
13C-Metabolic Flux Analysis: An Accurate Approach to Demystify Microbial Metabolism for Biochemical Production

Abstract
:1. Introduction
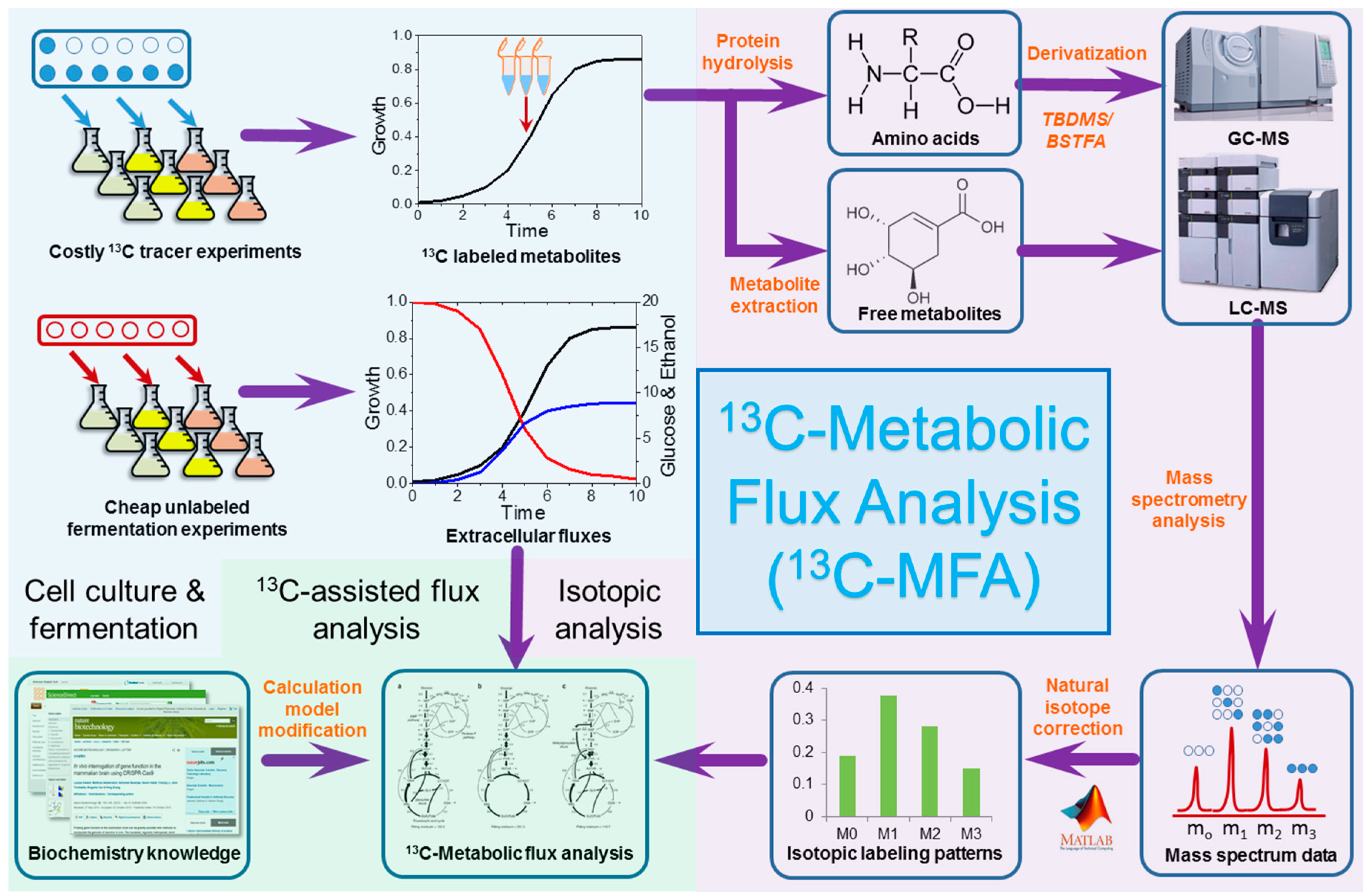
2. Techniques of 13C Metabolic Flux Analysis
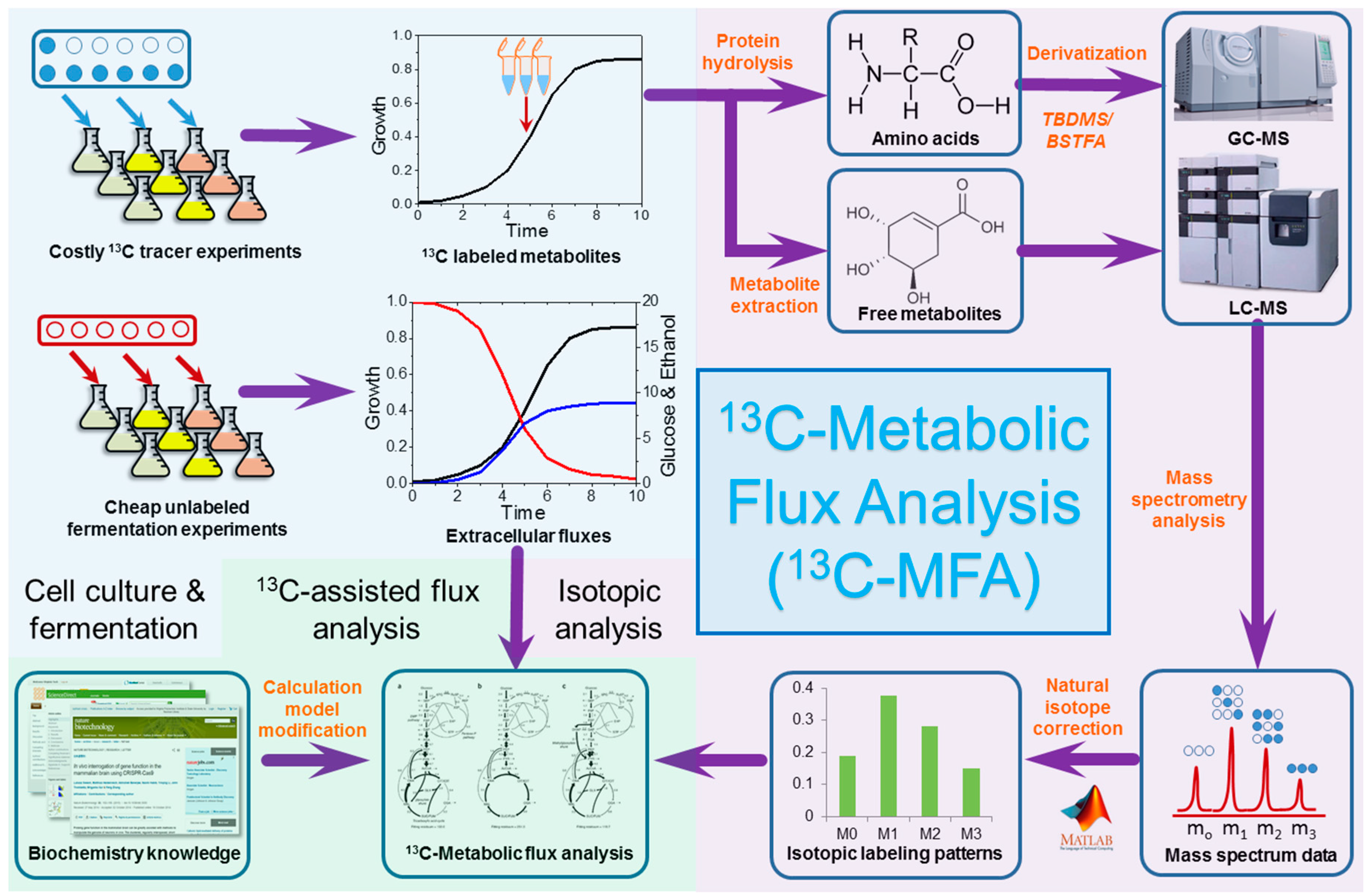
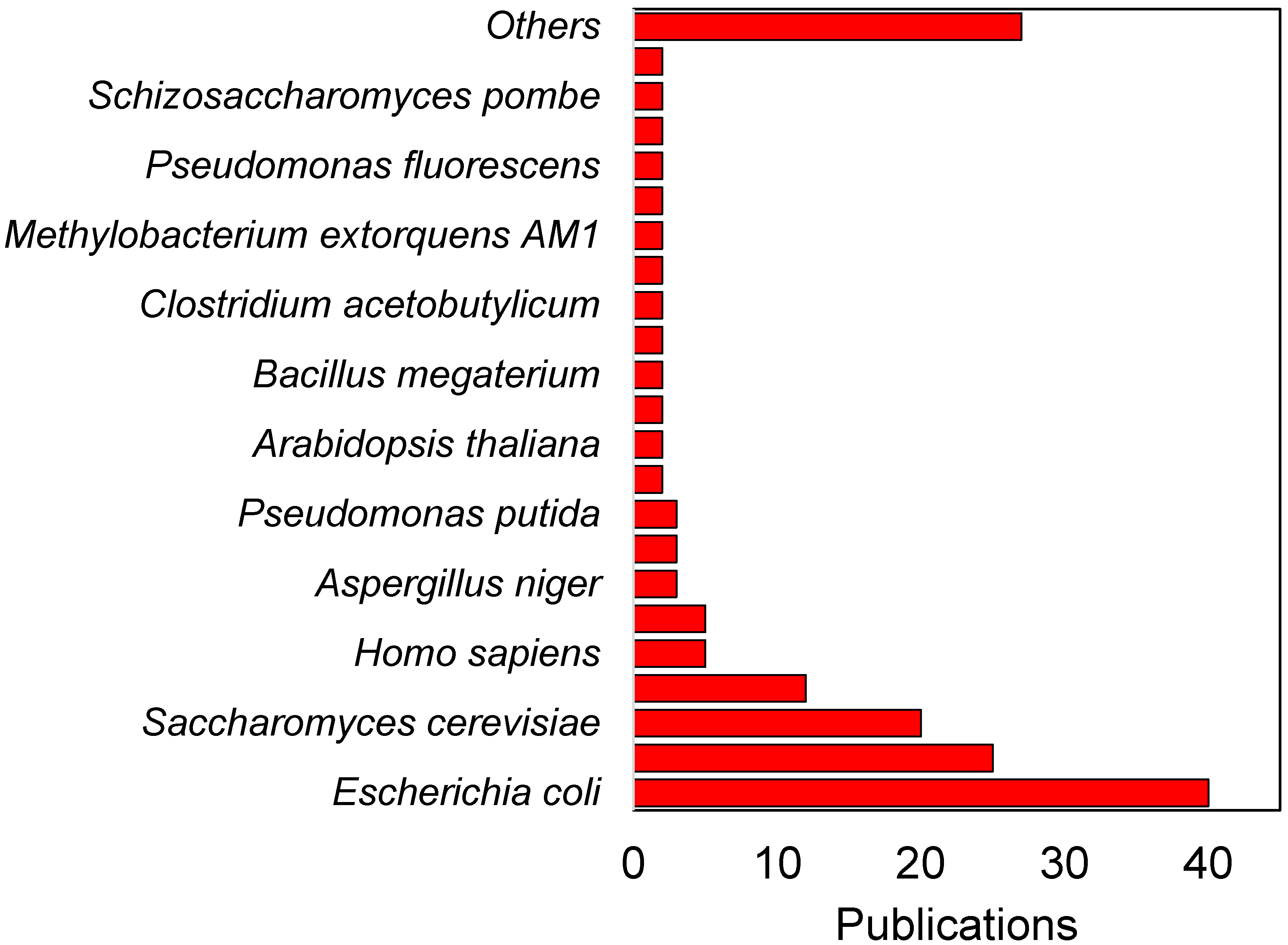
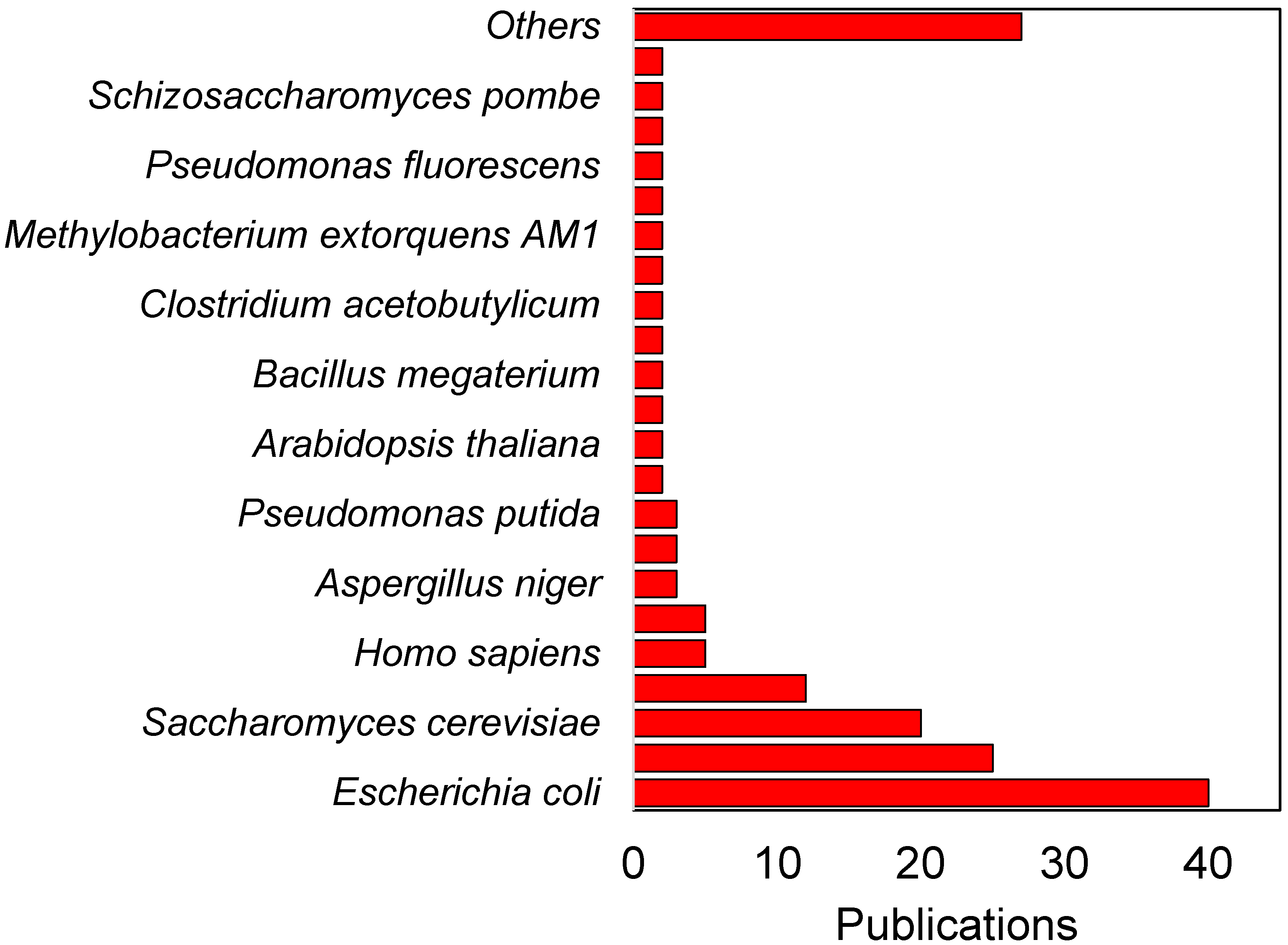
3. Integrating 13C Metabolic Flux Analysis and Metabolic Engineering for Different Industrial Microorganisms

{kind=link}
{kind=link}
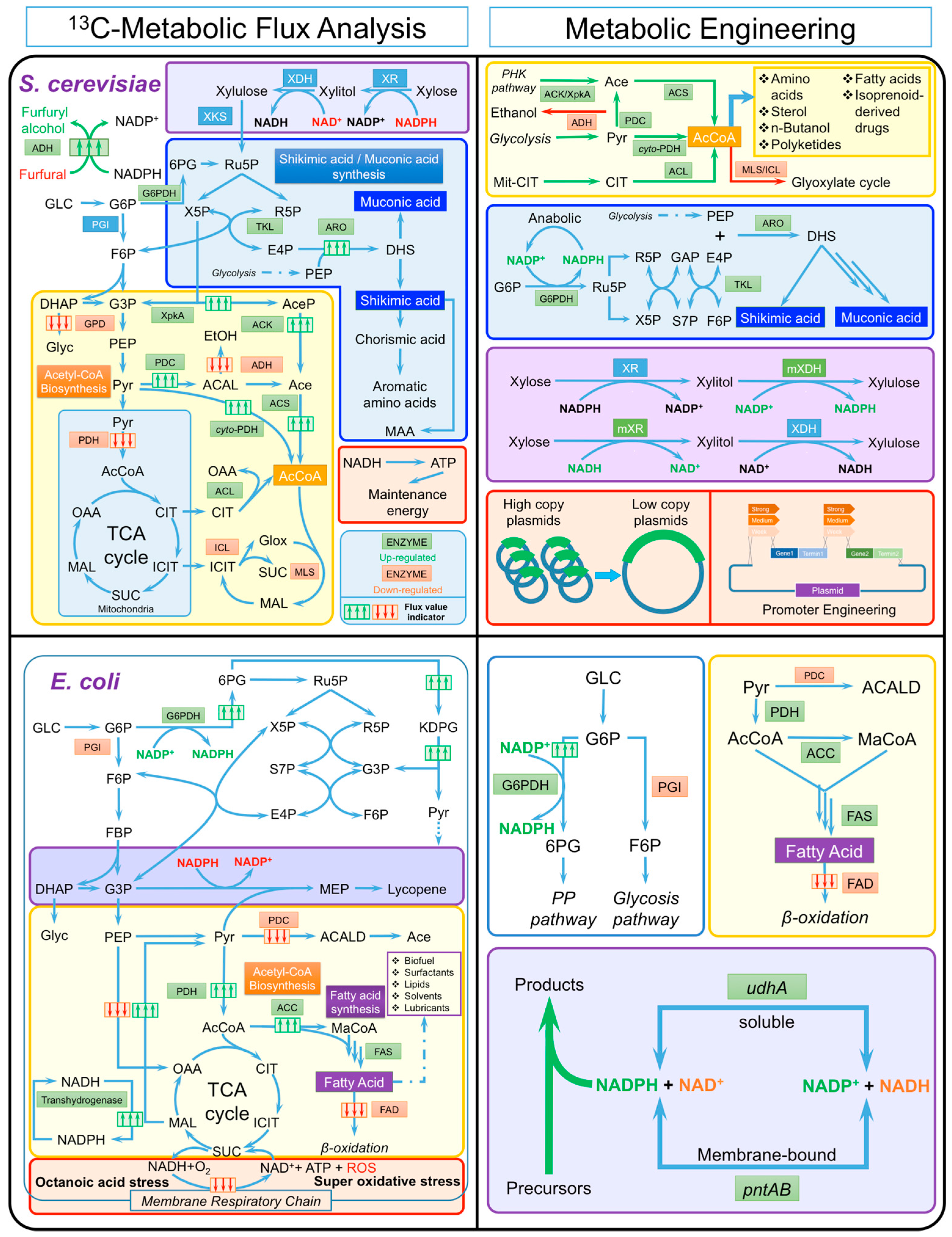
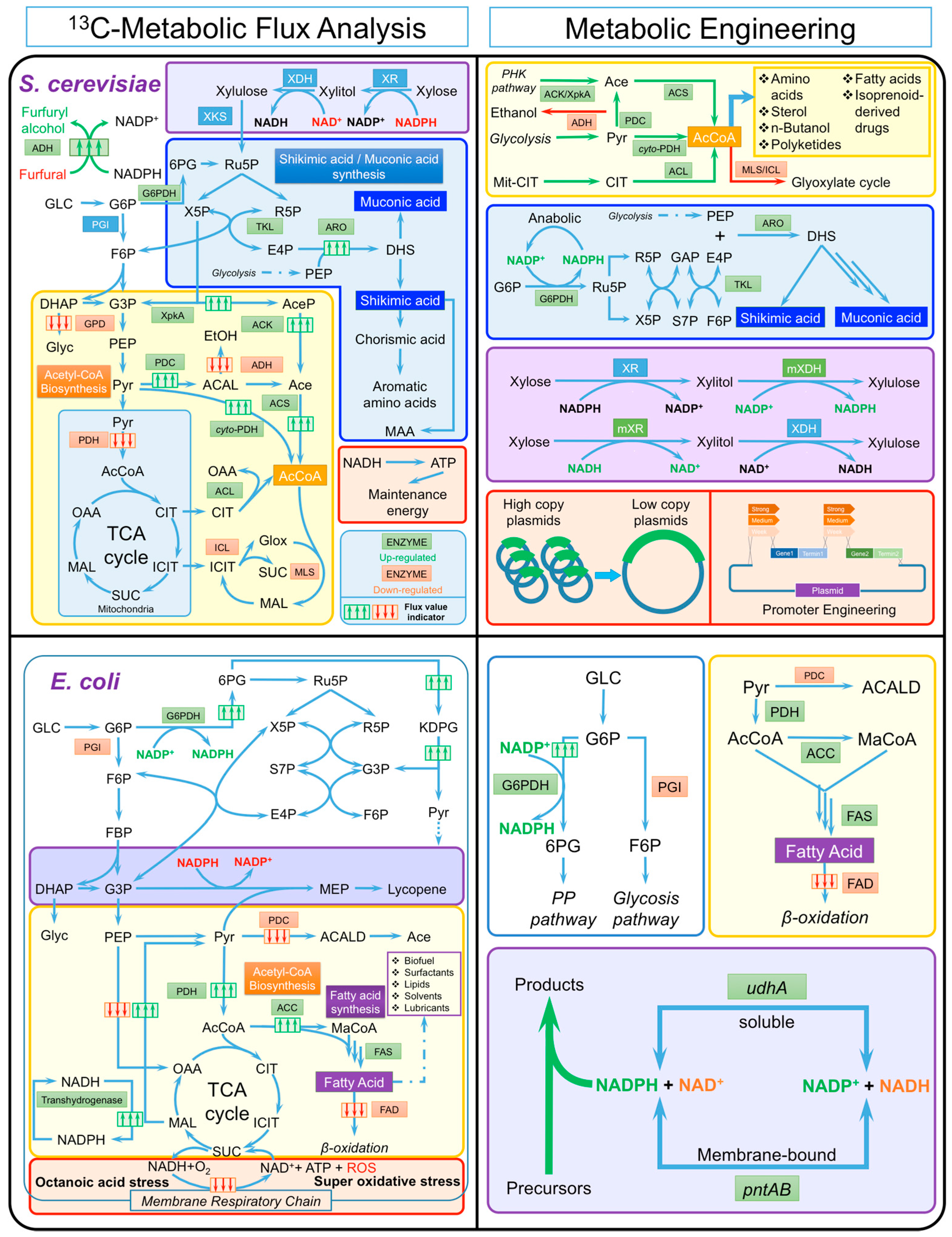{kind=link}
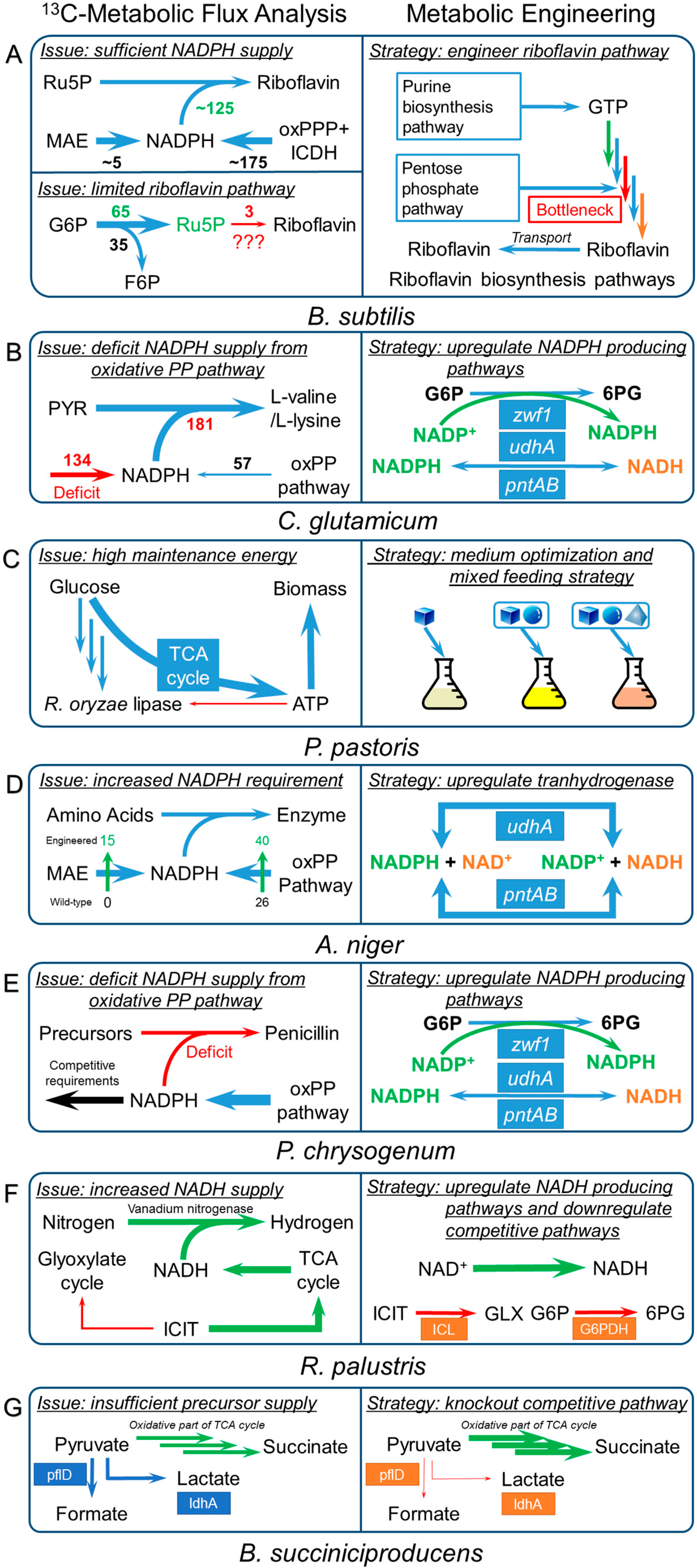{kind=link}
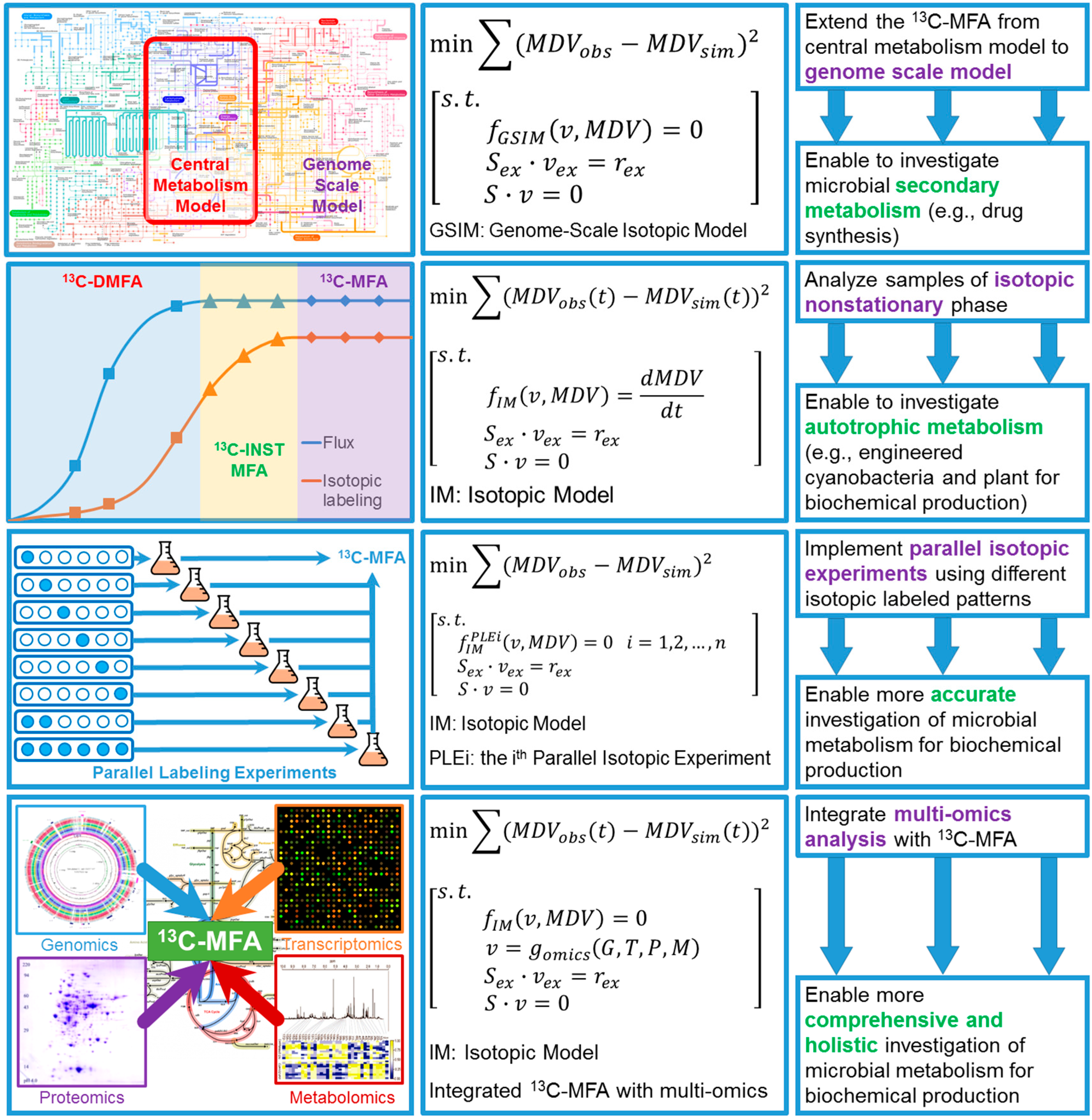{kind=link}
| Name | Capabilities | Labeled Pattern | Key Solver (Algorithm) | Platform | Developer |
|---|---|---|---|---|---|
| 13CFLUX2 [66] | Steady-state 13C-MFA | EMU [67] d | IPOPT | UNIX/Linux | Wiechert’s group |
| Metran [67] | Steady-state 13C-MFA | EMU | fmincon | MATLAB | Antoniewicz’s group |
| FIA [177] | Steady-state 13C-MFA | Fluxomer | SNOPT [178] | UNIX/Linux | Young’s group |
| influx_s [179] | Steady-state 13C-MFA | Cumomer | NLSIC [179] | UNIX/Linux | Portais’s group |
| C13 [180] | Steady-state 13C-MFA | SFL [181] e | fmincon | MATLAB | Nielsen’s group |
| OpenFLUX2 [65] | Steady-state 13C-MFA with PLEs a | EMU | fmincon | MATLAB | Mashko’s group |
| FiatFLUX [69] | METAFoRA b, steady-state 13C-MFA | MDV f | fmincon | MATLAB | Sauer’s group |
| INCA [68] | Steady-state 13C-MFA and INST-13C-MFA c | EMU | Customized Differential Equation Solver [71] | MATLAB | Young’s group |
| OpenMebius [182] | Steady-state 13C-MFA and INST-13C-MFA c | EMU | Levenberg-Marquardt method [183] | MATLAB | Shimizu’s group |
| Organism | Key Issues in Metabolic Engineering | Final Product (Objective) | Major Results of 13C-MFA | Strategies of Metabolic Engineering | Results of Metabolic Engineering |
|---|---|---|---|---|---|
| S. cerevisiae | Bottleneck step: Cytosolic acetyl-CoA supply | n-Butanol |
|
| |
| Bottleneck step: Cytosolic acetyl-CoA supply | Isoprenoid-derived drugs [184] |
|
|
| |
| Bottleneck step: Cytosolic acetyl-CoA supply | Various industrially relevant products |
|
|
| |
| Bottleneck step: Pentose phosphate pathway |
|
|
|
| |
| S. cerevisiae | Cofactor imbalance issue | Ethanol (Xylose utilization) [35] |
| ||
| S. cerevisiae | High maintenance energy | S-Adenosyl-L-methionine [34] |
|
|
|
| High maintenance energy | Xylose utilization [35] |
| |||
| S. cerevisiae | Stress response: Furfural [38] | Growth (Survival) |
|
|
|
| E. coli | Bottleneck step: Cytosolic acetyl-CoA supply, reduction power supply. | Fatty acid and fatty acid derived chemicals [56,57] |
|
|
|
| E. coli [186] | Cofactor imbalance issue | NADPH-dependent compounds [52,93,117] (Lycopene, fatty acid, etc.) |
|
|
|
| E. coli | Stress response: Octanoic acid [37] | Growth (Survival) |
|
|
|
| Stress response: Super-oxidative (paraquat induced) | Growth (Survival) |
| Suggested strategies: |
| |
| |||||
| B. subtilis | Bottleneck step: biosynthesis pathways [126] | Riboflavin | Suggested strategies: |
| |
| |||||
| B. subtilis | High maintenance energy [128,129] | Riboflavin |
|
|
|
| C. glutamicum | Cofactor imbalance issue | L-lysine |
|
| |
| Cofactor imbalance issue | L-valine [53] |
|
|
| |
| P. pastoris | High maintenance energy | R. oryzae lipase [185] |
|
| |
| A. niger | Cofactor imbalance issues | Fructofuranosidase [151] |
| Suggested strategies: |
|
| |||||
| P. chrysogenum | Cofactor imbalance issues[153] | Penicillin-G |
| Suggested strategies: |
|
| |||||
| R. palustris | Cofactor imbalance issues [154] | Hydrogen |
| Suggested strategies: |
|
| |||||
| B. succiniciproducens | Bottleneck steps in precursor supply [155] | Succinate |
|
|
|
3.1. Saccharomyces Cerevisiae
3.1.1. Bottleneck Steps
3.1.2. Cofactor Imbalance
3.1.3. Metabolic Burden and Microbial Stress
3.2. Escherichia coli
3.2.1. Bottleneck Steps
3.2.2. Cofactor Imbalance
3.2.3. Metabolic Burden and Microbial Stress
3.3. Bacillus Subtilis

3.4. Corynebacterium Glutamicum
3.5. Other Industrial Microorganisms
3.5.1. Pichia Pastoris
3.5.2. Aspergillus Niger
3.5.3. Penicillium Chrysogenum
3.5.4. Rhodopseudomonas Palustris
3.5.5. Basfia Succiniciproducens
4. Perspectives of Integrating 13C Metabolic Flux Analysis with Metabolic Engineering
4.1. Expand 13C-MFA into Genome Scale
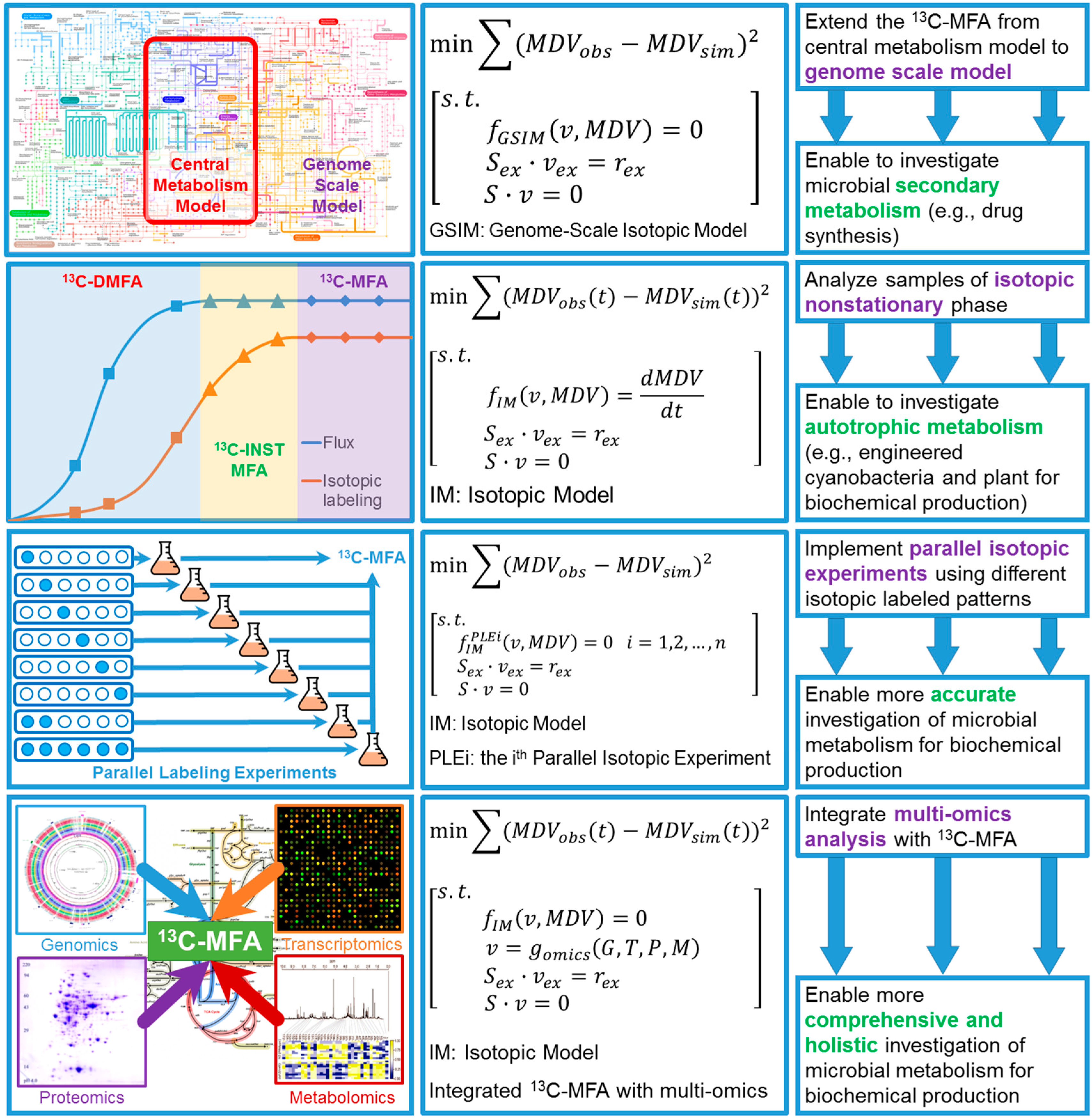
4.2. Isotopic Nonstationary 13C-MFA (13C-INST-MFA)
4.3. 13C-Based Dynamic Metabolic Flux Analysis (13C-DMFA)
4.4. Improve Accuracy of 13C-MFA via Parallel Labeling Experiments (PLE)
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sheng, J.; Feng, X. Metabolic engineering of yeast to produce fatty acid-derived biofuels: Bottlenecks and solutions. Front. Microbiol. 2015, 6, 554. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kim, H.U.; Kim, D.I.; Lee, S.Y. Production of bulk chemicals via novel metabolic pathways in microorganisms. Biotechnol. Adv. 2013, 31, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Weusthuis, R.A.; Lamot, I.; van der Oost, J.; Sanders, J.P.M. Microbial production of bulk chemicals: Development of anaerobic processes. Trends Biotechnol. 2011, 29, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Hermann, B.G.; Blok, K.; Patel, M.K. Producing bio-based bulk chemicals using industrial biotechnology saves energy and combats climate change. Environ. Sci. Technol. 2007, 41, 7915–7921. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, G. Challenges in engineering microbes for biofuels production. Science 2007, 315, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Yahya, P.P.; Keasling, J.D. Advanced biofuel production in microbes. Biotechnol. J. 2010, 5, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Yahya, P.P.; Zhang, F.; del Cardayre, S.B.; Keasling, J.D. Microbial engineering for the production of advanced biofuels. Nature 2012, 488, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Chou, H.; Ham, T.S.; Lee, T.S.; Keasling, J.D. Metabolic engineering of microorganisms for biofuels production: From bugs to synthetic biology to fuels. Curr. Opin. Biotechnol. 2008, 19, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, G. Metabolic engineering: Enabling technology for biofuels production. Metab. Eng. 2008, 10, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Xiao, W.-H.; Tyo, K.E.J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Kim, H.U.; Park, J.H.; Park, J.M.; Kim, T.Y. Metabolic engineering of microorganisms: General strategies and drug production. Drug Discov. Today 2009, 14, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.J.J.; Pitera, D.J.; Withers, S.T.; Newman, J.D.; Keasling, J.D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat. Biotech. 2003, 21, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.C.Y.; Keasling, J.D. Production of isoprenoid pharmaceuticals by engineered microbes. Nat. Chem. Biol. 2006, 2, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L.; Sanchez, S. Microbial drug discovery: 80 Years of progress. J. Antibiot. 2009, 62, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Miralles, N.; Domingo-Espín, J.; Corchero, J.L.; Vázquez, E.; Villaverde, A. Microbial factories for recombinant pharmaceuticals. Microb. Cell Fact. 2009, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Keasling, J.D. Manufacturing molecules through metabolic engineering. Science 2010, 330, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, S.; Cann, A.F.; Connor, M.R.; Shen, C.R.; Smith, K.M.; Brynildsen, M.P.; Chou, K.J.Y.; Hanai, T.; Liao, J.C. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008, 10, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli k-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Huang, C., Jr.; Lin, H.; Yang, X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 2012, 39, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Alper, H.; Miyaoku, K.; Stephanopoulos, G. Construction of lycopene-overproducing E. Coli strains by combining systematic and combinatorial gene knockout targets. Nat. Biotech. 2005, 23, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Farmer, W.R.; Liao, J.C. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat. Biotech. 2000, 18, 533–537. [Google Scholar]
- Clomburg, J.; Gonzalez, R. Biofuel production in Escherichia coli: The role of metabolic engineering and synthetic biology. Appl. Microbiol. Biotechnol. 2010, 86, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Borneman, A.R.; Desany, B.A.; Riches, D.; Affourtit, J.P.; Forgan, A.H.; Pretorius, I.S.; Egholm, M.; Chambers, P.J. Whole-genome comparison reveals novel genetic elements that characterize the genome of industrial strains of Saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1001287. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, S.; Olsson, L.; Nielsen, J. Metabolic engineering of saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2000, 64, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Veronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; Andre, B.; et al. Functional profiling of the saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Jewett, M.C. Impact of Systems Biology on Metabolic Engineering of Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Q. New challenges and opportunities for industrial biotechnology. Microb. Cell Fact. 2012, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R. Five hard truths for synthetic biology. Nature 2010, 463, 288. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M. Methods and advances in metabolic flux analysis: A mini-review. J. Ind. Microbiol. Biotechnol. 2015, 42, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D. 13c metabolic flux analysis of recombinant expression hosts. Curr. Opin. Biotechnol. 2014, 30, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; San, K.-Y.; Bennett, G.N. Cofactor engineering for advancing chemical biotechnology. Curr. Opin. Biotechnol. 2013, 24, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, T.M.; Ahn, W.S.; Stephanopoulos, G. The oxidative pentose phosphate pathway is the primary source of nadph for lipid overproduction from glucose in Yarrowia lipolytica. Metab. Eng. 2015, 30, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Hollinshead, W.D.; Henson, W.R.; Abernathy, M.; Moon, T.S.; Tang, Y.J. Rapid metabolic analysis of rhodococcus opacus pd630 via parallel 13c-metabolite fingerprinting. Biotechnol. Bioeng. 2015, 113, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Kajihata, S.; Matsuda, F.; Shimizu, H. 13c-metabolic flux analysis in s-adenosyl-l-methionine production by saccharomyces cerevisiae. J. Biosci. Bioeng. 2015. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhao, H. Investigating xylose metabolism in recombinant saccharomyces cerevisiae via 13c metabolic flux analysis. Microb. Cell Fact. 2013, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.H.; Ghaderi, A.; Fink, G.R.; Stephanopoulos, G. Engineering alcohol tolerance in yeast. Science 2014, 346, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yoon, J.; Jarboe, L.; Shanks, J. Metabolic flux analysis of Escherichia coli mg1655 under octanoic acid (c8) stress. Appl. Microbiol. Biotechnol. 2015, 99, 4397–4408. [Google Scholar] [CrossRef] [PubMed]
- Heer, D.; Heine, D.; Sauer, U. Resistance of Saccharomyces cerevisiae to high concentrations of furfural is based on nadph-dependent reduction by at least two oxireductases. Appl. Environ. Microbiol. 2009, 75, 7631–7638. [Google Scholar] [CrossRef] [PubMed]
- Çakar, Z.P.; Seker, U.O.S.; Tamerler, C.; Sonderegger, M.; Sauer, U. Evolutionary engineering of multiple-stress resistant Saccharomyces cerevisiae. FEMS Yeast Res. 2005, 5, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Heinzle, E. Mass spectrometry for metabolic flux analysis. Biotechnol. Bioeng. 1999, 62, 739–750. [Google Scholar] [CrossRef]
- Wiechert, W. 13c metabolic flux analysis. Metab. Eng. 2001, 3, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Dauner, M.; Sauer, U. Gc-ms analysis of amino acids rapidly provides rich information for isotopomer balancing. Biotechnol. Prog. 2000, 16, 642–649. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, A.A. Use of 13c labelling and nmr spectroscopy in metabolic flux analysis. In Nmr in Biotechnology: Theory and Applications; Barbotin, J.N., Portais, J.C., Eds.; Horizon Scientific Press: Norwich, UK, 2000; Chapter 4. [Google Scholar]
- Christensen, B.; Nielsen, J. Isotopomer analysis using gc-ms. Metab. Eng. 1999, 1, 282–290. [Google Scholar] [CrossRef] [PubMed]
- SZYPERSKI, T. 13c-nmr, ms and metabolic flux balancing in biotechnology research. Q. Rev. Biophys. 1998, 31, 41–106. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhuang, W.-Q.; Colletti, P.; Tang, Y. Metabolic pathway determination and flux analysis in nonmodel microorganisms through 13c-isotope labeling. In Microbial Systems Biology; Navid, A., Ed.; Humana Press: New York, NY, USA, 2012; Volume 881, pp. 309–330. [Google Scholar]
- You, L.; Page, L.; Feng, X.; Berla, B.; Pakrasi, H.B.; Tang, Y.J. Metabolic pathway confirmation and discovery through (13)c-labeling of proteinogenic amino acids. J. Vis. Exp. 2012, 59, e3583. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U. Metabolic Networks in Motion: 13c-Based Flux Analysis. Mol. Syst. Biol. 2006, 2, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Martin, H.G.; Myers, S.; Rodriguez, S.; Baidoo, E.E.K.; Keasling, J.D. Advances in analysis of microbial metabolic fluxes via 13c isotopic labeling. Mass Spectrom. Rev. 2009, 28, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Si, T.; Nair, N.U.; Zhao, H. Design and construction of acetyl-coa overproducing saccharomyces cerevisiae strains. Metab. Eng. 2014, 24, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Papini, M.; Nookaew, I.; Siewers, V.; Nielsen, J. Physiological characterization of recombinant saccharomyces cerevisiae expressing the aspergillus nidulans phosphoketolase pathway: Validation of activity through 13c-based metabolic flux analysis. Appl. Microbiol. Biotechnol. 2012, 95, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; San, K.-Y.; Bennett, G. Improvement of nadph bioavailability in Escherichia coli by replacing nad+-dependent glyceraldehyde-3-phosphate dehydrogenase gapa with nadp+-dependent gapb from bacillus subtilis and addition of nad kinase. J. Ind. Microbiol. Biotechnol. 2013, 40, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Bartek, T.; Blombach, B.; Lang, S.; Eikmanns, B.J.; Wiechert, W.; Oldiges, M.; Nöh, K.; Noack, S. Comparative 13c metabolic flux analysis of pyruvate dehydrogenase complex-deficient, l-valine-producing corynebacterium glutamicum. Appl. Environ. Microbiol. 2011, 77, 6644–6652. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Vemuri, G.; Bao, X.; Olsson, L. Impact of overexpressing nadh kinase on glucose and xylose metabolism in recombinant xylose-utilizing saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2009, 82, 909–919. [Google Scholar] [PubMed]
- Berríos-Rivera, S.J.; Bennett, G.N.; San, K.-Y. Metabolic engineering of Escherichia coli: Increase of nadh availability by overexpressing an nad+-dependent formate dehydrogenase. Metab. Eng. 2002, 4, 217–229. [Google Scholar]
- He, L.; Xiao, Y.; Gebreselassie, N.; Zhang, F.; Antoniewicz, M.R.; Tang, Y.J.; Peng, L. Central metabolic responses to the overproduction of fatty acids in Escherichia coli based on 13c-metabolic flux analysis. Biotechnol. Bioeng. 2014, 111, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, S.; Tee, T.W.; Chowdhury, A.; Zomorrodi, A.R.; Yoon, J.M.; Fu, Y.; Shanks, J.V.; Maranas, C.D. An integrated computational and experimental study for overproducing fatty acids in Escherichia coli. Metab. Eng. 2012, 14, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Fendt, S.-M.; Ruhl, M.; Sauer, U. 13c-based metabolic flux analysis. Nat. Protocols 2009, 4, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, F.; Tang, Y.; Kruppa, G.H.; Keasling, J.D. Analysis of amino acid isotopomers using ft-icr ms. Anal. Chem. 2007, 79, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Luo, S.; He, Z.; Feng, X. 13c pathway analysis of biofilm metabolism of shewanella oneidensis mr-1. RSC Adv. 2015, 5, 39840–39843. [Google Scholar] [CrossRef]
- Iwatani, S.; Van Dien, S.; Shimbo, K.; Kubota, K.; Kageyama, N.; Iwahata, D.; Miyano, H.; Hirayama, K.; Usuda, Y.; Shimizu, K.; et al. Determination of metabolic flux changes during fed-batch cultivation from measurements of intracellular amino acids by lc-ms/ms. J. Biotechnol. 2007, 128, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Letisse, F.; Sokol, S.; Portais, J.-C. Isocor: Correcting ms data in isotope labeling experiments. Bioinformatics 2012, 28, 1294–1296. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.A.; Dauner, M.; Wiechert, W. New tools for mass isotopomer data evaluation in 13c flux analysis: Mass isotope correction, data consistency checking, and precursor relationships. Biotechnol. Bioeng. 2004, 85, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, T.; Rui, B.; Zhou, W.; Zhou, X.; Shang, C.; Xin, C.; Liu, X.; Li, G.; Jiang, J.; et al. Cecafdb: A curated database for the documentation, visualization and comparative analysis of central carbon metabolic flux distributions explored by 13c-fluxomics. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Shupletsov, M.S.; Golubeva, L.I.; Rubina, S.S.; Podvyaznikov, D.A.; Iwatani, S.; Mashko, S.V. Openflux2: (13)c-mfa modeling software package adjusted for the comprehensive analysis of single and parallel labeling experiments. Microb. Cell Fact. 2014, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, M.; Nöh, K.; Dalman, T.; Niedenführ, S.; Stute, B.; Wiechert, W. 13cflux2—high-performance software suite for 13c-metabolic flux analysis. Bioinformatics 2013, 29, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Antoniewicz, M.R.; Kelleher, J.K.; Stephanopoulos, G. Elementary metabolite units (emu): A novel framework for modeling isotopic distributions. Metab. Eng. 2007, 9, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D. Inca: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Fischer, E.; Sauer, U. Fiatflux—A software for metabolic flux analysis from (13)c-glucose experiments. BMC Bioinf. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Albornoz, M.; Thankaswamy-Kosalai, S.; Nilsson, A.; Väremo, L.; Nookaew, I.; Nielsen, J. Biomet toolbox 2.0: Genome-wide analysis of metabolism and omics data. Nucleic Acids Res. 2014, 42, W175–W181. [Google Scholar] [CrossRef] [PubMed]
- Young, J.D.; Walther, J.L.; Antoniewicz, M.R.; Yoo, H.; Stephanopoulos, G. An elementary metabolite unit (emu) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 2008, 99, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Chopra, P.; Kamma, A. Engineering life through synthetic biology. Silico Biol. 2006, 6, 401–410. [Google Scholar]
- Hong, K.-K.; Nielsen, J. Metabolic engineering of saccharomyces cerevisiae: A key cell factory platform for future biorefineries. Cell. Mol. Life Sci. 2012, 69, 2671–2690. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Jewett, M.C. Impact of systems biology on metabolic engineering of saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Bro, C.; Regenberg, B.; Förster, J.; Nielsen, J. In silico aided metabolic engineering of saccharomyces cerevisiae for improved bioethanol production. Metab. Eng. 2006, 8, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Overkamp, K.M.; Bakker, B.M.; Kötter, P.; Luttik, M.A.H.; van Dijken, J.P.; Pronk, J.T. Metabolic engineering of glycerol production in saccharomyces cerevisiae. Appl. Environ. Microbiol. 2002, 68, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Lian, J.; Zhao, H. Metabolic engineering of saccharomyces cerevisiae to improve 1-hexadecanol production. Metab. Eng. 2015, 27, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Runguphan, W.; Keasling, J.D. Metabolic engineering of saccharomyces cerevisiae for production of fatty acid-derived biofuels and chemicals. Metab. Eng. 2014, 21, 103–113. [Google Scholar] [CrossRef] [PubMed]
- DeJong, J.M.; Liu, Y.; Bollon, A.P.; Long, R.M.; Jennewein, S.; Williams, D.; Croteau, R.B. Genetic engineering of taxol biosynthetic genes in saccharomyces cerevisiae. Biotechnol. Bioeng. 2006, 93, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Yamano, S.; Ishii, T.; Nakagawa, M.; Ikenaga, H.; Misawa, N. Metabolic engineering for production of β-carotene and lycopene in saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 1994, 58, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.A.; Leavitt, J.M.; Karim, A.S.; Alper, H.S. Metabolic engineering of muconic acid production in saccharomyces cerevisiae. Metab. Eng. 2013, 15, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wood, K.V.; Morgan, J.A. Metabolic engineering of the phenylpropanoid pathway in saccharomyces cerevisiae. Appl. Environ. Microbiol. 2005, 71, 2962–2969. [Google Scholar] [CrossRef] [PubMed]
- Frick, O.; Wittmann, C. Characterization of the metabolic shift between oxidative and fermentative growth in saccharomyces cerevisiae by comparative 13c flux analysis. Microb. Cell Fact. 2005, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. Atp-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed]
- Meile, L.; Rohr, L.M.; Geissmann, T.A.; Herensperger, M.; Teuber, M. Characterization of the d-xylulose 5-phosphate/d-fructose 6-phosphate phosphoketolase gene (xfp) from bifidobacterium lactis. J. Bacteriol. 2001, 183, 2929–2936. [Google Scholar] [CrossRef] [PubMed]
- Panagiotou, G.; Andersen, M.R.; Grotkjaer, T.; Regueira, T.B.; Hofmann, G.; Nielsen, J.; Olsson, L. Systems analysis unfolds the relationship between the phosphoketolase pathway and growth in Aspergillus nidulans. PLoS ONE 2008, 3, e3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, B.W.; Shi, S.; Siewers, V.; Nielsen, J. Improved production of fatty acid ethyl esters in saccharomyces cerevisiae through up-regulation of the ethanol degradation pathway and expression of the heterologous phosphoketolase pathway. Microb. Cell Fact. 2014, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, M.; Johansson, B.; Hahn-Hägerdal, B.; Gorwa-Grauslund, M.F. Reduced oxidative pentose phosphate pathway flux in recombinant xylose-utilizing saccharomyces cerevisiae strains improves the ethanol yield from xylose. Appl. Environ. Microbiol. 2002, 68, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Abu Saleh, A.; Pack, S.P.; Annaluru, N.; Kodaki, T.; Makino, K. Ethanol production from xylose by recombinant saccharomyces cerevisiae expressing protein-engineered nadh-preferring xylose reductase from pichia stipitis. Microbiology 2007, 153, 3044–3054. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, M.; Bengtsson, O.; Franke, K.; Lee, H.; Hahn-Hägerdal, B.; Gorwa-Grauslund, M.F. The expression of a pichia stipitis xylose reductase mutant with higher km for nadph increases ethanol production from xylose in recombinant saccharomyces cerevisiae. Biotechnol. Bioeng. 2006, 93, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Pack, S.P.; Saleh, A.A.; Annaluru, N.; Kodaki, T.; Makino, K. The positive effect of the decreased nadph-preferring activity of xylose reductase from pichia stipitis on ethanol production using xylose-fermenting recombinant saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2007, 71, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Runquist, D.; Hahn-Hägerdal, B.; Bettiga, M. Increased expression of the oxidative pentose phosphate pathway and gluconeogenesis in anaerobically growing xylose-utilizing saccharomyces cerevisiae. Microb. Cell Fact. 2009, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Petschacher, B.; Nidetzky, B. Altering the coenzyme preference of xylose reductase to favor utilization of nadh enhances ethanol yield from xylose in a metabolically engineered strain of saccharomyces cerevisiae. Microb. Cell Fact. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, O.; Hahn-Hägerdal, B.; Gorwa-Grauslund, M.F. Xylose reductase from pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant saccharomyces cerevisiae. Biotechnol. Biofuels 2009, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Runquist, D.; Hahn-Hägerdal, B.; Bettiga, M. Increased ethanol productivity in xylose-utilizing saccharomyces cerevisiae via a randomly mutagenized xylose reductase. Appl. Environ. Microbiol. 2010, 76, 7796–7802. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Saleh, A.A.; Pack, S.P.; Annaluru, N.; Kodaki, T.; Makino, K. Ethanol production from xylose by recombinant saccharomyces cerevisiae expressing protein engineered nadp+-dependent xylitol dehydrogenase. J. Biotechnol. 2007, 130, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Matsushika, A.; Watanabe, S.; Kodaki, T.; Makino, K.; Inoue, H.; Murakami, K.; Takimura, O.; Sawayama, S. Expression of protein engineered nadp+-dependent xylitol dehydrogenase increases ethanol production from xylose in recombinant saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2008, 81, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Krahulec, S.; Klimacek, M.; Nidetzky, B. Engineering of a matched pair of xylose reductase and xylitol dehydrogenase for xylose fermentation by saccharomyces cerevisiae. Biotechnol. J. 2009, 4, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Matsushika, A.; Inoue, H.; Watanabe, S.; Kodaki, T.; Makino, K.; Sawayama, S. Efficient bioethanol production by a recombinant flocculent saccharomyces cerevisiae strain with a genome-integrated nadp(+)-dependent xylitol dehydrogenase gene. Appl. Environ. Microbiol. 2009, 75, 3818–3822. [Google Scholar] [CrossRef] [PubMed]
- Verho, R.; Londesborough, J.; Penttilä, M.; Richard, P. Engineering redox cofactor regeneration for improved pentose fermentation in saccharomyces cerevisiae. Appl. Environ. Microbiol. 2003, 69, 5892–5897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-C.; Liu, J.-J.; Ding, W.-T. Decreased xylitol formation during xylose fermentation in saccharomyces cerevisiae due to overexpression of water-forming nadh oxidase. Appl. Environ. Microbiol. 2012, 78, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, T.M.; Stephanopoulos, G. Metabolomic and 13c-metabolic flux analysis of a xylose-consuming saccharomyces cerevisiae strain expressing xylose isomerase. Biotechnol. Bioeng. 2015, 112, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.; Bailey, J.E. Plasmid presence changes the relative levels of many host cell proteins and ribosome components in recombinant Escherichia coli. Biotechnol. Bioeng. 1991, 37, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Guyot, S.; Gervais, P.; Young, M.; Winckler, P.; Dumont, J.; Davey, H.M. Surviving the heat: Heterogeneity of response in saccharomyces cerevisiae provides insight into thermal damage to the membrane. Environ. Microbiol. 2015, 17, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Nugroho, R.H.; Yoshikawa, K.; Shimizu, H. Metabolomic analysis of acid stress response in saccharomyces cerevisiae. J. Biosci. Bioeng. 2015, 120, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Parawira, W.; Tekere, M. Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: Review. Crit. Rev. Biotechnol. 2011, 31, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Fillet, S.; Gibert, J.; Suárez, B.; Lara, A.; Ronchel, C.; Adrio, J. Fatty alcohols production by oleaginous yeast. J. Ind. Microbiol. Biotechnol. 2015, 42, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Cronan, J.E. Escherichia coli unsaturated fatty acid synthesis: Complex transcription of the faba gene and in vivo identification of the essential reaction catalyzed by fabb. J. Biol. Chem. 2009, 284, 29526–29535. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.S.; Solbiati, J.; Cronan, J.E., Jr. Overproduction of acetyl-coa carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J. Biol. Chem. 2000, 275, 28593–28598. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Vora, H.; Khosla, C. Overproduction of free fatty acids in E. Coli: Implications for biodiesel production. Metab. Eng. 2008, 10, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ouellet, M.; Batth, T.S.; Adams, P.D.; Petzold, C.J.; Mukhopadhyay, A.; Keasling, J.D. Enhancing fatty acid production by the expression of the regulatory transcription factor fadr. Metab. Eng. 2012, 14, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Subrahmanyam, S.; Cronan, J.E., Jr. Overproduction of a functional fatty acid biosynthetic enzyme blocks fatty acid synthesis in Escherichia coli. J. Bacteriol. 1998, 180, 4596–4602. [Google Scholar] [PubMed]
- Kim, Y.M.; Cho, H.-S.; Jung, G.Y.; Park, J.M. Engineering the pentose phosphate pathway to improve hydrogen yield in recombinant Escherichia coli. Biotechnol. Bioeng. 2011, 108, 2941–2946. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.; Park, J.-B.; Park, K.; Kim, M.-D.; Seo, J.-H. Enhanced production of ɛ-caprolactone by overexpression of nadph-regenerating glucose 6-phosphate dehydrogenase in recombinant Escherichia coli harboring cyclohexanone monooxygenase gene. Appl. Microbiol. Biotechnol. 2007, 76, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Cirino, P.C. Improved nadph supply for xylitol production by engineered Escherichia coli with glycolytic mutations. Biotechnol. Prog. 2011, 27, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; San, K.-Y.; Bennett, G. Improvement of nadph bioavailability in Escherichia coli through the use of phosphofructokinase deficient strains. Appl. Microbiol. Biotechnol. 2013, 97, 6883–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemler, J.A.; Fowler, Z.L.; McHugh, K.P.; Koffas, M.A.G. Improving nadph availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab. Eng. 2010, 12, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, C.H.; Nam, S.W.; Kim, P. Alteration of reducing powers in an isogenic phosphoglucose isomerase (pgi)-disrupted Escherichia coli expressing nad(p)-dependent malic enzymes and nadp-dependent glyceraldehyde 3-phosphate dehydrogenase. Lett. Appl. Microbiol. 2011, 52, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.M.; Andrews, J.; Hussein, I.; Bennett, G.N.; San, K.-Y. Effect of overexpression of a soluble pyridine nucleotide transhydrogenase (udha) on the production of poly(3-hydroxybutyrate) in Escherichia coli. Biotechnol. Prog. 2006, 22, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-H.; Marx, C.J.; Sauer, U. Transhydrogenase promotes the robustness and evolvability of E. Coli deficient in nadph production. PLoS Genet. 2015, 11, e1005007. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Kim, S.W.; Keasling, J.D. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab. Eng. 2000, 2, 328–338. [Google Scholar] [CrossRef] [PubMed]
- King, T.; Lucchini, S.; Hinton, J.C.D.; Gobius, K. Transcriptomic analysis of Escherichia coli o157:H7 and k-12 cultures exposed to inorganic and organic acids in stationary phase reveals acidulant- and strain-specific acid tolerance responses. Appl. Environ. Microbiol. 2010, 76, 6514–6528. [Google Scholar] [CrossRef] [PubMed]
- Rui, B.; Shen, T.; Zhou, H.; Liu, J.; Chen, J.; Pan, X.; Liu, H.; Wu, J.; Zheng, H.; Shi, Y. A systematic investigation of Escherichia coli central carbon metabolism in response to superoxide stress. BMC Syst. Biology 2010, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.B.; Sloma, A.; Hermann, T.; Theriault, K.; Zachgo, E.; Erdenberger, T.; Hannett, N.; Chatterjee, N.P.; Williams, V., II; Rufo, G.A., Jr.; et al. Genetic engineering of bacillus subtilis for the commercial production of riboflavin. J. Ind. Microbiol. Biotech. 1999, 22, 8–18. [Google Scholar] [CrossRef]
- Sauer, U.; Hatzimanikatis, V.; Bailey, J.E.; Hochuli, M.; Szyperski, T.; Wuthrich, K. Metabolic fluxes in riboflavin-producing bacillus subtilis. Nat. Biotech. 1997, 15, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Dauner, M.; Bailey, J.E.; Sauer, U. Metabolic flux analysis with a comprehensive isotopomer model in bacillus subtilis. Biotechnol. Bioeng. 2001, 76, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Sauer, U. Large-scale in vivo flux analysis shows rigidity and suboptimal performance of bacillus subtilis metabolism. Nat. Genet. 2005, 37, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Tannler, S.; Decasper, S.; Sauer, U. Maintenance metabolism and carbon fluxes in bacillus species. Microb. Cell Fact. 2008, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Sauer, U.; Hatzimanikatis, V.; Hohmann, H.P.; Manneberg, M.; van Loon, A.P.; Bailey, J.E. Physiology and metabolic fluxes of wild-type and riboflavin-producing bacillus subtilis. Appl. Environ. Microbiol. 1996, 62, 3687–3696. [Google Scholar] [PubMed]
- Wiechert, W.; de Graaf, A.A. In vivo stationary flux analysis by 13c labeling experiments. In Metabolic Engineering; Sahm, H., Wandrey, C., Eds.; Springer: Berlin, Germany, 1996; Volume 54, pp. 109–154. [Google Scholar]
- Wittmann, C.; Heinzle, E. Application of maldi-tof ms to lysine-producing corynebacterium glutamicum. Eur. J. Biochem. 2001, 268, 2441–2455. [Google Scholar] [CrossRef] [PubMed]
- Klapa, M.I.; Aon, J.-C.; Stephanopoulos, G. Systematic quantification of complex metabolic flux networks using stable isotopes and mass spectrometry. Eur. J. Biochem. 2003, 270, 3525–3542. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.-E.; Wittmann, C.; Nielsen, L.; Kromer, J. Openflux: Efficient modelling software for 13c-based metabolic flux analysis. Microb. Cell Fact. 2009, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Krömer, J.O.; Sorgenfrei, O.; Klopprogge, K.; Heinzle, E.; Wittmann, C. In-depth profiling of lysine-producing corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 2004, 186, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Klopprogge, C.; Herold, A.; Zelder, O.; Bolten, C.J.; Wittmann, C. Metabolic flux engineering of l-lysine production in corynebacterium glutamicum—Over expression and modification of g6p dehydrogenase. J. Biotechnol. 2007, 132, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Zelder, O.; Häfner, S.; Schröder, H.; Wittmann, C. From zero to hero—Design-based systems metabolic engineering of corynebacterium glutamicum for l-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Klopprogge, C.; Zelder, O.; Heinzle, E.; Wittmann, C. Amplified expression of fructose 1,6-bisphosphatase in corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl. Environ. Microbiol. 2005, 71, 8587–8596. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, R.R.; Chen, Z.; Rappert, S.; Zeng, A.-P. A de novo nadph generation pathway for improving lysine production of corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab. Eng. 2014, 25, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Sreekrishna, K.; Brankamp, R.G.; Kropp, K.E.; Blankenship, D.T.; Tsay, J.-T.; Smith, P.L.; Wierschke, J.D.; Subramaniam, A.; Birkenberger, L.A. Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast pichia pastoris. Gene 1997, 190, 55–62. [Google Scholar] [CrossRef]
- Cregg, J.; Cereghino, J.; Shi, J.; Higgins, D. Recombinant protein expression in pichia pastoris. Mol. Biotechnol. 2000, 16, 23–52. [Google Scholar] [CrossRef]
- Cereghino, J.L.; Cregg, J.M. Heterologous protein expression in the methylotrophic yeast pichia pastoris. FEMS Microbiol. Rev. 2000, 24, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Sreekrishna, K.; Potenz, R.H.; Cruze, J.A.; McCombie, W.R.; Parker, K.A.; Nelles, L.; Mazzaferro, P.K.; Holden, K.A.; Harrison, R.G.; Wood, P.J. High level expression of heterologous proteins in methylotrophic yeast pichia pastoris. J. Basic Microbiol. 1988, 28, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein expression in pichia pastoris: Recent achievements and perspectives for heterologous protein production. Appl. Microbiol. Biotechnol. 2014, 98, 5301–5317. [Google Scholar] [CrossRef] [PubMed]
- Daly, R.; Hearn, M.T.W. Expression of heterologous proteins in pichia pastoris: A useful experimental tool in protein engineering and production. J. Mol. Recognit. 2005, 18, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, G.; Diano, A.; Nielsen, J. Recombinant bacterial hemoglobin alters metabolism of aspergillus niger. Metab. Eng. 2009, 11, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Karube, I.; Suzuki, S. Penicillin g production by immobilized whole cells of penicillium chrysogenum. Biotechnol. Bioeng. 1979, 21, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Ahmad, T.; Aryal, S.; Rana, B.; Sapkota, B. Penicillin production and history: An overview. Int. J. Microbiol. Allied Sci. 2014, 1, 5. [Google Scholar]
- Hu, X.-Q.; Chu, J.; Zhang, S.-L.; Zhuang, Y.-P.; Wang, Y.-H.; Zhu, S.; Zhu, Z.-G.; Yuan, Z.-Y. A novel feeding strategy during the production phase for enhancing the enzymatic synthesis of s-adenosyl-l-methionine by methylotrophic pichia pastoris. Enzym. Microb. Technol. 2007, 40, 669–674. [Google Scholar] [CrossRef]
- Hu, X.-Q.; Chu, J.; Zhang, Z.; Zhang, S.-L.; Zhuang, Y.-P.; Wang, Y.-H.; Guo, M.-J.; Chen, H.-X.; Yuan, Z.-Y. Effects of different glycerol feeding strategies on s-adenosyl-l-methionine biosynthesis by pgap-driven pichia pastoris overexpressing methionine adenosyltransferase. J. Biotechnol. 2008, 137, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Driouch, H.; Melzer, G.; Wittmann, C. Integration of in vivo and in silico metabolic fluxes for improvement of recombinant protein production. Metab. Eng. 2012, 14, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Christensen, B.; Hjort, C.; Nielsen, J. Construction and characterization of an oxalic acid nonproducing strain of aspergillus niger. Metab. Eng. 2000, 2, 34–41. [Google Scholar] [CrossRef] [PubMed]
- van Gulik, W.M.; de Laat, W.T.A.M.; Vinke, J.L.; Heijnen, J.J. Application of metabolic flux analysis for the identification of metabolic bottlenecks in the biosynthesis of penicillin-g. Biotechnol. Bioeng. 2000, 68, 602–618. [Google Scholar] [CrossRef]
- McKinlay, J.B.; Oda, Y.; Rühl, M.; Posto, A.L.; Sauer, U.; Harwood, C.S. Non-growing rhodopseudomonas palustris increases the hydrogen gas yield from acetate by shifting from the glyoxylate shunt to the tricarboxylic acid cycle. J. Biol. Chem. 2014, 289, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Reinefeld, J.; Stellmacher, R.; Schäfer, R.; Lange, A.; Meyer, H.; Lalk, M.; Zelder, O.; von Abendroth, G.; Schröder, H.; et al. Systems-wide analysis and engineering of metabolic pathway fluxes in bio-succinate producing basfia succiniciproducens. Biotechnol. Bioeng. 2013, 110, 3013–3023. [Google Scholar] [CrossRef] [PubMed]
- Ravikirthi, P.; Suthers, P.F.; Maranas, C.D. Construction of an E. Coli genome-scale atom mapping model for mfa calculations. Biotechnol. Bioeng. 2011, 108, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Sauer, U. Novel biological insights through metabolomics and 13c-flux analysis. Curr. Opin. Microbiol. 2009, 12, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Büscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Christen, S.; Sauer, U. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13c flux analysis and metabolomics. FEMS Yeast Res. 2011, 11, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Maranas, C.D. Clca: Maximum common molecular substructure queries within the metrxn database. J. Chem. Inf. Model. 2014, 54, 3417–3438. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Suthers, P.; Maranas, C. Metrxn: A knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinf. 2012, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Maranas, C.D. 13c metabolic flux analysis at a genome-scale. Metab. Eng. 2015, 32, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Jazmin, L.; O’Grady, J.; Ma, F.; Allen, D.; Morgan, J.; Young, J. Isotopically nonstationary mfa (inst-mfa) of autotrophic metabolism. In Plant Metabolic Flux Analysis; Dieuaide-Noubhani, M., Alonso, A.P., Eds.; Humana Press: New York, NY, USA, 2014; Volume 1090, pp. 181–210. [Google Scholar]
- Murphy, T.A.; Dang, C.V.; Young, J.D. Isotopically nonstationary 13c flux analysis of myc-induced metabolic reprogramming in b-cells. Metab. Eng. 2013, 15, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Jazmin, L.; Young, J. Isotopically nonstationary 13c metabolic flux analysis. In Systems Metabolic Engineering; Alper, H.S., Ed.; Humana Press: New York, NY, USA, 2013; Volume 985, pp. 367–390. [Google Scholar]
- Wiechert, W.; Nöh, K. Isotopically non-stationary metabolic flux analysis: Complex yet highly informative. Curr. Opin. Biotechnol. 2013, 24, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W.; Nöh, K. From stationary to instationary metabolic flux analysis. In Technology Transfer in Biotechnology; Kragl, U., Ed.; Springer: Berlin, Germany, 2005; Volume 92, pp. 145–172. [Google Scholar]
- Young, J.D.; Shastri, A.A.; Stephanopoulos, G.; Morgan, J.A. Mapping photoautotrophic metabolism with isotopically nonstationary 13c flux analysis. Metab. Eng. 2011, 13, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Jazmin, L.J.; Young, J.D.; Allen, D.K. Isotopically nonstationary 13c flux analysis of changes in arabidopsis thaliana leaf metabolism due to high light acclimation. Proc. Natl. Acad. Sci. USA 2014, 111, 16967–16972. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.; Owende, P. Biofuels from microalgae—A review of technologies for production, processing, and extractions of biofuels and co-products. Renew. Sustain. Energy Rev. 2010, 14, 557–577. [Google Scholar] [CrossRef]
- Varman, A.; Yu, Y.; You, L.; Tang, Y. Photoautotrophic production of d-lactic acid in an engineered cyanobacterium. Microb. Cell Fact. 2013, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y. Engineering cyanobacteria for fuels and chemicals production. Protein Cell 2010, 1, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Leighty, R.W.; Antoniewicz, M.R. Complete-mfa: Complementary parallel labeling experiments technique for metabolic flux analysis. Metab. Eng. 2013, 20, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Long, C.P.; Antoniewicz, M.R. Integrated 13c-metabolic flux analysis of 14 parallel labeling experiments in Escherichia coli. Metab. Eng. 2015, 28, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Leighty, R.W.; Antoniewicz, M.R. Parallel labeling experiments with [u-13c]glucose validate E. Coli metabolic network model for 13c metabolic flux analysis. Metab. Eng. 2012, 14, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.B.; Antoniewicz, M.R. Parallel labeling experiments and metabolic flux analysis: Past, present and future methodologies. Metab. Eng. 2013, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Srour, O.; Young, J.D.; Eldar, Y.C. Fluxomers: A new approach for (13)c metabolic flux analysis. BMC Syst. Biol. 2011, 5, 129. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.E.; Murray, W.; Saunders, M.A. Snopt: An sqp algorithm for large-scale constrained optimization. SIAM Rev. 2005, 47, 99–131. [Google Scholar] [CrossRef]
- Sokol, S.; Millard, P.; Portais, J.-C. Influx_s: Increasing numerical stability and precision for metabolic flux analysis in isotope labelling experiments. Bioinformatics 2012, 28, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Cvijovic, M.; Olivares-Hernández, R.; Agren, R.; Dahr, N.; Vongsangnak, W.; Nookaew, I.; Patil, K.R.; Nielsen, J. Biomet toolbox: Genome-wide analysis of metabolism. Nucleic Acids Res. 2010, 38, W144–W149. [Google Scholar] [CrossRef] [PubMed]
- Gombert, A.K.; Moreira dos Santos, M.; Christensen, B.; Nielsen, J. Network identification and flux quantification in the central metabolism of saccharomyces cerevisiae under different conditions of glucose repression. J. Bacteriol. 2001, 183, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Kajihata, S.; Furusawa, C.; Matsuda, F.; Shimizu, H. Openmebius: An open source software for isotopically nonstationary 13c-based metabolic flux analysis. BioMed Res. Int. 2014, 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T. Numerical Recipes in C: The Art of Scientific Computing; Cambridge University Press: Cambridge, UK, 1988; p. 735. [Google Scholar]
- Shiba, Y.; Paradise, E.M.; Kirby, J.; Ro, D.-K.; Keasling, J.D. Engineering of the pyruvate dehydrogenase bypass in saccharomyces cerevisiae for high-level production of isoprenoids. Metab. Eng. 2007, 9, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Jordà, J.; Jouhten, P.; Cámara, E.; Maaheimo, H.; Albiol, J.; Ferrer, P. Metabolic flux profiling of recombinant protein secreting pichia pastoris growing on glucose:Methanol mixtures. Microb. Cell Fact. 2012, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, T.; Sauer, U. Different biochemical mechanisms ensure network-wide balancing of reducing equivalents in microbial metabolism. J. Bacteriol. 2009, 191, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, W.; Sheng, J.; Feng, X. 13C-Metabolic Flux Analysis: An Accurate Approach to Demystify Microbial Metabolism for Biochemical Production. Bioengineering 2016, 3, 3. https://doi.org/10.3390/bioengineering3010003
Guo W, Sheng J, Feng X. 13C-Metabolic Flux Analysis: An Accurate Approach to Demystify Microbial Metabolism for Biochemical Production. Bioengineering. 2016; 3(1):3. https://doi.org/10.3390/bioengineering3010003
Chicago/Turabian StyleGuo, Weihua, Jiayuan Sheng, and Xueyang Feng. 2016. "13C-Metabolic Flux Analysis: An Accurate Approach to Demystify Microbial Metabolism for Biochemical Production" Bioengineering 3, no. 1: 3. https://doi.org/10.3390/bioengineering3010003