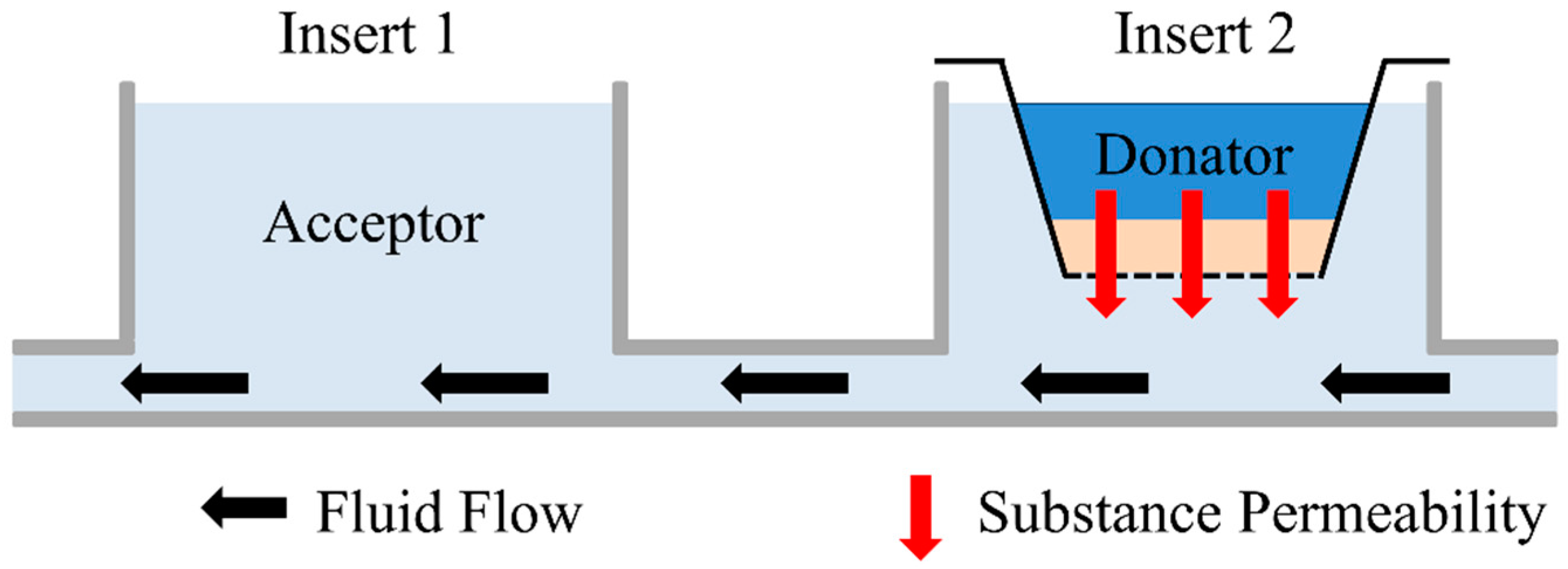
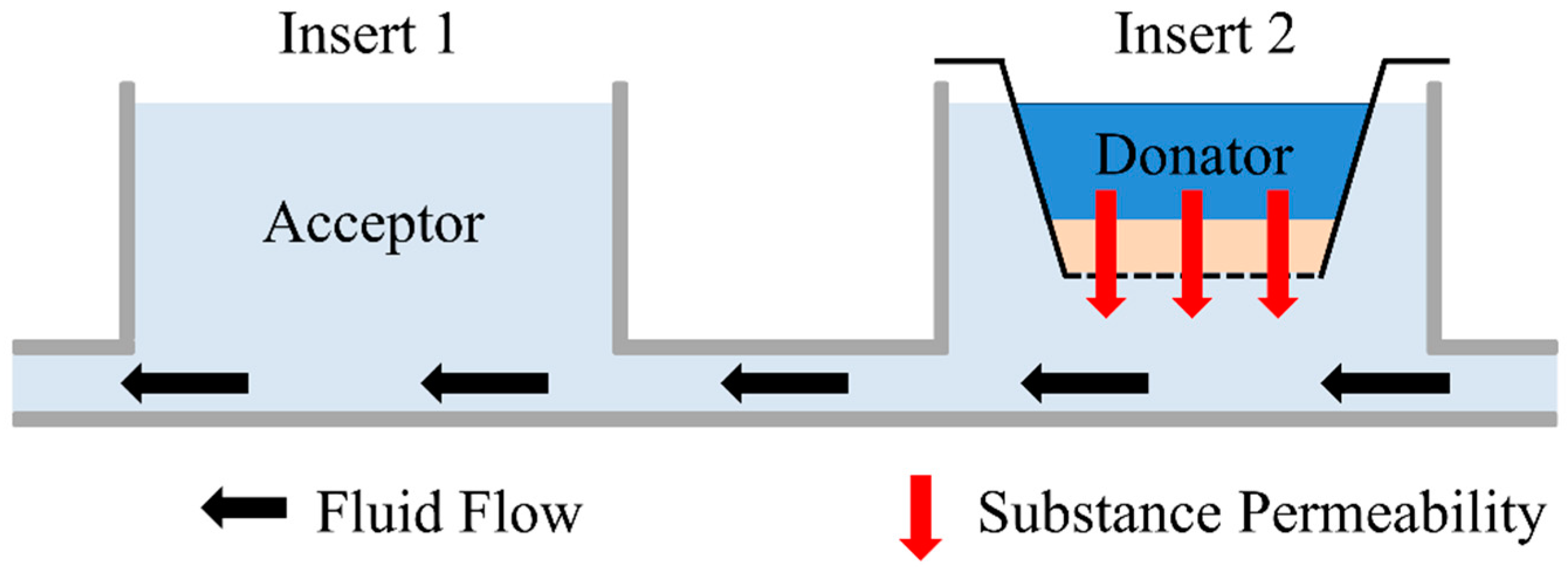
2.5. Permeation Measurement
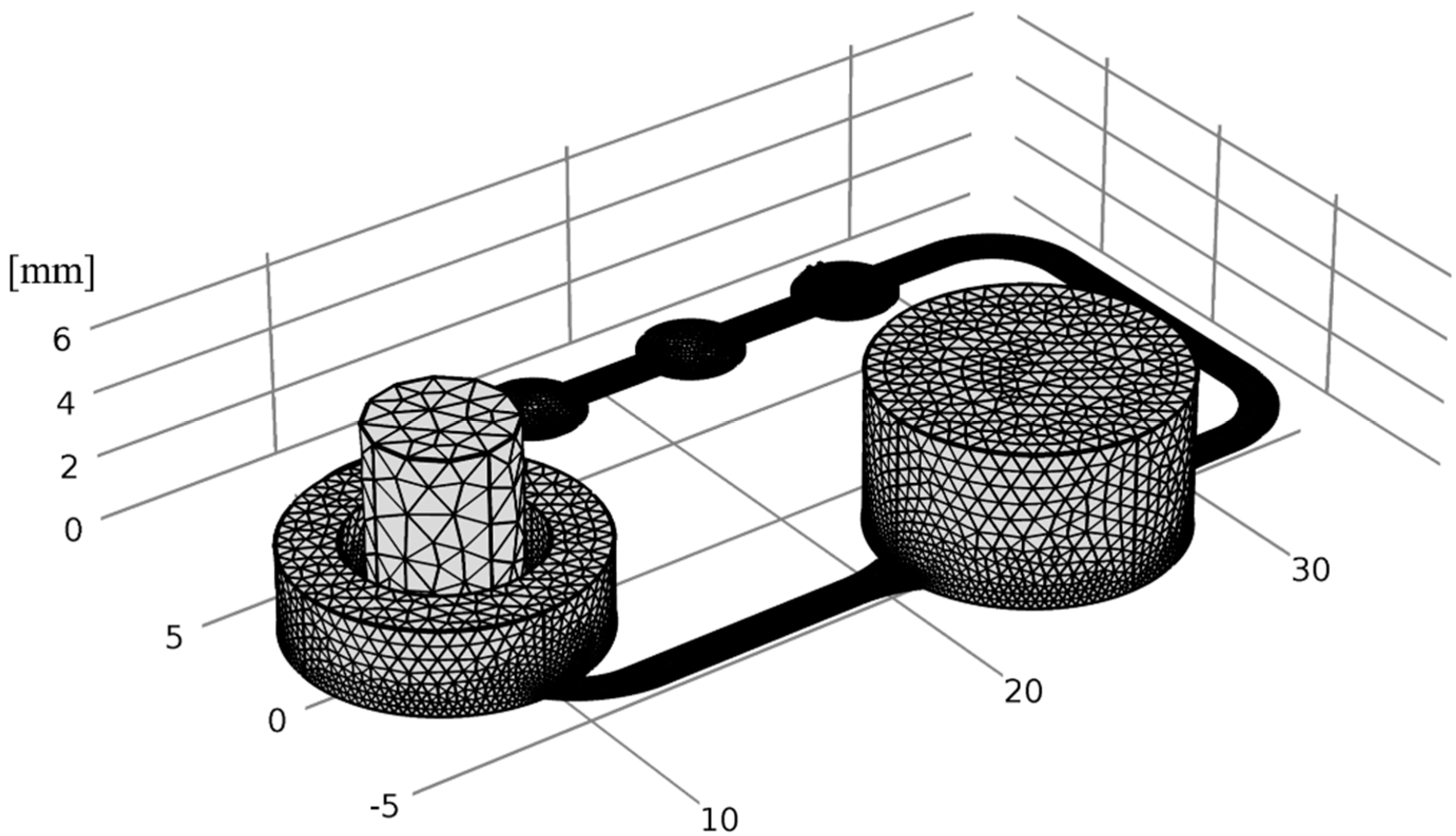
2.6. Numerical Simulation
- (a)
- The diffusion coefficient of fluorescein sodium salt in H2O was set to a value of 1 × 10−9 m2/s, which is approximately 100× higher than the diffusion coefficient through the ftSE.
- (b)
- In the experiment, the substance diffuses through the ftSE, and a membrane of the CCI system. This is considered to be one homogenous phase.
- (c)
- For the computation fluid dynamic (CFD) simulation, all physical boundary effects on walls were neglected.
- (d)
- The fluid flow in the 2OC was assumed to be a laminar. The experimentally measured mean value was used as flow velocity.
3. Results
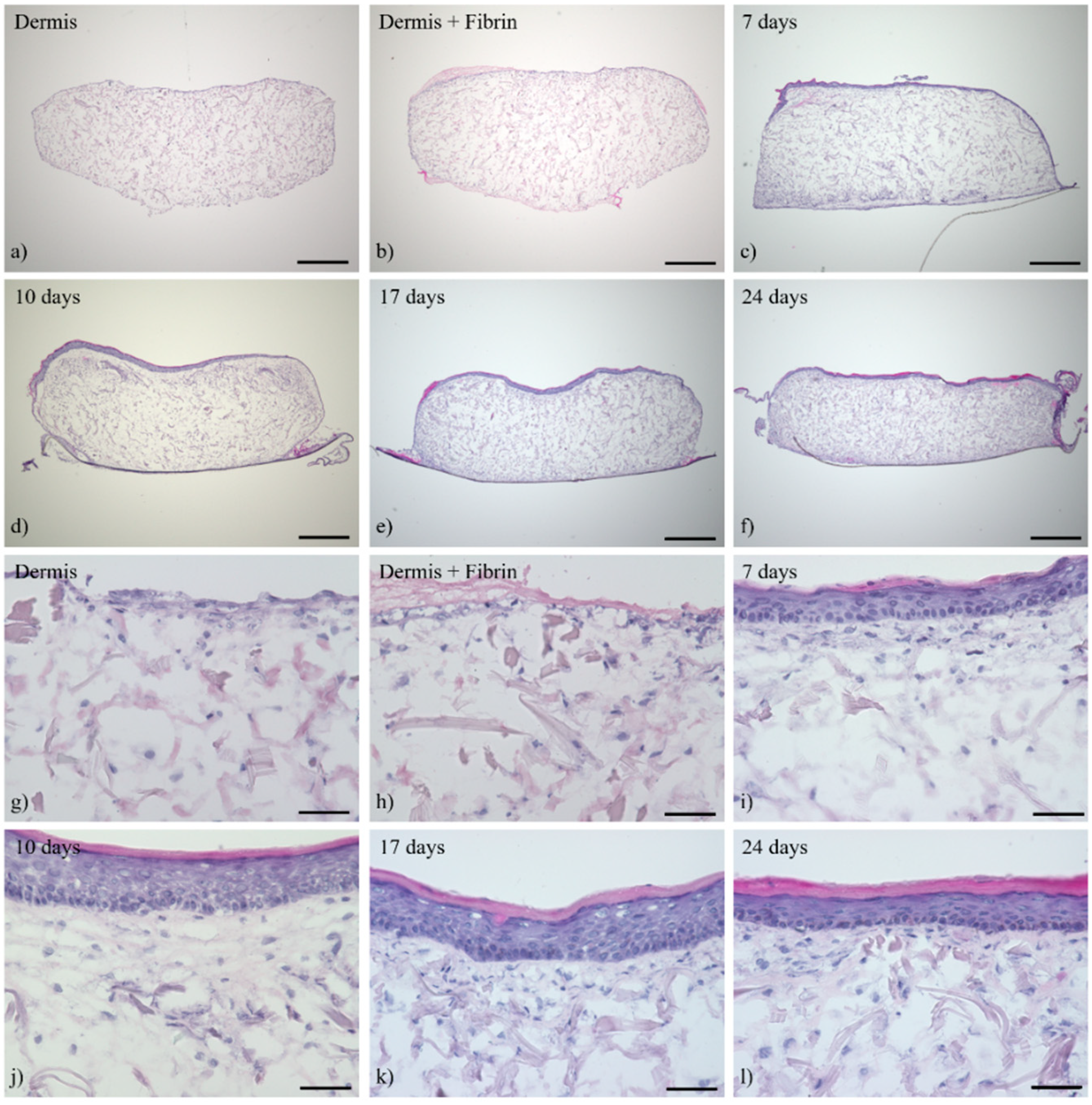
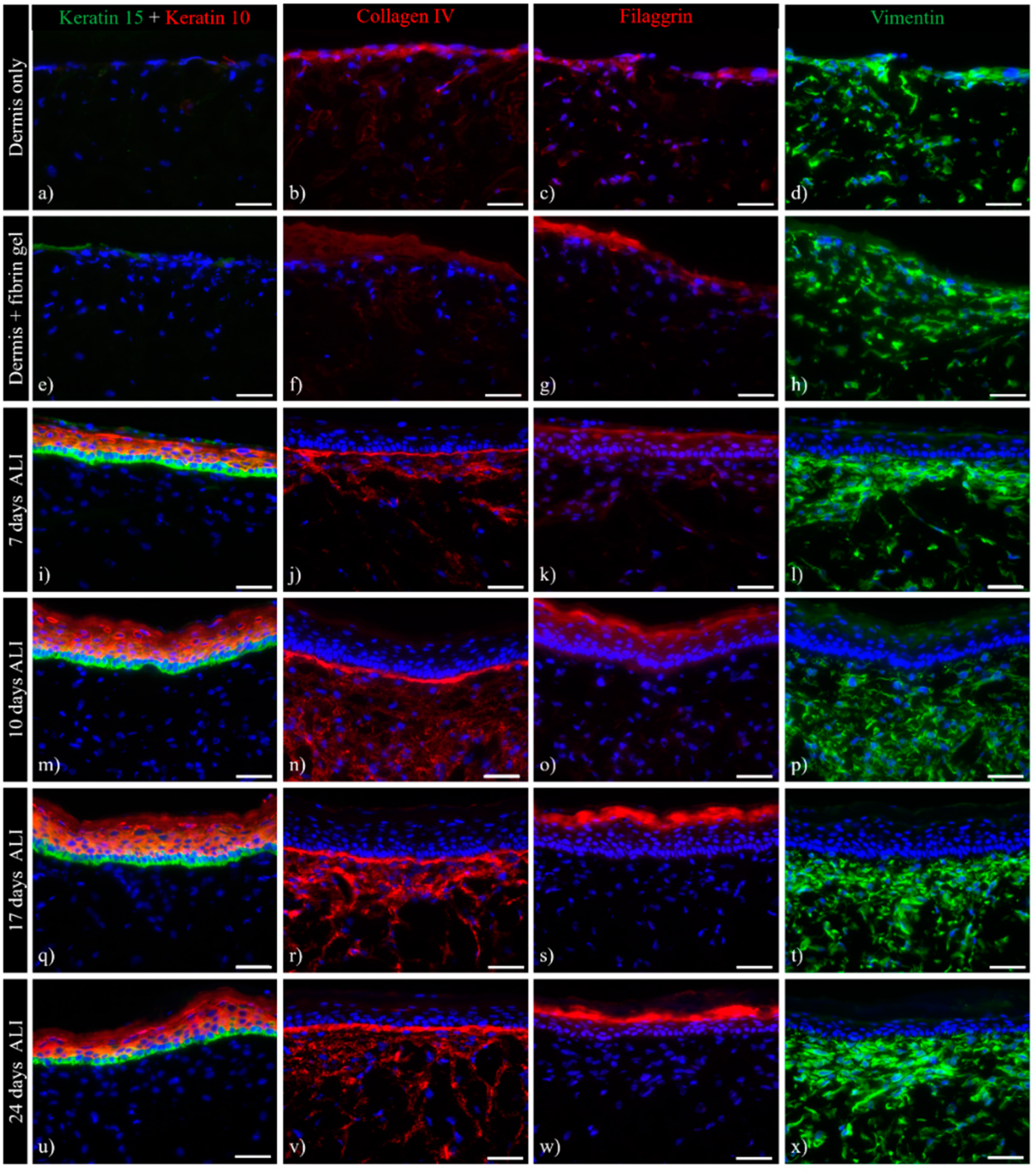
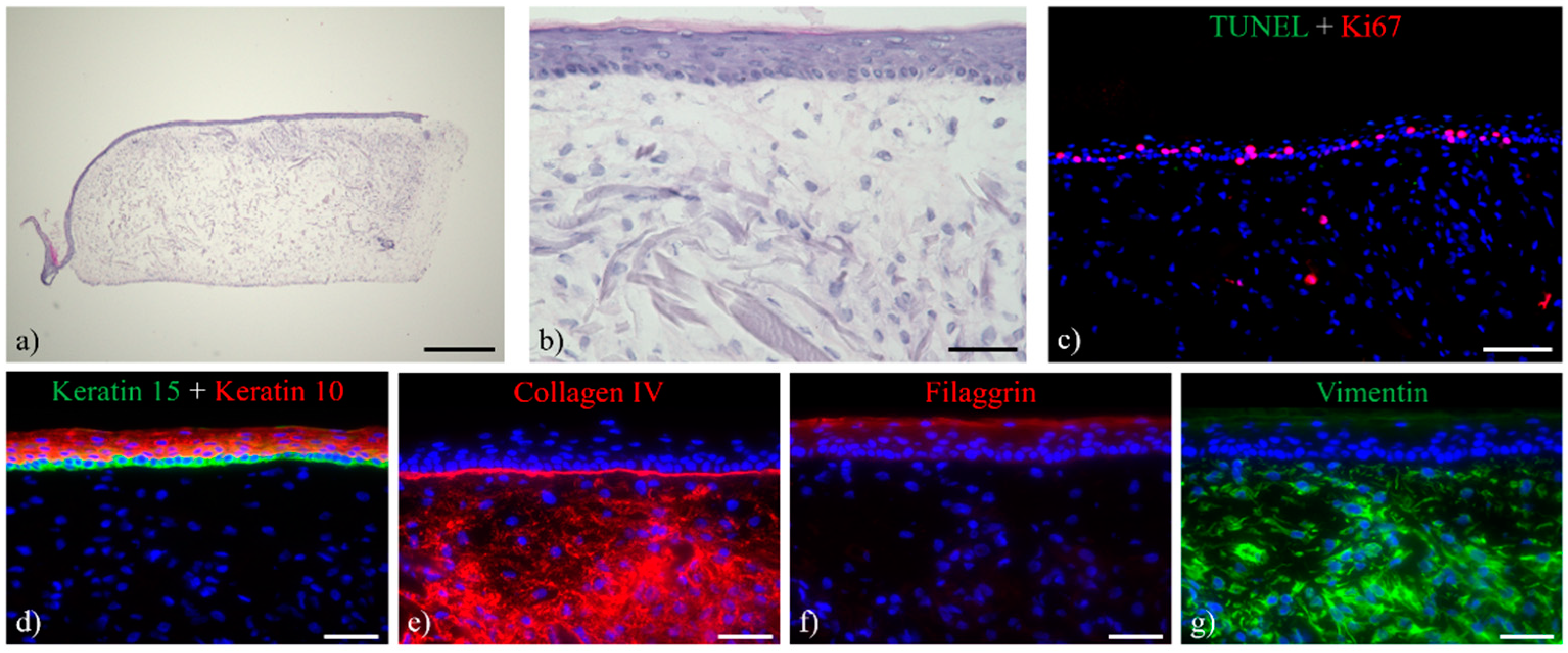
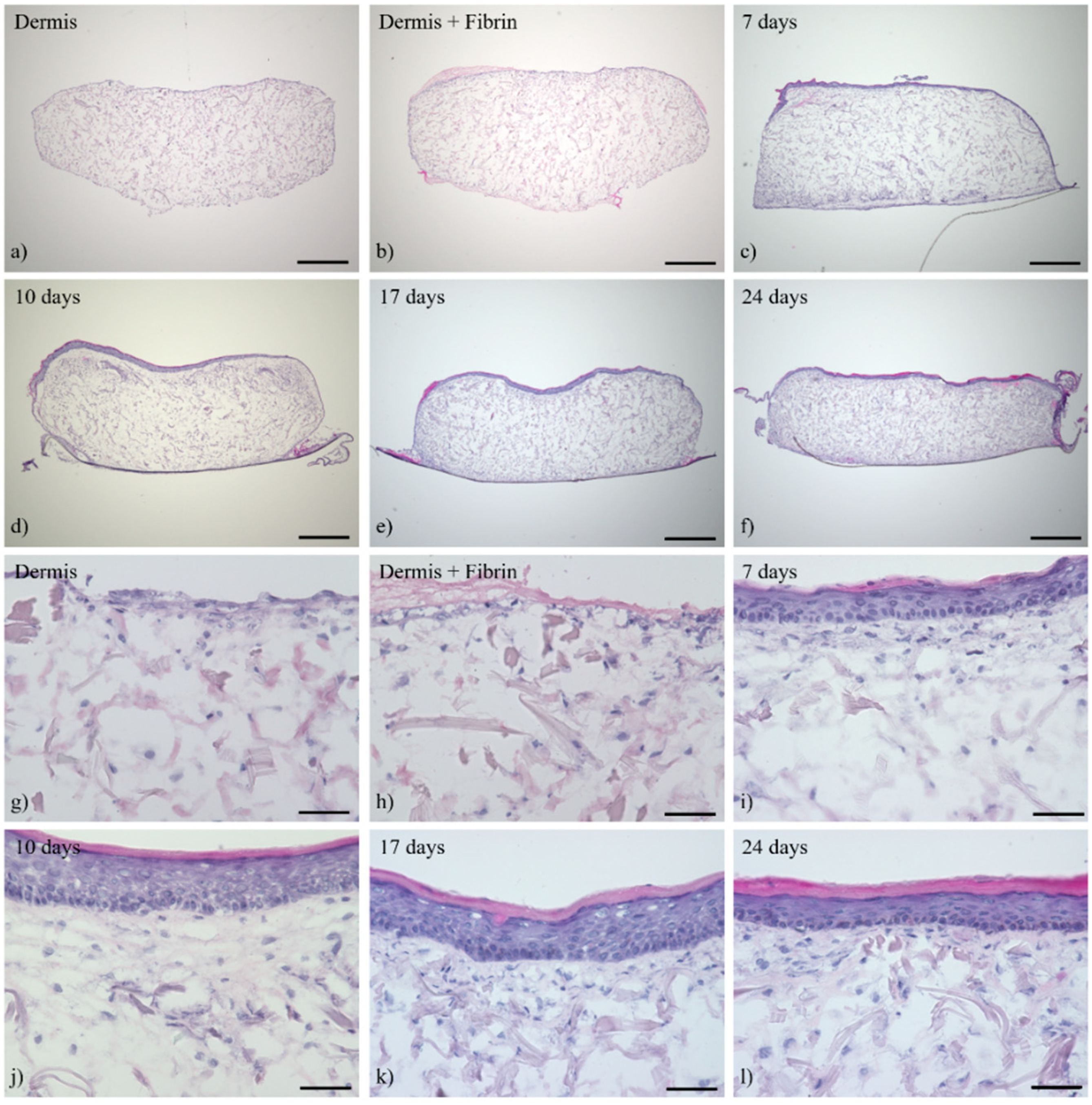
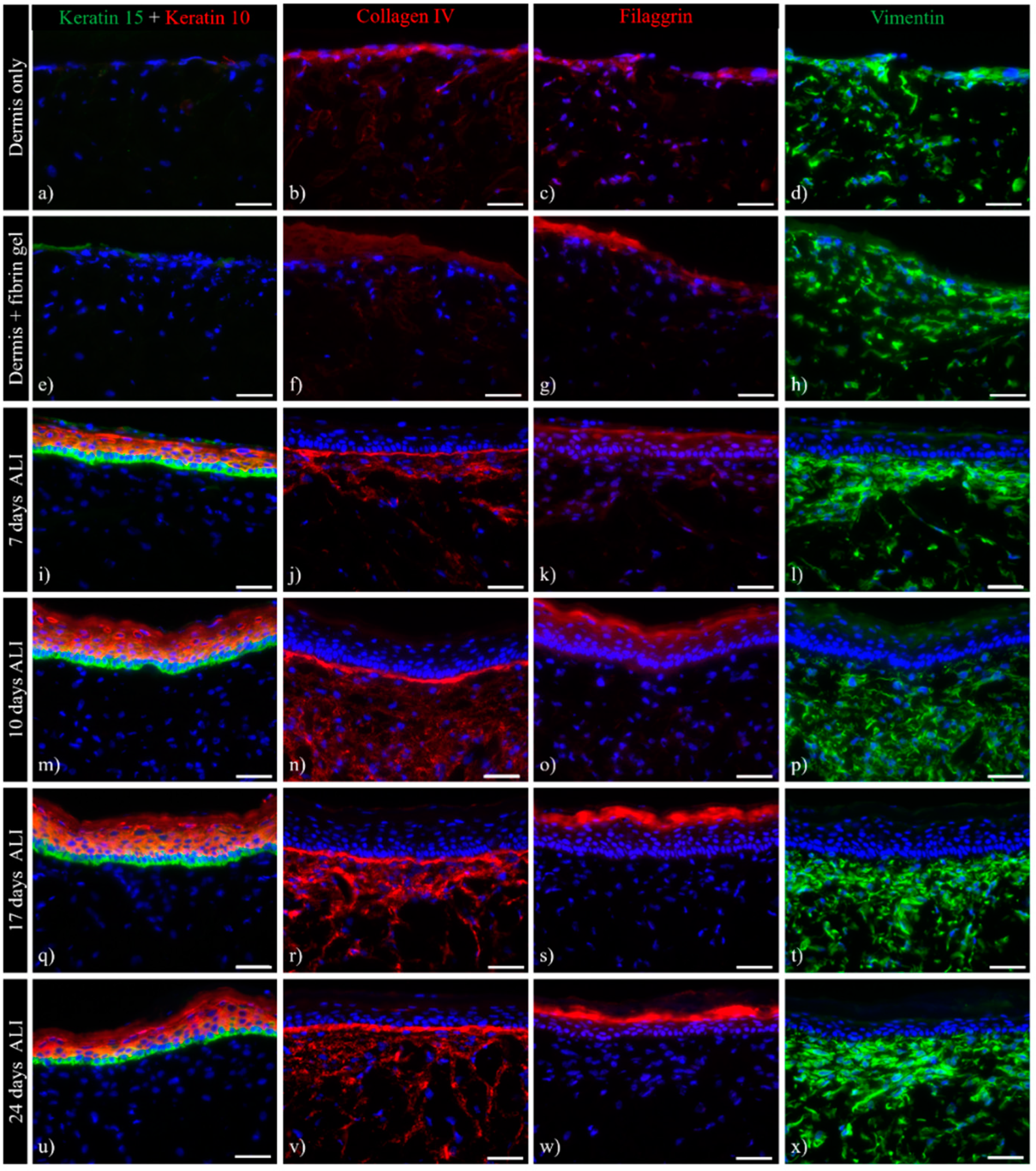
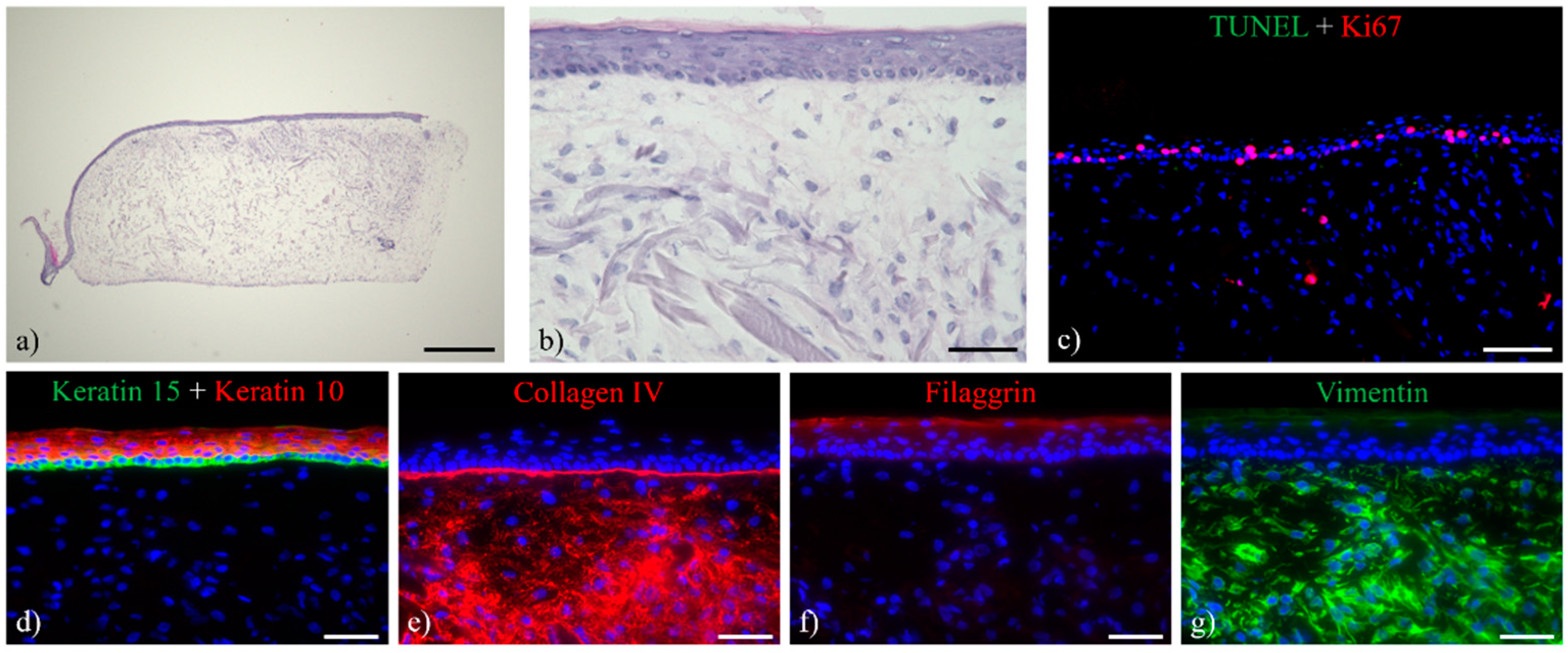
3.1. Histological Analysis of the Full-Thickness Skin Equivalents
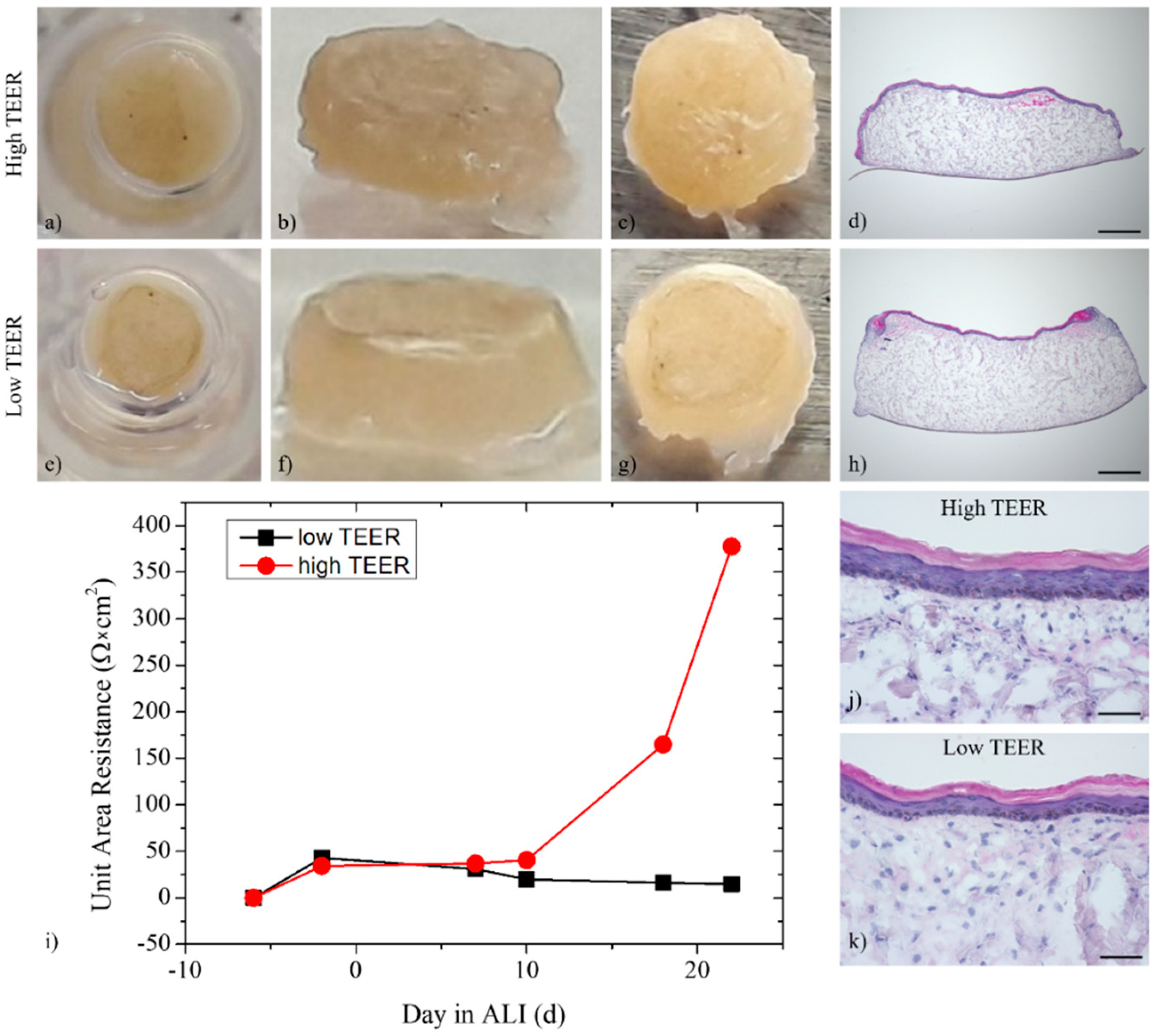
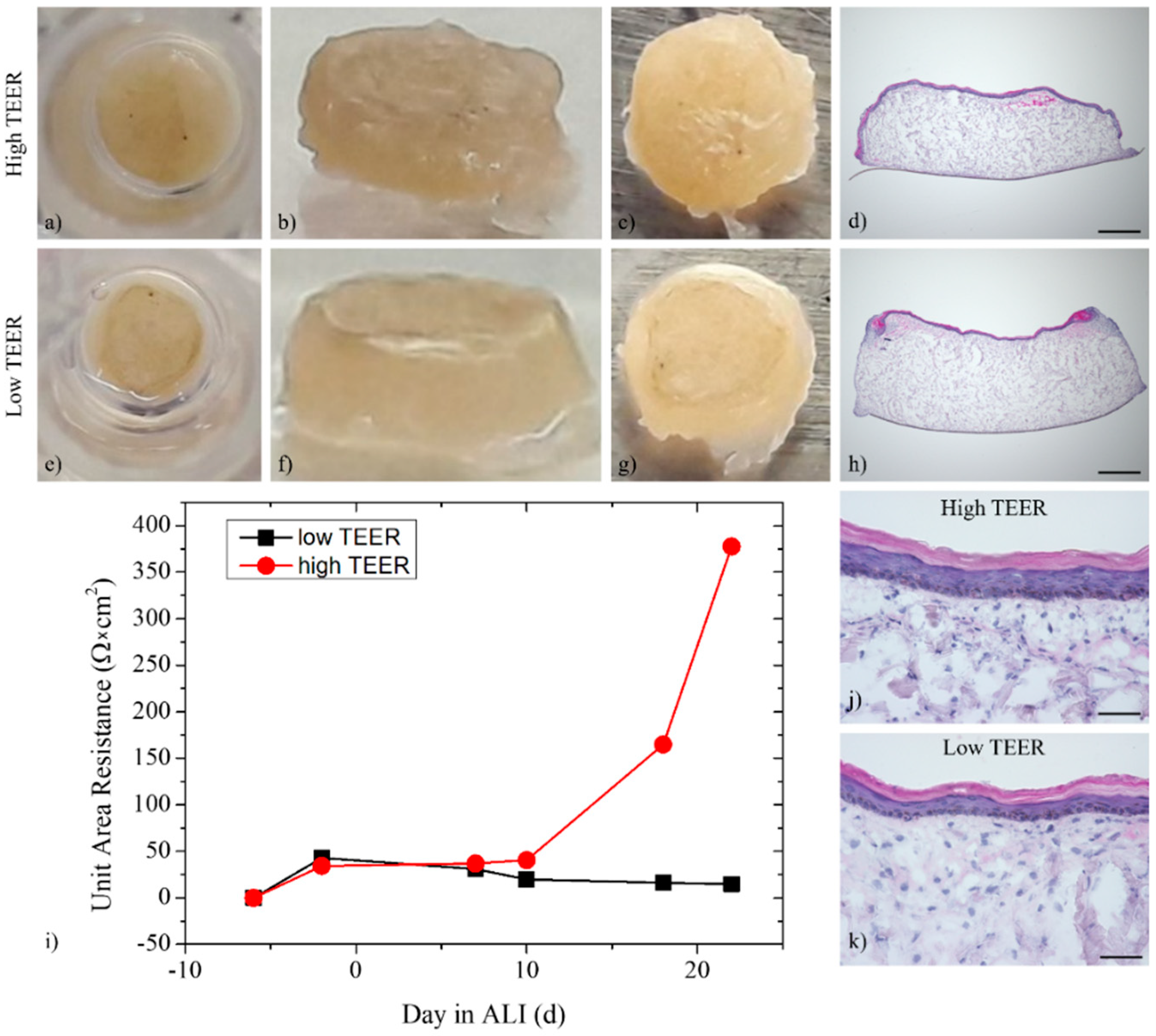
3.2. Multi-Organ-Chip Culture
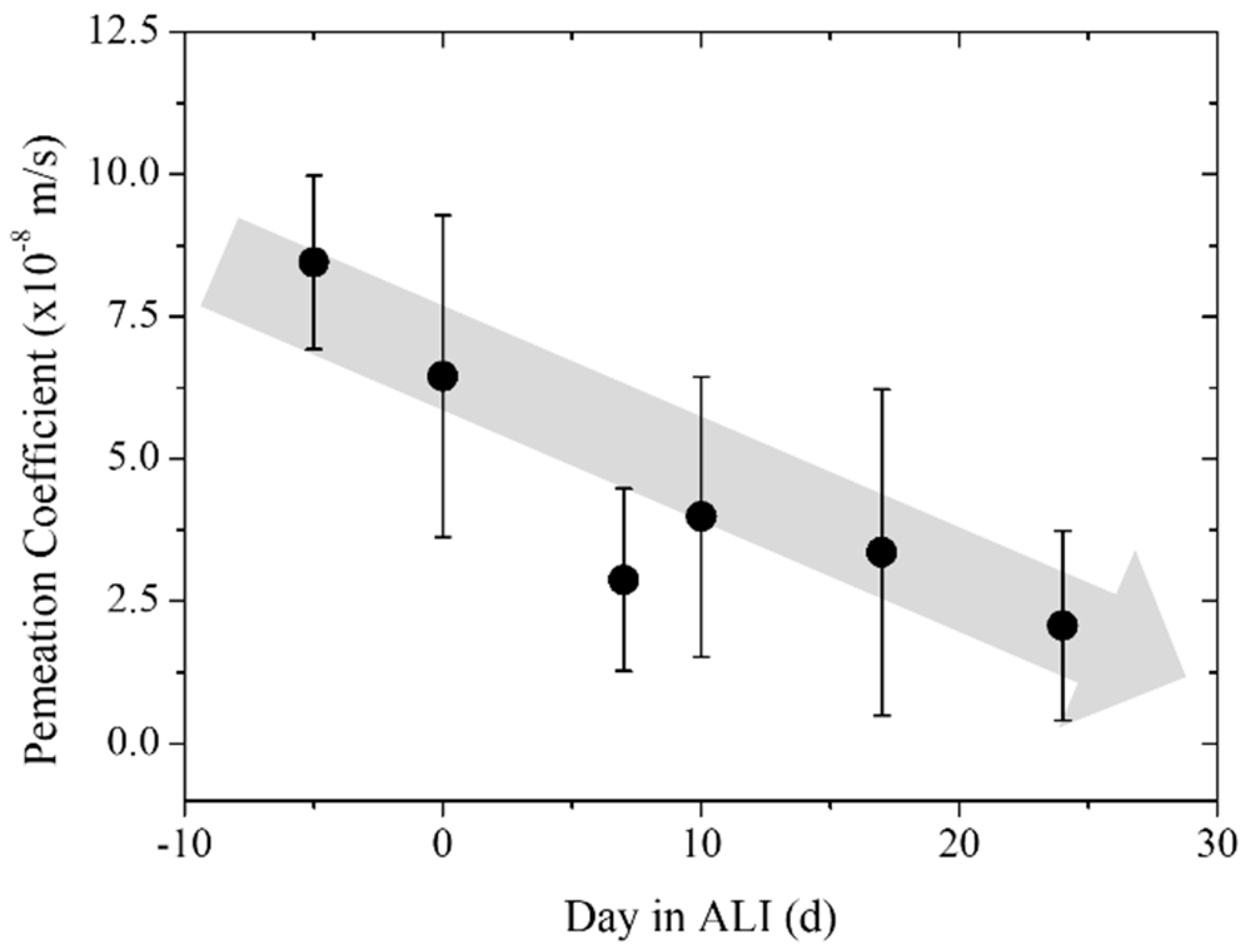
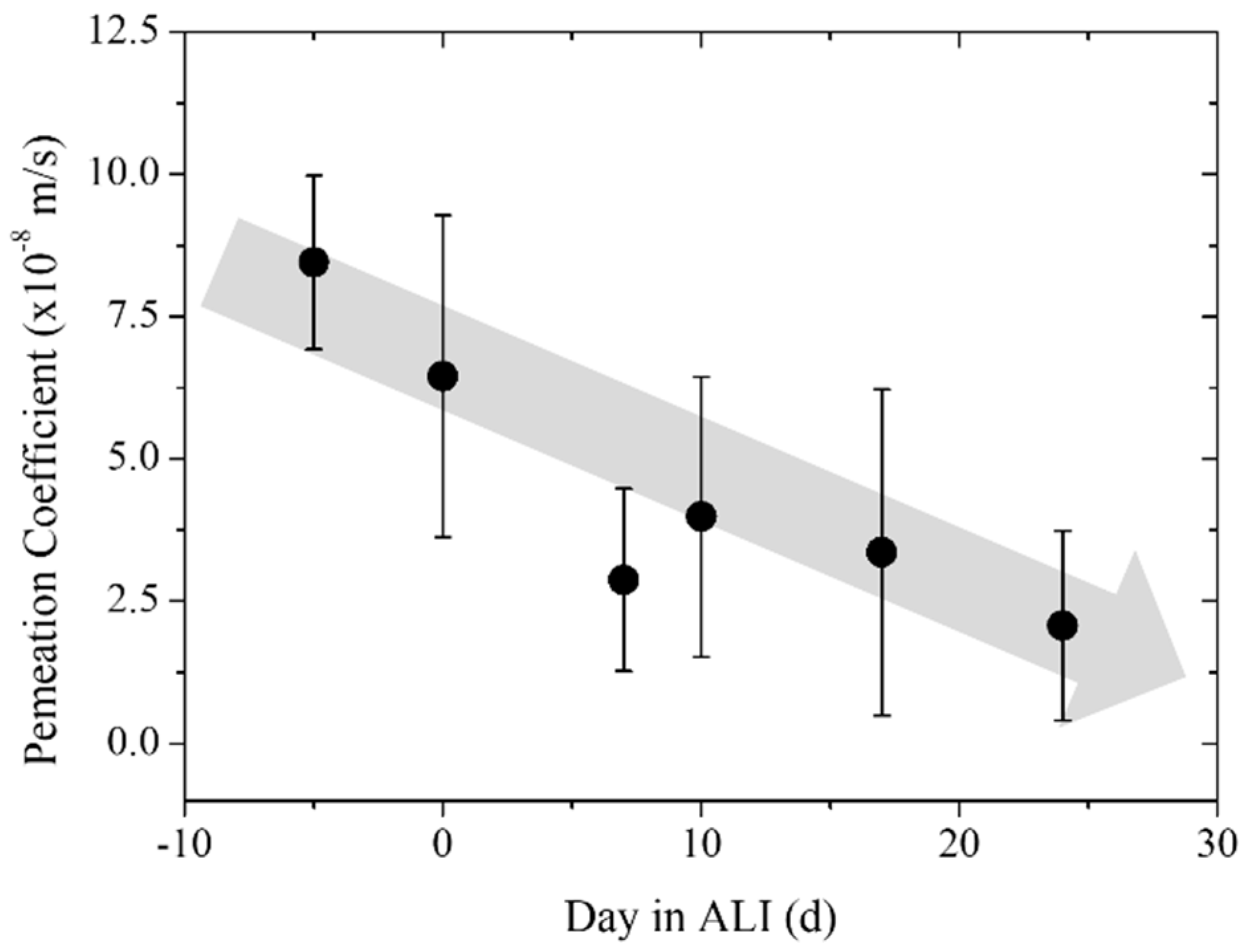
3.3. Trans-Epithelial Electrical Resistance and Permeation
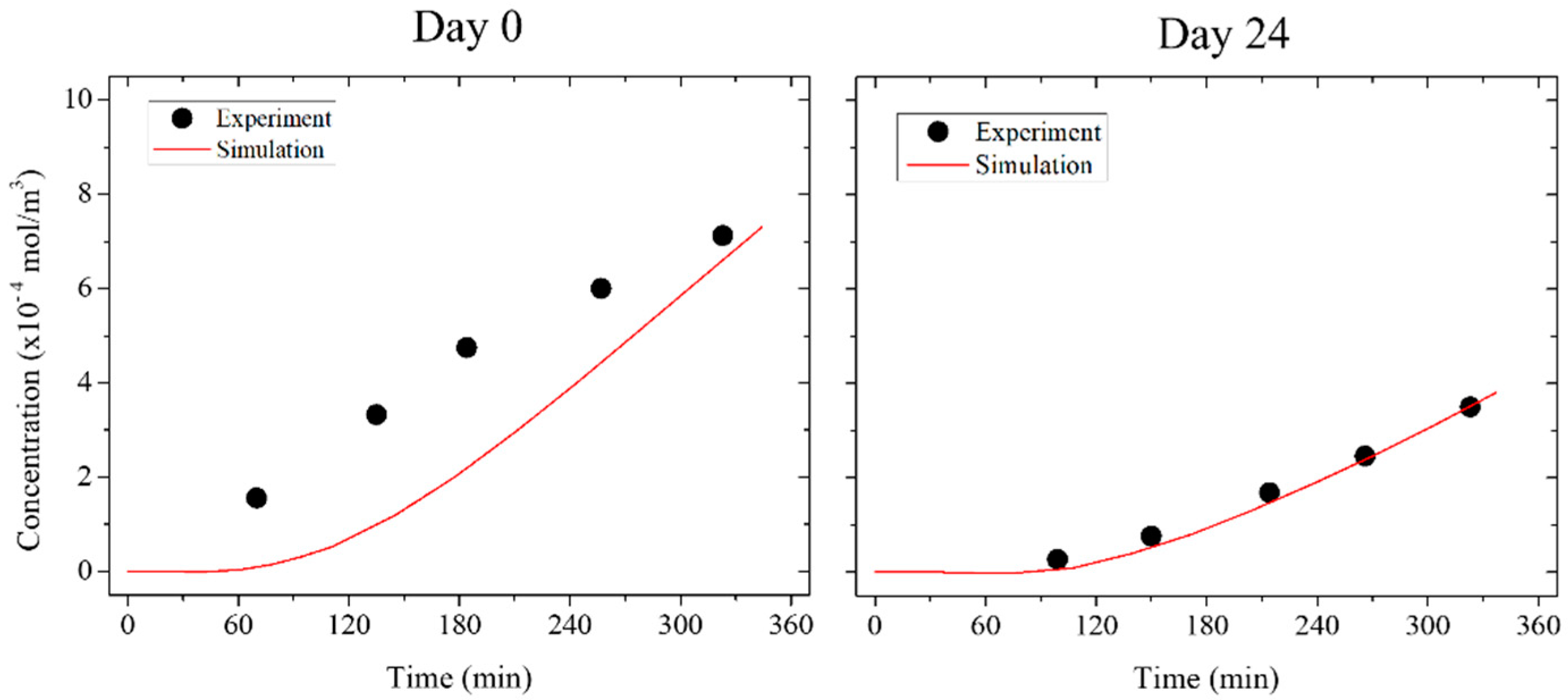
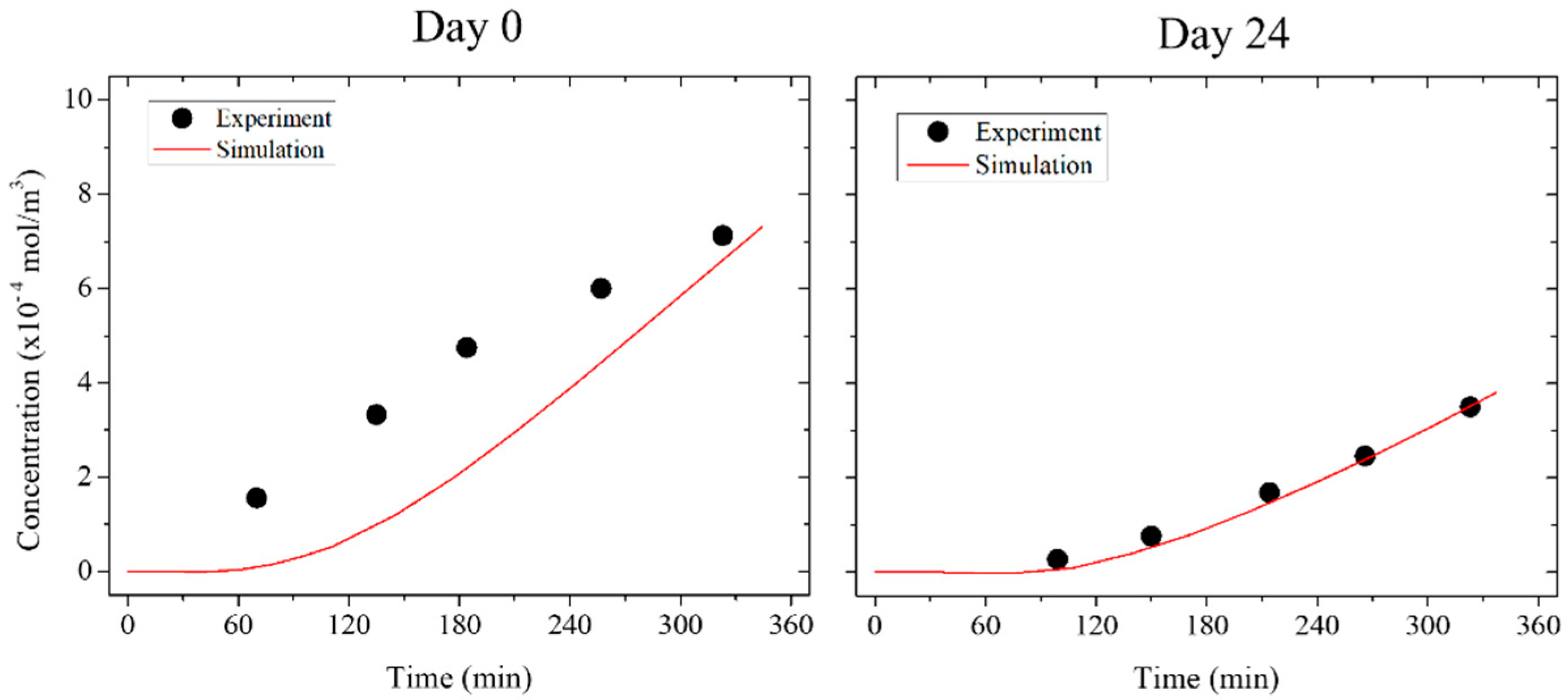
3.4. Numerical Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chuong, C.M.; Nickoloff, B.J.; Elias, P.M.; Goldsmith, L.A.; Macher, E.; Maderson, P.A.; Sundberg, J.P.; Tagami, H.; Plonka, P.M.; Therstrup-Pederson, K.; et al. What is the ‘true’ function of skin? Exp. Dermatol. 2002, 11, 159–187. [Google Scholar] [PubMed]
- McNamee, P.; Hibatallah, J.; Costabel-Farkas, M.; Goebel, C.; Araki, D.; Dufour, E.; Hewitt, N.J.; Jones, P.; Kirst, A.; Le Varlet, B.; et al. A tiered approach to the use of alternatives to animal testing for the safety assessment of cosmetics: Eye irritation. Regul. Toxicol. Pharmacol. 2009, 54, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basketter, D.; Jírova, D.; Kandárová, H. Review of skin irritation/corrosion hazards on the basis of human data: A regulatory perspective. Interdiscip. Toxicol. 2012, 5, 98–104. [Google Scholar] [CrossRef] [PubMed]
- York, M.; Griffiths, H.A.; Whittle, E.; Basketter, D.A. Evaluation of a human patch test for the identification and classification of skin irritation potential. Contact Dermat. 1996, 34, 204–212. [Google Scholar] [CrossRef]
- Ackermann, K.; Lombardi Borgia, S.; Korting, H.C.; Mewes, K.R.; Schäfer-Korting, M. The Phenion® full-thickness skin model for percutaneous absorption testing. Skin Pharmacol. Physiol. 2010, 23, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asbill, C. Evaluation of a human bio-engineered skin equivalent for drug permeation studies. Pharm. Res. 2000, 17, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Dreher, F.; Patouillet, C.; Fouchard, F.; Zanini, M.; Messager, A.; Roguet, R.; Cottin, M.; Leclaire, J.; Benech-Kieffer, F. Improvement of the experimental setup to assess cutaneous bioavailability on human skin models: Dynamic protocol. Skin Pharmacol. Appl. Skin Physiol. 2002, 15, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Netzlaff, F.; Kaca, M.; Bock, U.; Haltner-Ukomadu, E.; Meiers, P.; Lehr, C.-M.; Schaefer, U.F. Permeability of the reconstructed human epidermis model Episkin® in comparison to various human skin preparations. Eur. J. Pharm. Biopharm. 2007, 66, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Bram, B.; Jente, B.; Christian, B.; Nathalie, R.; Filip, S.; Philipe, B.; van Jan, B.; de Bart, S. A new discriminative criterion for the development of Franz diffusion tests for transdermal pharmaceuticals. J. Pharm. Pharm. Sci. 2010, 13, 218–230. [Google Scholar]
- Bonferoni, M.C.; Rossi, S.; Ferrari, F.; Caramella, C. A modified Franz diffusion cell for simultaneous assessment of drug release and washability of mucoadhesive gels. Pharm. Dev. Technol. 1999, 4, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Mehta, G.; Lesher-Perez, S.C.; Takayama, S. Organs-on-a-chip: A focus on compartmentalized microdevices. Ann. Biomed. Eng. 2012, 40, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Tissue models: A living system on a chip. Nature 2011, 471, 661–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, U.; Walles, H.; Hoffmann, S.; Lindner, G.; Horland, R.; Sonntag, F.; Klotzbach, U.; Sakharov, D.; Tonevitsky, A.; Lauster, R. ‘Human-on-a-chip’ developments: A translational cutting-edge alternative to systemic safety assessment and efficiency evaluation of substances in laboratory animals and man? Altern. Lab. Anim. 2012, 40, 235–257. [Google Scholar] [PubMed]
- Abaci, H.E.; Gledhill, K.; Guo, Z.; Christiano, A.M.; Shuler, M.L. Pumpless microfluidic platform for drug testing on human skin equivalents. Lab Chip 2015, 15, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, M.; Dancik, Y.; Sriram, G.; Wu, B.; Teo, Y.L.; Feng, Z.; Bigliardi-Qi, M.; Wu, R.G.; Wang, Z.P.; Bigliardi, P.L. Multi-chamber microfluidic platform for high-precision skin permeation testing. Lab Chip 2017, 17, 1625–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Morimoto, Y.; Takeuchi, S. Skin integrated with perfusable vascular channels on a chip. Biomaterials 2017, 116, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Sriram, G.; Alberti, M.; Dancik, Y.; Wu, B.; Wu, R.; Feng, Z.; Ramasamy, S.; Bigliardi, P.L.; Bigliardi-Qi, M.; Wang, Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Mater. Today 2018, 21, 326–340. [Google Scholar] [CrossRef]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.-M.; Maschmeyer, I.; Lorenz, A.K.; Horland, R.; Schimek, K.M.S.; Busek, M.; Sonntag, F.; Lauster, R.; Marx, U. The Multi-organ chip—A microfluidic platform for long-term multi-tissue coculture. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wagner, I.; Materne, E.-M.; Brincker, S.; Süßbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.; Haluza, D.; Burjak, S.; Eisenbock, B.; Kamolz, L.-P.; Frey, M. Cultivation of keratinocytes and preadipocytes on a collagen-elastin scaffold (Matriderm®): First results of an in vitro study. Eur. Surg. 2009, 41, 189–193. [Google Scholar] [CrossRef]
- Hsu, H.-H.; Kracht, J.-K.; Harder, L.E.; Rudnik, K.; Lindner, G.; Schimek, K.; Marx, U.; Pörtner, R. A method for determination and simulation of permeability and diffusion in a 3D tissue model in a membrane insert system for multi-well plates. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-H.; Schimek, K.; Marx, U.; Pörtner, R. Measurement and simulation of permeation and diffusion in native and cultivated tissue constructs. In Biomaterials in Regenerative Medicine; Dobrzański, L.A., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Marx, U.; Atac, B.; Brincker, S.; Busek, M.; Drewell, C.; Eger, R.; Fischer, M.; Frädrich, C.; Geßner, D.; Hasenberg, T.; et al. Multi-Organ-Chips: Developing a Human Chip-Based Plattform for Repeated Does Toxicity Testing; Technische Universität Berlin: Berlin, Germany, 2012. [Google Scholar]
- Schimek, K.; Busek, M.; Brincker, S.; Groth, B.; Hoffmann, S.; Lauster, R.; Lindner, G.; Lorenz, A.; Menzel, U.; Sonntag, F.; et al. Integrating biological vasculature into a multi-organ-chip microsystem. Lab Chip 2013, 13, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.-M.; Ramme, A.P.; Terrasso, A.P.; Serra, M.; Alves, P.M.; Brito, C.; Sakharov, D.A.; Tonevitsky, A.G.; Lauster, R.; Marx, U. A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing. J. Biotechnol. 2015, 205, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.-M.; Tonevitsky, A.G.; Marx, U. Chip-based liver equivalents for toxicity testing—Organotypicalness vs. cost-efficient high throughput. Lab Chip 2013, 13, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, F.; Gruchow, M.; Wagner, I.; Lindner, G.; Marx, U. Miniaturisierte humane organtypische Zell-und Gewebekulturen. Biospektrum 2011, 17, 418–421. [Google Scholar] [CrossRef]
- Busek, M.; Nötzel, M.; Polk, C.; Sonntag, F. Characterization and simulation of peristaltic micropumps. J. Sens. Sens. Syst. 2013, 2, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.-A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver–intestine and liver–skin co-cultures—A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Bose, A.; Teh, M.-T.; Mackenzie, I.C.; Waseem, A. Keratin k15 as a biomarker of epidermal stem cells. Int. J. Mol. Sci. 2013, 14, 19385–19398. [Google Scholar] [CrossRef] [PubMed]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, D.; Koxholt, I.; Thiemann, K.; Nischt, R. Skin basement membrane: The foundation of epidermal integrity—BM functions and diverse roles of bridging molecules nidogen and perlecan. BioMed Res. Int. 2013, 2013, 179784. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrell, J.M.; Baber, M.A.; Caplan, A.I. Clonal characterization of fibroblasts in the superficial layer of the adult human dermis. Cell Tissue Res. 2007, 327, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Groeber, F.; Holeiter, M.; Hampel, M.; Hinderer, S.; Schenke-Layland, K. Skin tissue engineering—In vivo and in vitro applications. Adv. Drug Deliv. Rev. 2011, 63, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Horch, R. Single-cell suspensions of cultured human keratinocytes in fibrin–glue reconstitute the epidermis. Cell Transpl. 1998, 7, 309–317. [Google Scholar] [CrossRef]
- Pellegrini, G.; Ranno, R.; Stracuzzi, G.; Bondanza, S.; Guerra, L.; Zambruno, G.; Micali, G.; de Luca, M. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin. Transplantation 1999, 68, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Ronfard, V.; Rives, J.M.; Neveux, Y.; Carsin, H.; Barrandon, Y. Long-term regeneration of human epidermis on third degree burns transplanted with autologous cultured epithelium grown on a fibrin matrix. Transplantation 2000, 70, 1588–1598. [Google Scholar] [CrossRef] [PubMed]
- Guth, K.; Schäfer-Korting, M.; Fabian, E.; Landsiedel, R.; van Ravenzwaay, B. Suitability of skin integrity tests for dermal absorption studies in vitro. Toxicol. Vitro 2015, 29, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Netzlaff, F.; Lehr, C.-M.; Wertz, P.W.; Schaefer, U.F. The human epidermis models EpiSkin, SkinEthic and EpiDerm: An evaluation of morphology and their suitability for testing phototoxicity, irritancy, corrosivity, and substance transport. Eur. J. Pharm. Biopharm. 2005, 60, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Groeber, F.; Engelhardt, L.; Egger, S.; Werthmann, H.; Monaghan, M.; Walles, H.; Hansmann, J. Impedance spectroscopy for the non-destructive evaluation of in vitro epidermal models. Pharm Res. 2015, 32, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Paul, A.; Vrana, N.E.; Zhao, X.; Memic, A.; Hwang, Y.-S.; Dokmeci, M.R.; Khademhosseini, A. Microfluidic techniques for development of 3D vascularized tissue. Biomaterials 2014, 35, 7308–7325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.E.; Blankschtein, D.; Langer, R. Evaluation of solute permeation through the stratum corneum: Lateral bilayer diffusion as the primary transport mechanism. J. Pharm. Sci. 1997, 86, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Edwards, D.A.; Blankschtein, D.; Langer, R. A mechanistic study of ultrasonically-enhanced transdermal drug delivery. J. Pharm. Sci. 1995, 84, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Metzler, R.; Klafter, J. The random walk’s guide to anomalous diffusion: A fractional dynamics approach. Phys. Rep. 2000, 339, 1–77. [Google Scholar] [CrossRef]
- Saxton, M.J. Anomalous subdiffusion in fluorescence photobleaching recovery: A monte carlo study. Biophys. J. 2001, 81, 2226–2240. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Dilution in PBS | Manufacturer |
|---|---|---|
| Collagen I, mouse anti-human, 5.9 mg/mL | 1:100 | Sigma 1 |
| Cytokeratin 10, mouse anti-human, 1 mg/mL | 1:100 | Chemicon 2 |
| Cytokeratin 15, rabbit anti-human, 100 µg/mL | 1:100 | Chemicon 2 |
| Filaggrin, mouse anti-human, 0.2 mg/mL | 1:50 | Thermo Fisher 3 |
| IgG–CF 488, goat anti-rabbit, 2 mg/mL | 1:200 | Biotium 4 |
| IgG–CF 594, goat anti-mouse, 2 mg/mL | 1:200 | Biotium 4 |
| Vimentin, IgG rabbit anti-human, 200 µg/mL | 1:100 | Santa Cruz 5 |
| Time (Day in ALI) | Permeation Coefficient (×10−8 m/s) | Error (×10−8 m/s) | Number of Samples (N) |
|---|---|---|---|
| Pre-culture | 8.23 | 1.53 | 3 |
| Day 0 | 6.45 | 2.83 | 4 |
| Day 7 | 2.88 | 1.60 | 5 |
| Day 10 | 3.99 | 2.46 | 5 |
| Day 17 | 3.36 | 2.86 | 5 |
| Day 24 | 2.07 | 1.66 | 5 |
| Blank | 0.03 | 0.02 | 5 |
| Time | Diffusion Coefficient (×10−11 m2/s) | r2 |
|---|---|---|
| Preculter | 20.45 | −19.15 |
| Day 0 | 4.41 | 0.32 |
| Day 7 | 2.55 | <0.01 |
| Day 10 | 2.88 | 0.49 |
| Day 17 | 2.66 | 0.83 |
| Day 24 | 1.65 | 0.98 |
| Blank | 0.03 | 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schimek, K.; Hsu, H.-H.; Boehme, M.; Kornet, J.J.; Marx, U.; Lauster, R.; Pörtner, R.; Lindner, G. Bioengineering of a Full-Thickness Skin Equivalent in a 96-Well Insert Format for Substance Permeation Studies and Organ-On-A-Chip Applications. Bioengineering 2018, 5, 43. https://doi.org/10.3390/bioengineering5020043
Schimek K, Hsu H-H, Boehme M, Kornet JJ, Marx U, Lauster R, Pörtner R, Lindner G. Bioengineering of a Full-Thickness Skin Equivalent in a 96-Well Insert Format for Substance Permeation Studies and Organ-On-A-Chip Applications. Bioengineering. 2018; 5(2):43. https://doi.org/10.3390/bioengineering5020043
Chicago/Turabian StyleSchimek, Katharina, Hao-Hsiang Hsu, Moritz Boehme, Jacob Jan Kornet, Uwe Marx, Roland Lauster, Ralf Pörtner, and Gerd Lindner. 2018. "Bioengineering of a Full-Thickness Skin Equivalent in a 96-Well Insert Format for Substance Permeation Studies and Organ-On-A-Chip Applications" Bioengineering 5, no. 2: 43. https://doi.org/10.3390/bioengineering5020043