Trends in Upstream and Downstream Process Development for Antibody Manufacturing


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
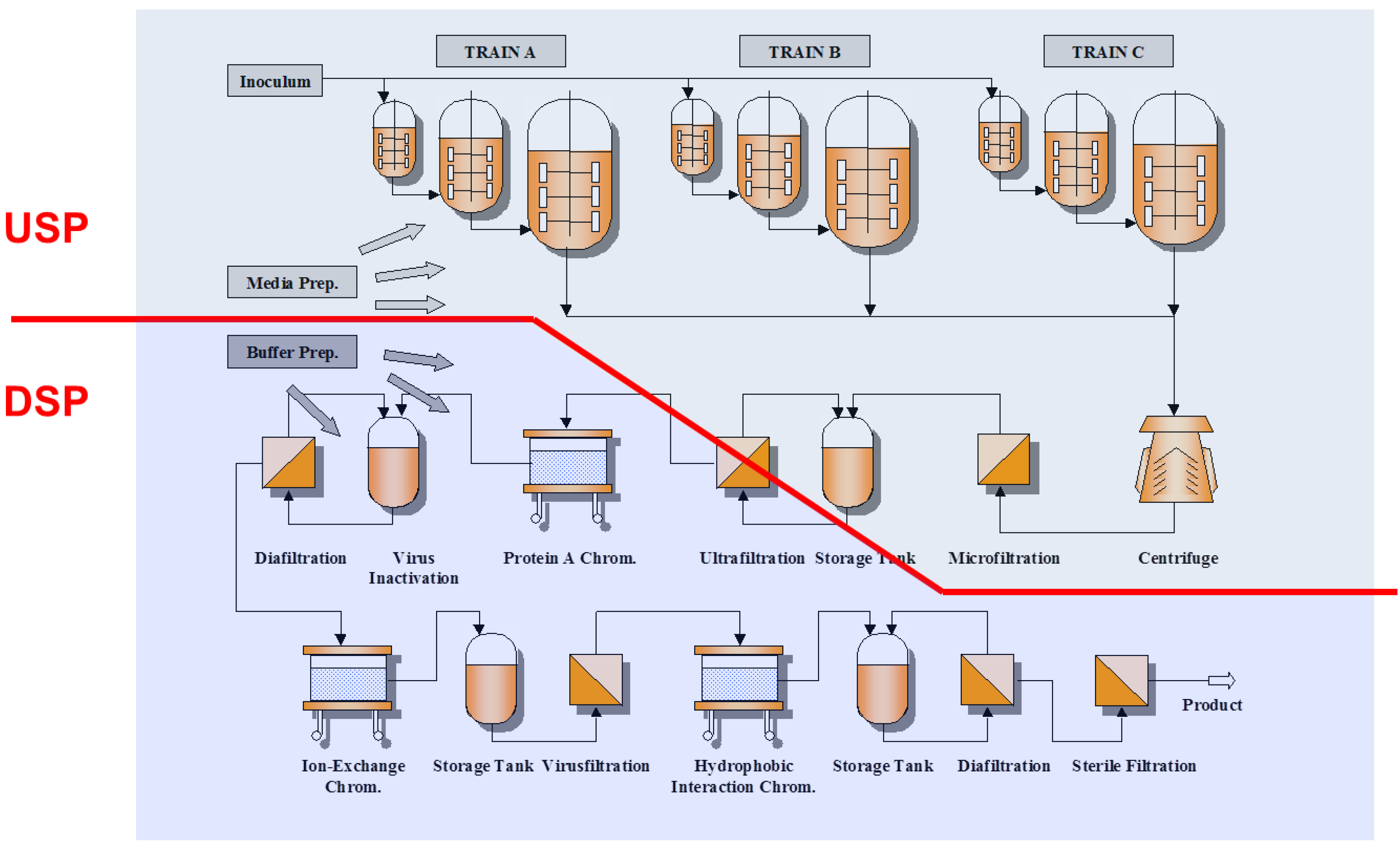
2. State of the Art in Process Development and Optimization
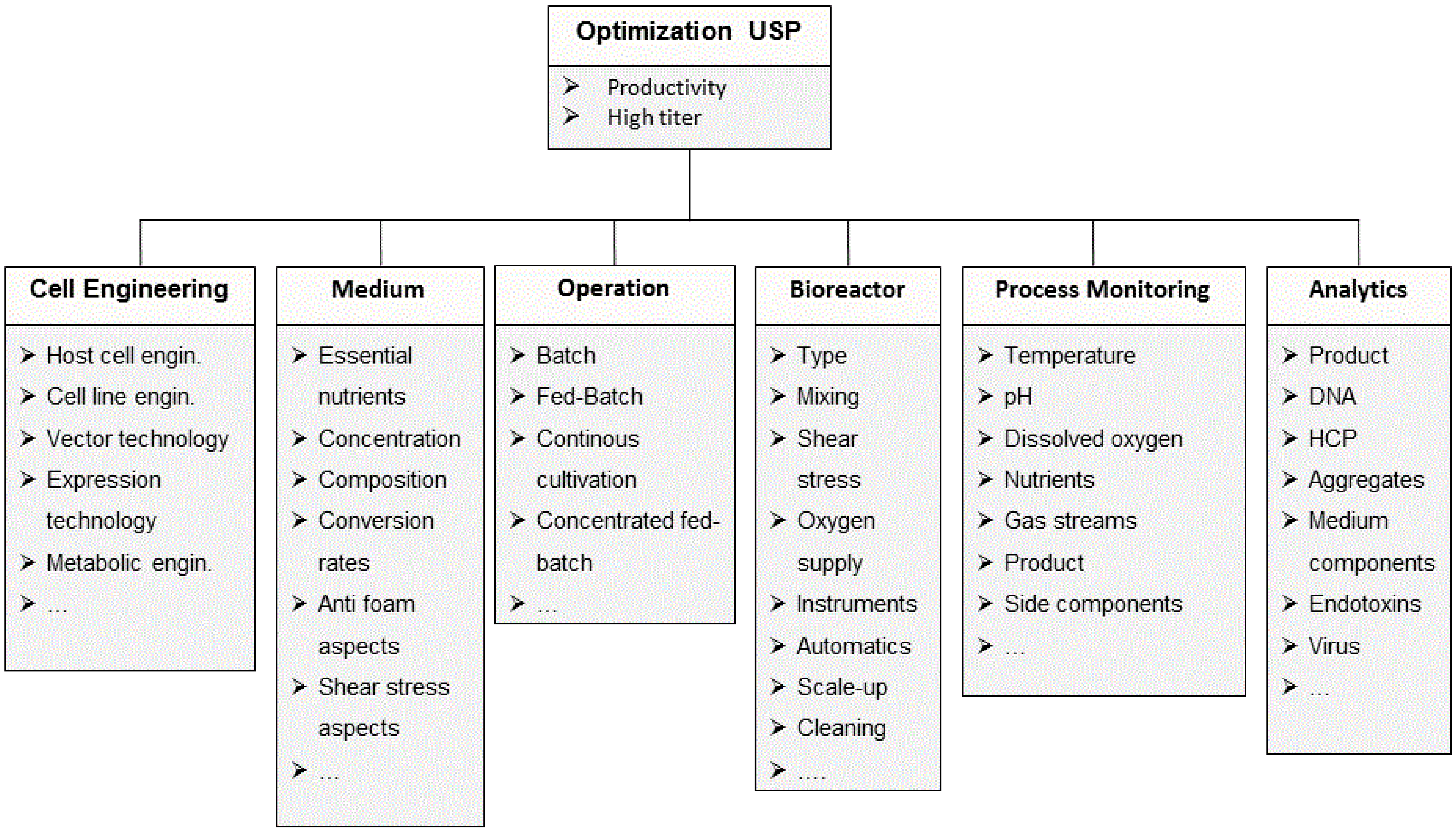
2.1. Process Development in Upstream Processing
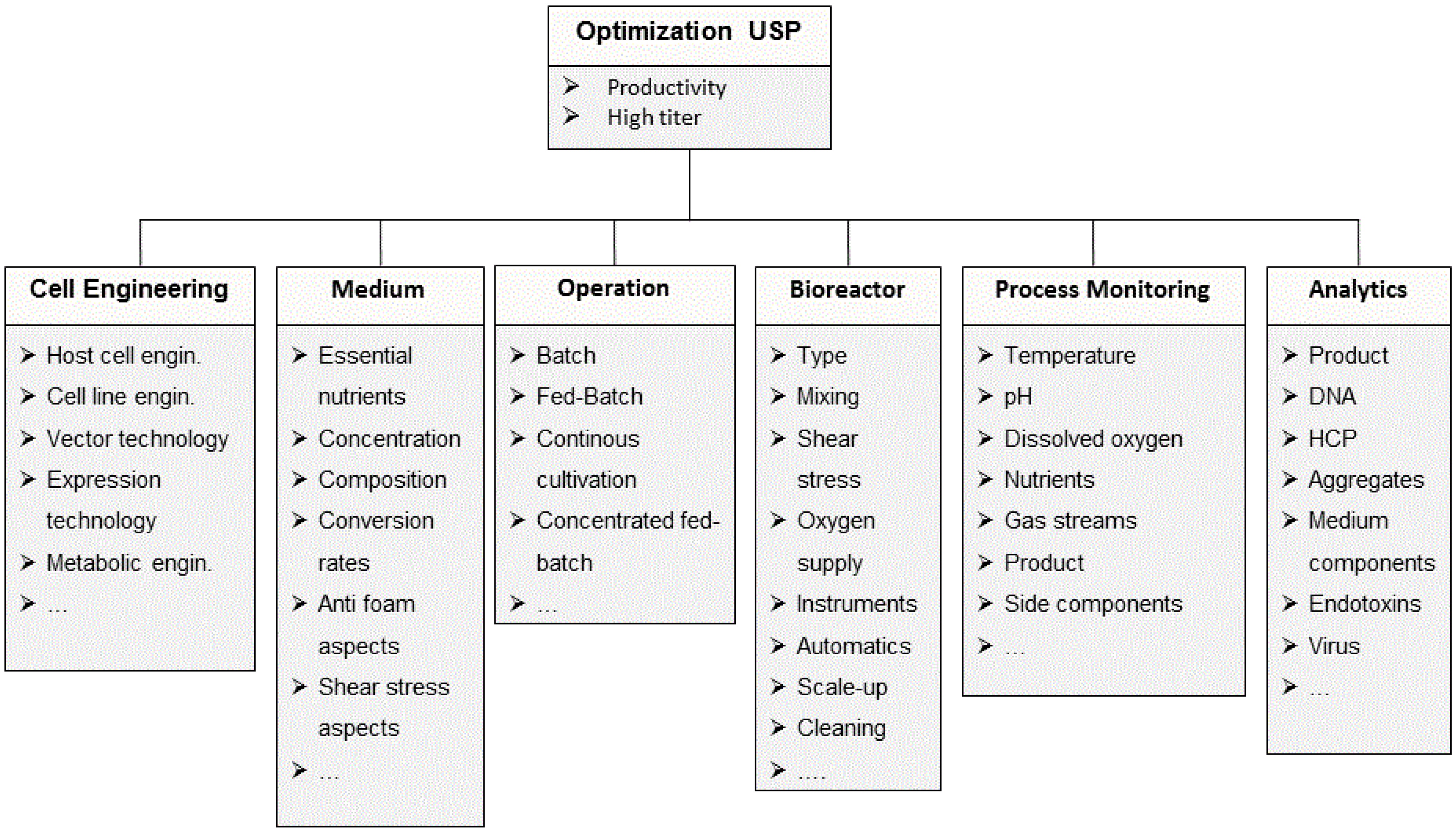
2.1.1. Cell Line Development and Clone Selection
2.1.2. Media Development and Optimization
2.1.3. Development of Process Strategies
2.1.4. Optimization of Bioreactor Systems
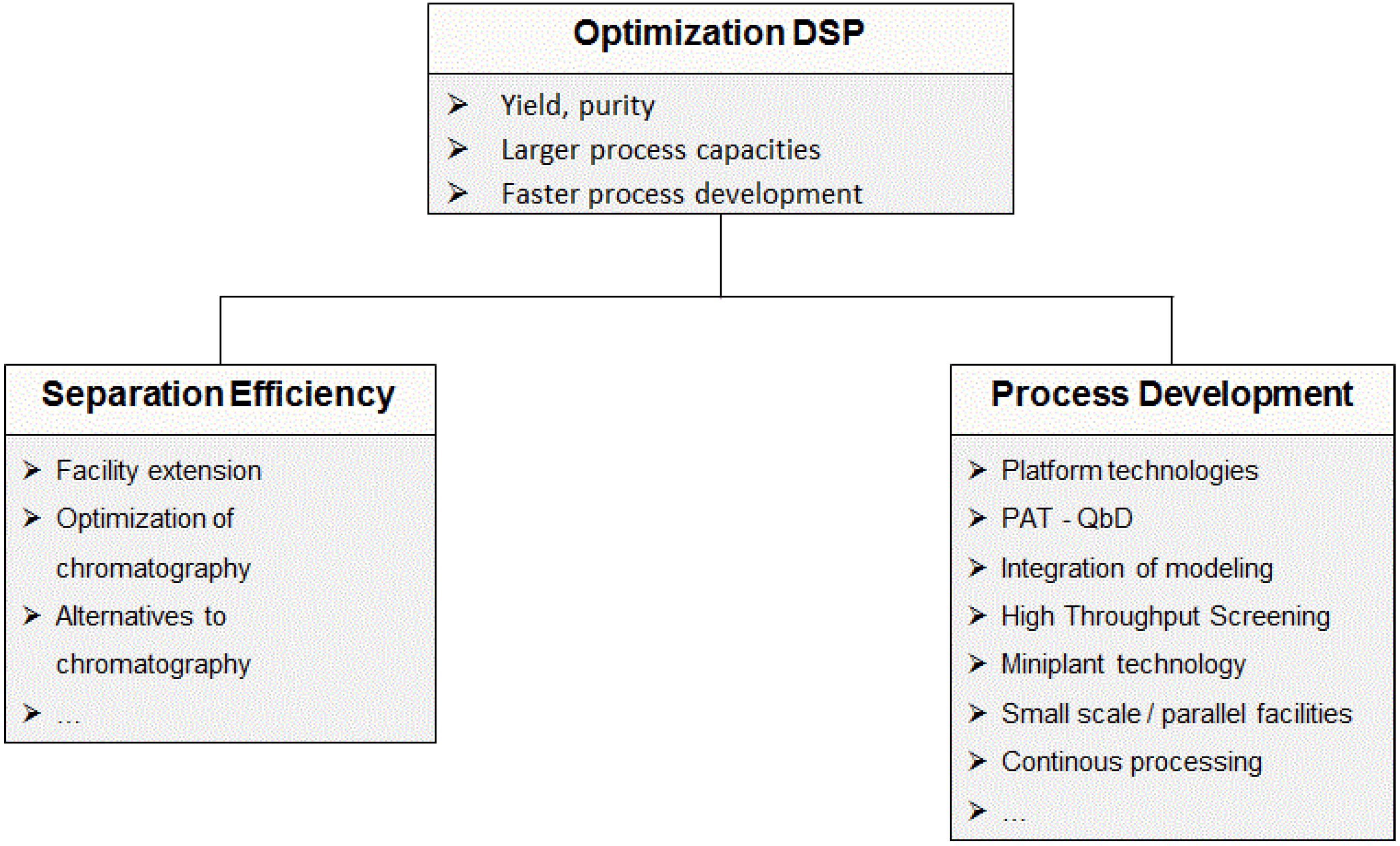
2.2. Process Development in Downstream Processing
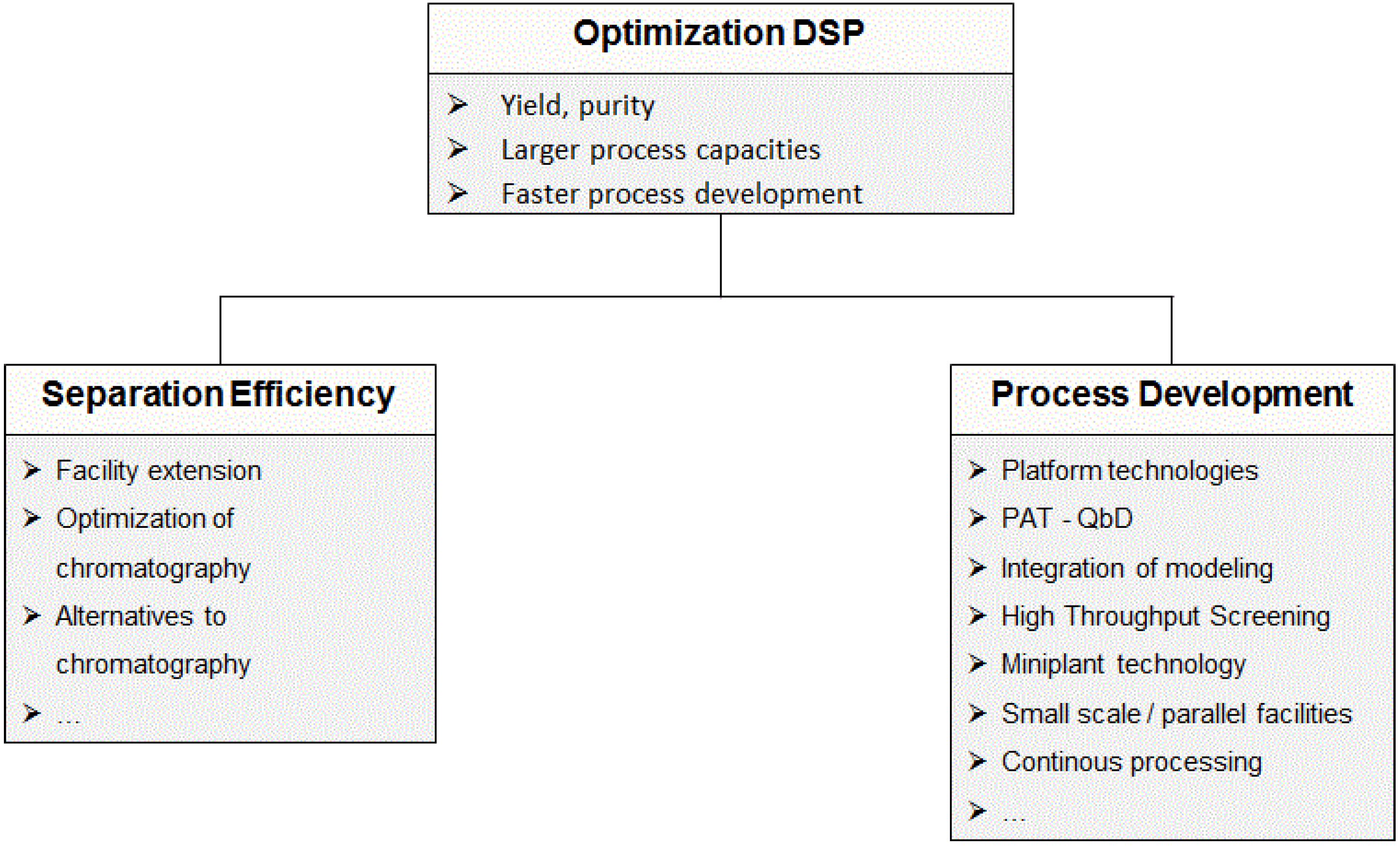
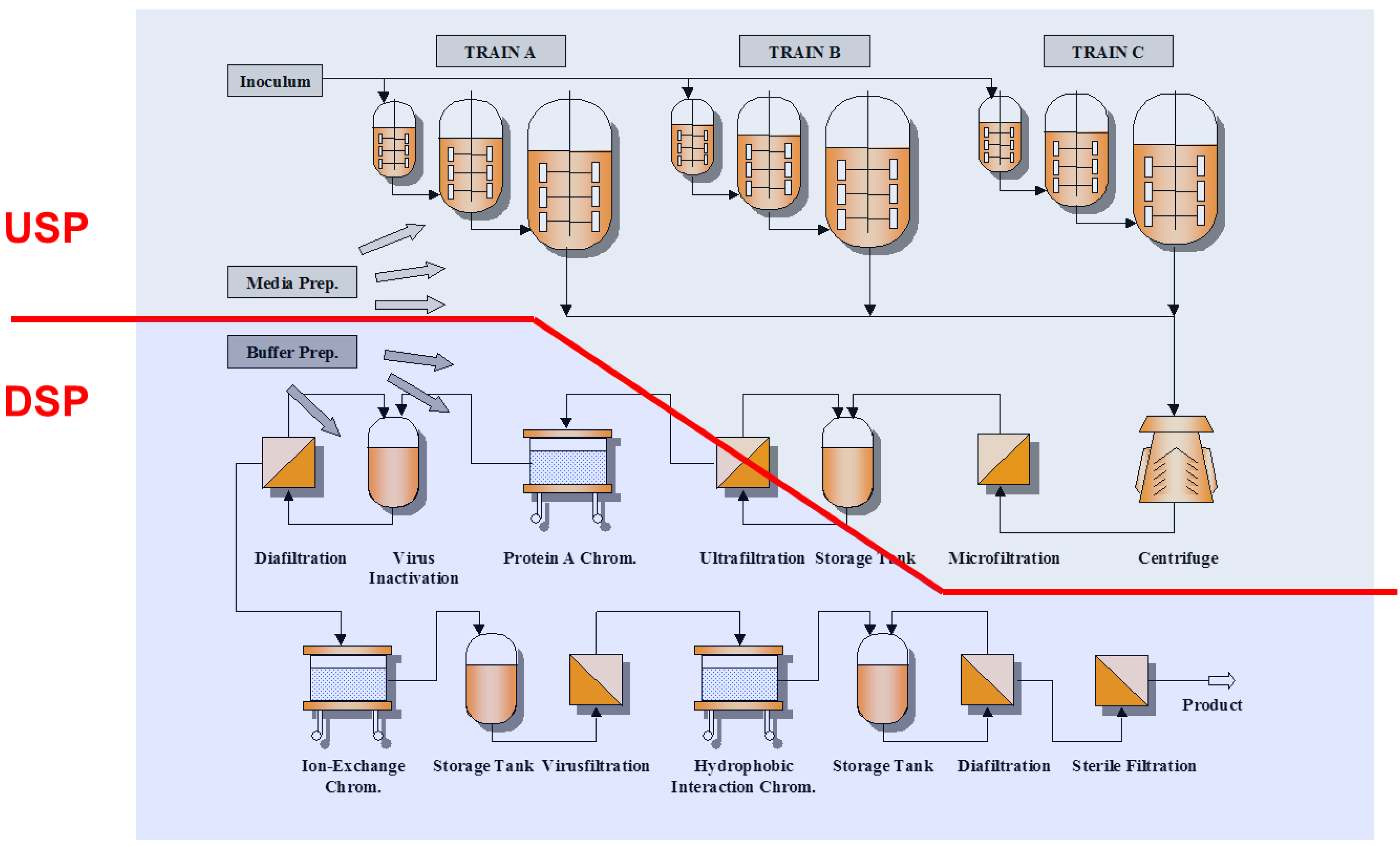
2.2.1. Chromatographic Separations
2.2.2. Non-Chromatographic Separations
3. Critical Parameters in Process Development
Impurities in Biopharmaceutical Manufacturing
4. Trends in Process Development and Optimization Strategies
5. Concluding Remarks and Outlook
Acknowledgments
Conflicts of Interest
References
- Jain, E.; Kumar, A. Upstream processes in antibody production: Evaluation of critical parameters. Biotechnol. Adv. 2008, 26, 46–72. [Google Scholar] [CrossRef]
- Shukla, A.A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef]
- Elvin, J.G.; Couston, R.G.; van der Walle; Christopher, F. Therapeutic antibodies: Market considerations, disease targets and bioprocessing. Int. J. Pharm. 2013, 440, 83–98. [Google Scholar] [CrossRef]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. Pharm. Sci. Encycl. 2010, 2, 466–479. [Google Scholar]
- Kelley, B. Very large scale monoclonal antibody purification: The case for conventional unit operations. Biotechnol. Progr. 2007, 23, 995–1008. [Google Scholar]
- Chon, J.H.; Zarbis-Papastoitsis, G. Advances in the production and downstream processing of antibodies. New Biotechnol. 2011, 28, 458–463. [Google Scholar] [CrossRef]
- Kelley, B. Industrialization of mAb production technology: The bioprocessing industry at a crossroads. MAbs 2009, 1, 443–452. [Google Scholar] [CrossRef]
- Butler, M.; Meneses-Acosta, A. Recent advances in technology supporting biopharmaceutical production from mammalian cells. Appl. Microbiol. Biotechnol. 2012, 96, 885–894. [Google Scholar] [CrossRef]
- Strube, J.; Grote, F.; Ditz, R. Bioprocess Design and Production Technology for the Future. In Biopharmaceutical Production Technology, 1st ed.; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany, 2012; Volume 2. [Google Scholar]
- Low, D.; O’Leary, R.; Pujar, N.S. Future of antibody purification. J. Chromatogr. B 2007, 848, 48–63. [Google Scholar] [CrossRef]
- Tait, A.S.; Hogwood, C.E.M.; Smales, C.M.; Bracewell, D.G. Host cell protein dynamics in the supernatant of a mAb producing CHO cell line. Biotechnol. Bioeng. 2012, 109, 971–982. [Google Scholar] [CrossRef]
- Hogwood, C.E.M.; Tait, A.S.; Koloteva-Levine, N.; Bracewell, D.G.; Smales, C. Mark the dynamics of the CHO host cell protein profile during clarification and protein A capture in a platform antibody purification process. Biotechnol. Bioeng. 2013, 110, 240–251. [Google Scholar] [CrossRef]
- Jin, M.; Szapiel, N.; Zhang, J.; Hickey, J.; Ghose, S. Profiling of host cell proteins by two-dimensional difference gel electrophoresis (2D-DIGE): Implications for downstream process development. Biotechnol. Bioeng. 2010, 105, 306–316. [Google Scholar] [CrossRef]
- Tscheliessnig, A.L.; Konrath, J.; Bates, R.; Jungbauer, A. Host cell protein analysis in therapeutic protein bioprocessing—Methods and applications. Biotechnol. J. 2013, 8, 655–670. [Google Scholar] [CrossRef]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. MAbs 2010, 2, 480–499. [Google Scholar] [CrossRef]
- Birch, J.R.; Racher, A.J. Antibody production. Adv. Drug Deliver Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies—Application of platform approaches. J. Chromatogr. B 2007, 848, 28–39. [Google Scholar] [CrossRef]
- Nfor, B.K.; Verhaert, P.D.E.M.; van der Wielen, L.A.M.; Hubbuch, J.; Ottens, M. Rational and systematic protein purification process development: The next generation. Trends Biotechnol. 2009, 27, 673–679. [Google Scholar] [CrossRef]
- Vogel, J.H.; Nguyen, H.; Giovanni, R.; Ignowski, J.; Garger, S.; Salgotra, A.; Tom, J. A new large-scale manufacturing platform for complex biopharmaceuticals. Biotechnol. Bioeng. 2012, 109, 3049–3058. [Google Scholar] [CrossRef]
- Bhambure, R.; Kumar, K.; Rathore, A.S. High-throughput process development for biopharmaceutical drug substances. Trends Biotechnol. 2011, 29, 127–135. [Google Scholar] [CrossRef]
- Amanullah, A.; Otero, J.M.; Mikola, M.; Hsu, A.; Zhang, J.; Aunins, J.; Schreyer, H.B.; Hope, J.A.; Russo, A.P. Novel micro-bioreactor high throughput technology for cell culture process development: Reproducibility and scalability assessment of fed-batch CHO cultures. Biotechnol. Bioeng. 2010, 106, 57–67. [Google Scholar]
- Maria, J.; Girard, P.; Bourgeois, M.; Baumgartner, G.; Jacko, B.; Amstutz, H.; Wurm, F.M. TubeSpin satellites: A fast track approach for process development with animal cells using shaking technology. Biochem. Eng. J. 2004, 17, 217–223. [Google Scholar] [CrossRef]
- Bareither, R.; Bargh, N.; Oakeshott, R.; Watts, K.; Pollard, D. Automated disposable small scale reactor for high throughput bioprocess development: A proof of concept study. Biotechnol. Bioeng. 2013, 110, 3126–3138. [Google Scholar] [CrossRef]
- De Jesus, Maria; Wurm, F.M. Manufacturing recombinant proteins in kg-ton quantities using animal cells in bioreactors. Eur. J. Pharm Biopharm. 2011, 78, 184–188. [Google Scholar] [CrossRef]
- Jordan, M.; Voisard, D.; Berthoud, A.; Tercier, L.; Kleuser, B.; Baer, G.; Broly, H. Cell culture medium improvement by rigorous shuffling of components using media blending. Cytotechnology 2013, 65, 31–40. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Liu, M.; Zhang, T.; Zhang, J.; Wang, X.; Xiang, W. Rational development of a serum-free medium and fed-batch process for a GS-CHO cell line expressing recombinant antibody. Cytotechnology 2013, 65, 363–378. [Google Scholar] [CrossRef]
- Sen, S.; Roychoudhury, P. Development of optimal medium for production of commercially important monoclonal antibody 520C9 by hybridoma cell. Cytotechnology 2013, 65, 233–252. [Google Scholar] [CrossRef]
- Jiang, Z.; Droms, K.; Geng, Z.; Casnocha, S.; Xiao, Z.; Gorfien, S. Fed-batch cell culture process optimization. A rationally integrated approach. BioProcess Int. 2012, 10, 40–45. [Google Scholar]
- Heckathorn, R.; Adams, D.; Hunter, J.; Frieden, E. Increasing Upstream Process Development Efficiency by Implementing Platform Glutamine Synthetase Cell Culture Processes. In Cells and Culture; Noll, T., Ed.; Springer Netherlands: Heidelberg, Germany, 2010; pp. 245–251. [Google Scholar]
- Castro, P.L.; Hayter, P.; Ison, A.; Bull, A. Application of a statistical design to the optimization of culture medium for recombinant interferon-gamma production by Chinese hamster ovary cells. Appl. Microbiol. Biotechnol. 1992, 38, 84–90. [Google Scholar] [CrossRef]
- Hammett, K.; Kuchibhatla, J.; Hunt, C.; Holdread, S.; Brooks, J. Developing Chemically Defined Media through DOE: Complete Optimization with Increased Protein Production in Less than 8 Months. In Cell Technology for Cell Products; Smith, R., Ed.; Springer Netherlands: Heidelberg, Germany, 2007; pp. 683–691. [Google Scholar]
- Michels, D.A.; Parker, M.; Salas-Solano, O. Quantitative impurity analysis of monoclonal antibody size heterogeneity by CE-LIF: Example of development and validation through a quality-by-design framework. Electrophoresis 2012, 33, 815–826. [Google Scholar] [CrossRef]
- Pathak, M.; Dutta, D.; Rathore, A. Analytical QbD: Development of a native gel electrophoresis method for measurement of monoclonal antibody aggregates. Electrophoresis 2014, 35, 2163–2171. [Google Scholar]
- Jiang, C.; Flansburg, L.; Ghose, S.; Jorjorian, P.; Shukla, A.A. Defining process design space for a hydrophobic interaction chromatography (HIC) purification step: Application of quality by design (QbD) principles. Biotechnol. Bioeng. 2010, 107, 985–997. [Google Scholar] [CrossRef]
- Del Val, I.J.; Kontoravdi, C.; Nagy, J.M. Towards the implementation of quality by design to the production of therapeutic monoclonal antibodies with desired glycosylation patterns. Biotechnol. Prog. 2010, 26, 1505–1527. [Google Scholar] [CrossRef]
- Martin-Moe, S.; Lim, F.J.; Wong, R.L.; Sreedhara, A.; Sundaram, J.; Sane, S.U. A new roadmap for biopharmaceutical drug product development: Integrating development, validation, and quality by design. J. Pharm Sci. 2011, 100, 3031–3043. [Google Scholar] [CrossRef]
- Rathore, A.S. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol. 2009, 27, 546–553. [Google Scholar] [CrossRef]
- Horvath, B.; Mun, M.; Laird, M. Characterization of a monoclonal antibody cell culture production process using a quality by design approach. Mol. Biotechnol. 2010, 45, 203–206. [Google Scholar] [CrossRef]
- Harms, J.; Wang, X.; Kim, T.; Yang, X.; Rathore, A. Defining process design space for biotech products: Case study of pichia pastoris fermentation. Biotechnol. Prog. 2008, 24, 655–662. [Google Scholar] [CrossRef]
- Abu-Absi, S.F.; Yang, L.; Thompson, P.; Jiang, C.; Kandula, S.; Schilling, B.; Shukla, A.A. Defining process design space for monoclonal antibody cell culture. Biotechnol. Bioeng. 2010, 106, 894–905. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kiu, X. Cell culture processes for biologics manufacturing: Recent developments and trends. Pharm. Bioprocess. 2013, 1, 133–136. [Google Scholar] [CrossRef]
- Rita Costa, A.; Elisa Rodrigues, M.; Henriques, M.; Azeredo, J.; Oliveira, R. Guidelines to cell engineering for monoclonal antibody production. Eur. J. Pharm Biopharm. 2010, 74, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol. Adv. 2012, 30, 1158–1170. [Google Scholar] [CrossRef]
- Jayapal, K.P.; Wlaschin, K.F.; Hu, W.; Yap, M.G. Recombinant protein therapeutics from CHO cells-20 years and counting. Chem. Eng. Prog. 2007, 103, 40–47. [Google Scholar]
- Durocher, Y.; Butler, M. Expression systems for therapeutic glycoprotein production. Curr. Opin. Biotech. 2009, 20, 700–707. [Google Scholar] [CrossRef]
- Hossler, P.; Khattak, S.F.; Li, Z.J. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009, 19, 936–949. [Google Scholar] [CrossRef]
- Kanda, Y.; Imai-Nishiya, H.; Kuni-Kamochi, R.; Mori, K.; Inoue, M.; Kitajima-Miyama, K.; Okazaki, A.; Iida, S.; Shitara, K.; Satoh, M. Establishment of a GDP-mannose 4,6-dehydratase (GMD) knockout host cell line: A new strategy for generating completely non-fucosylated recombinant therapeutics. J. Biotechnol. 2007, 130, 300–310. [Google Scholar] [CrossRef]
- Mori, K.; Kuni-Kamochi, R.; Yamane-Ohnuki, N.; Wakitani, M.; Yamano, K.; Imai, H.; Kanda, Y.; Niwa, R.; Iida, S.; Uchida, K.; Shitara, K.; Satoh, M. Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnol. Bioeng. 2004, 88, 901–908. [Google Scholar] [CrossRef]
- Kim, S.; Lee, G. Down-regulation of lactate dehydrogenase-A by siRNAs for reduced lactic acid formation of Chinese hamster ovary cells producing thrombopoietin. Appl. Microbiol. Biotechnol. 2007, 74, 152–159. [Google Scholar] [CrossRef]
- Chen, K.; Liu, Q.; Xie, L.; Sharp, P.A.; Wang, D.I.C. Engineering of a mammalian cell line for reduction of lactate formation and high monoclonal antibody production. Biotechnol. Bioeng. 2001, 72, 55–61. [Google Scholar] [CrossRef]
- Jeong, D.-W.; Cho, I.; Kim, T.; Bae, G.; Kim, I.-H.; Kim, I. Effects of lactate dehydrogenase suppression and glycerol-3-phosphate dehydrogenase overexpression on cellular metabolism. Mol. Cell. Biochem. 2006, 284, 1–8. [Google Scholar] [CrossRef]
- Sauerwald, T.M.; Figueroa, B.; Hardwick, J.M.; Oyler, G.A.; Betenbaugh, M.J. Combining caspase and mitochondrial dysfunction inhibitors of apoptosis to limit cell death in mammalian cell cultures. Biotechnol. Bioeng. 2006, 94, 362–372. [Google Scholar] [CrossRef]
- Lee, C.J.; Seth, G.; Tsukuda, J.; Hamilton, R.W. A clone screening method using mRNA levels to determine specific productivity and product quality for monoclonal antibodies. Biotechnol. Bioeng. 2009, 102, 1107–1118. [Google Scholar] [CrossRef]
- Noh, S.M.; Sathyamurthy, M.; Lee, G.M. Development of recombinant Chinese hamster ovary cell lines for therapeutic protein production. Curr. Opin. Chem. Eng. 2013, 2, 391–397. [Google Scholar] [CrossRef]
- Baldi, L.; Hacker, D.L.; Adam, M.; Wurm, F.M. Recombinant protein production by large-scale transient gene expression in mammalian cells: State of the art and future perspectives. Biotechnol. Lett. 2007, 29, 677–684. [Google Scholar] [CrossRef]
- Girard, P.; Derouazi, M.; Baumgartner, G.; Bourgeois, M.; Jordan, M.; Jacko, B.; Wurm, F.M. 100-liter transient transfection. Cytotechnology 2002, 38, 15–21. [Google Scholar] [CrossRef]
- Rajendra, Y.; Kiseljak, D.; Baldi, L.; Hacker, D.L.; Wurm, F.M. A simple high-yielding process for transient gene expression in CHO cells. J. Biotechnol. 2011, 153, 22–26. [Google Scholar] [CrossRef]
- Fletcher, T. Designing culture media for recombinant protein production: A rational approach. BioProcess Int. 2005, 3, 30–36. [Google Scholar]
- Kim, S.; Lee, G. Development of serum-free medium supplemented with hydrolysates for the production of therapeutic antibodies in CHO cell cultures using design of experiments. Appl. Microbiol. Biotechnol. 2009, 83, 639–648. [Google Scholar] [CrossRef]
- Lu, F.; Toh, P.C.; Burnett, I.; Li, F.; Hudson, T.; Amanullah, A.; Li, J. Automated dynamic fed-batch process and media optimization for high productivity cell culture process development. Biotechnol. Bioeng. 2013, 110, 191–205. [Google Scholar] [CrossRef]
- Ma, N.; Ellet, J.; Okediadi, C.; Hermes, P.; McCormick, E.; Casnocha, S. A single nutrient feed supports both chemically defined NS0 and CHO fed-batch processes: Improved productivity and lactate metabolism. Biotechnol. Prog. 2009, 25, 1353–1363. [Google Scholar]
- Zhou, W.; Chen, C.-C.; Buckland, B.; Aunins, J. Fed-batch culture of recombinant NS0 myeloma cells with high monoclonal antibody production. Biotechnol. Bioeng. 1997, 55, 783–792. [Google Scholar] [CrossRef]
- Voisard, D.; Meuwly, F.; Ruffieux, P.-A.; Baer, G.; Kadouri, A. Potential of cell retention techniques for large-scale high-density perfusion culture of suspended mammalian cells. Biotechnol. Bioeng. 2003, 82, 751–765. [Google Scholar] [CrossRef]
- Henzler, H.-J. Kontinuierliche Fermentation mit tierischen Zellen. Teil 1. Aspekte der kontinuierlichen Prozessführung. Chem. Ing. Tech. 2012, 84, 1469–1481. [Google Scholar] [CrossRef]
- Clincke, M.-F.; Mölleryd, C.; Samani, P.K.; Lindskog, E.; Fäldt, E.; Walsh, K.; Chotteau, V. Very high density of Chinese hamster ovary cells in perfusion by alternating tangential flow or tangential flow filtration in WAVE Bioreactor™-part II: Applications for antibody production and cryopreservation. Biotechnol. Prog. 2013, 29, 768–777. [Google Scholar] [CrossRef]
- Clincke, M.-F.; Mölleryd, C.; Zhang, Y.; Lindskog, E.; Walsh, K.; Chotteau, V. Very high density of CHO cells in perfusion by ATF or TFF in WAVE bioreactor™. Part I. Effect of the cell density on the process. Biotechnol. Prog. 2013, 29, 754–767. [Google Scholar] [CrossRef]
- Pollock, J.; Ho, S.V.; Farid, S.S. Fed-batch and perfusion culture processes: Economic, environmental, and operational feasibility under uncertainty. Biotechnol. Bioeng. 2013, 110, 206–219. [Google Scholar] [CrossRef]
- Bonham-Carter, J.; Shevitz, J. A brief history of perfusion biomanufacturing. BioProcess Int. 2011, 9, 24–32. [Google Scholar]
- Castilho, L.R.; Anspach, F.B.; Deckwer, W.-D. An integrated process for mammalian cell perfusion cultivation and product purification using a dynamic filter. Biotechnol. Prog. 2002, 18, 776–781. [Google Scholar] [CrossRef]
- Henzler, H.-J. Kontinuierliche Fermentation mit tierischen Zellen. Teil 2. Techniken und Methoden der Zellrückhaltung. Chem. Ing. Tech. 2012, 84, 1482–1496. [Google Scholar] [CrossRef]
- Gomez, N.; Ouyang, J.; Nguyen, M.D.H.; Vinson, A.R.; Lin, A.A.; Yuk, I.H. Effect of temperature, pH, dissolved oxygen, and hydrolysate on the formation of triple light chain antibodies in cell culture. Biotechnol. Prog. 2010, 26, 1438–1445. [Google Scholar] [CrossRef]
- Jing, Y.; Borys, M.; Nayak, S.; Egan, S.; Qian, Y.; Pan, S.-H.; Li, Z.J. Identification of cell culture conditions to control protein aggregation of IgG fusion proteins expressed in Chinese hamster ovary cells. Process. Biochem. 2012, 47, 69–75. [Google Scholar] [CrossRef]
- Ye, J.; Kober, V.; Tellers, M.; Naji, Z.; Salmon, P.; Markusen, J.F. High-level protein expression in scalable CHO transient transfection. Biotechnol. Bioeng. 2009, 103, 542–551. [Google Scholar] [CrossRef]
- Diekmann, S.; Dürr, C.; Herrmann, A.; Lindner, I.; Jozic, D. Single use bioreactors for the clinical production of monoclonal antibodies—A study to analyze the performance of a CHO cell line and the quality of the produced monoclonal antibody. BMC Proc. 2011, 5 (Suppl. 8). [Google Scholar] [CrossRef]
- Whitford, W.G. Single-use systems as principal components in bioproduction. BioProcess Int. 2010, 8, 34–42. [Google Scholar]
- Langer, E.S.; Rader, R.A. Single-use technologies in biopharmaceutical manufacturing: A 10-year review of trends and the future. Eng. Life Sci. 2014, 14, 238–243. [Google Scholar] [CrossRef]
- Minow, B.; Seidemann, J.; Tschoepe, S.; Gloeckner, A.; Neubauer, P. Harmonization and characterization of different single-use bioreactors adopting a new sparger design. Eng. Life Sci. 2014, 14, 272–282. [Google Scholar] [CrossRef]
- Löffelholz, C.; Husemann, U.; Greller, G.; Meusel, W.; Kauling, J.; Ay, P.; Kraume, M.; Eibl, R.; Eibl, D. Bioengineering parameters for single-use bioreactors: Overview and evaluation of suitable methods. Chem. Ing. Tech. 2013, 85, 40–56. [Google Scholar] [CrossRef]
- Shukla, A.A.; Gottschalk, U. Single-use disposable technologies for biopharmaceutical manufacturing. Trends Biotechnol. 2013, 31, 147–154. [Google Scholar] [CrossRef]
- Reclari, M.; Dreyer, M.; Tissot, S.; Obreschkow, D.; Wurm, F.M.; Farhat, M. Surface wave dynamics in orbital shaken cylindrical containers. Phys. Fluids 2014, 26. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotech. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Gottschalk, U. Process Scale Purification of Antibodies: Downstream Processing of Monoclonal Antibodies: Current Practices and Future Opportunities, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—Generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hinckley, P. Host cell protein clearance during protein A chromatography: Development of an improved column wash step. Biotechnol. Prog. 2008, 24, 1115–1121. [Google Scholar] [CrossRef]
- Tarrant, R.D.R.; Velez-Suberbie, M.L.; Tait, A.S.; Smales, C.M.; Bracewell, D.G. Host cell protein adsorption characteristics during protein A chromatography. Biotechnol. Prog. 2012, 28, 1037–1044. [Google Scholar] [CrossRef]
- Royce, J. High-capacity protein A chromatography medium for MAb coapture from high-titer feedds. BioProcess Int. 2014, 12, 40–41. [Google Scholar]
- Lain, B.; Cacciuttolo, M.A.; Zarbis-Papastoitsis, G. Development of a high-capacity Mab capture step based on cation-exchange chromatography. BioProcess Int. 2009, 26–34. [Google Scholar]
- Lain, B. Protein A: The life of disruptive technology. BioProcess Int. 2013, 11, 29–38. [Google Scholar]
- Ghose, S.; Hubbard, B.; Cramer, S.M. Evaluation and comparison of alternatives to Protein A chromatography: Mimetic and hydrophobic charge induction chromatographic stationary phases. J. Chromatogr. A 2006, 1122, 144–152. [Google Scholar] [CrossRef]
- Gagnon, P. How to choose an industrial cation exchanger for IgG purification. BioProcess Int. 2010, 8, 22–34. [Google Scholar]
- Jackewitz, A. Reducing elution volumes with high capacity and improved mass transfer ion-exchange resins. BioProcess Int. 2008, 6, 108–110. [Google Scholar]
- El Khoury, G.; Lowe, C.R. A biomimetic Protein G affinity adsorbent: An Ugi ligand for immunoglobulins and Fab fragments based on the third IgG-binding domain of Protein G. J. Mol. Recognit. 2013, 26, 190–200. [Google Scholar] [CrossRef]
- Arakawa, T.; Kita, Y.; Sato, H.; Ejima, D. MEP chromatography of antibody and Fc-fusion protein using aqueous arginine solution. Protein Expres. Purif. 2009, 63, 158–163. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Liu, Z. Restricted access boronate affinity porous monolith as a protein A mimetic for the specific capture of immunoglobulin G. Chem. Sci. 2012, 3, 1467–1471. [Google Scholar] [CrossRef]
- Qian, J.; El Khoury, G.; Issa, H.; Al-Qaoud, K.; Shihab, P.; Lowe, C.R. A synthetic Protein G adsorbent based on the multi-component Ugi reaction for the purification of mammalian immunoglobulins. J. Chromatogr. B. 2012, 898, 15–23. [Google Scholar] [CrossRef]
- Roque, A.C.A.; Taipa, M.Â.; Lowe, C.R. An artificial protein L for the purification of immunoglobulins and Fab fragments by affinity chromatography. J. Chromatogr. A 2005, 1064, 157–167. [Google Scholar] [CrossRef]
- Teng, S.F.; Sproule, K.; Husain, A.; Lowe, C.R. Affinity chromatography on immobilized “biomimetic” ligands: Synthesis, immobilization and chromatographic assessment of an immunoglobulin G-binding ligand. J. Chromatogr. B 2000, 740, 1–15. [Google Scholar] [CrossRef]
- Azevedo, A.M.; Rosa, P.A.J.; Ferreira, I.F.; Aires-Barros, M. Raquel chromatography-free recovery of biopharmaceuticals through aqueous two-phase processing. Trends Biotechnol. 2009, 27, 240–247. [Google Scholar] [CrossRef]
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B 2007, 848, 40–47. [Google Scholar] [CrossRef]
- Pezzini, J.; Joucla, G.; Gantier, R.; Toueille, M.; Lomenech, A.-M.; Le Sénéchal, C.; Garbay, B.; Santarelli, X.; Cabanne, C. Antibody capture by mixed-mode chromatography: A comprehensive study from determination of optimal purification conditions to identification of contaminating host cell proteins. J. Chromatogr. A 2011, 1218, 8197–8208. [Google Scholar] [CrossRef]
- Fischer-Fruhholz, S.; Zhou, D.; Hirai, M. Sartobind STIC® salt-tolerant membrane chromatography. Nat. Meth. 2010, 7, an12–an13. [Google Scholar] [CrossRef]
- Toueille, M.; Uzel, A.; Depoisier, J.-F.; Gantier, R. Designing new monoclonal antibody purification processes using mixed-mode chromatography sorbents. J. Chromatogr. B 2011, 879, 836–843. [Google Scholar] [CrossRef]
- Helling, C.; Borrmann, C.; Strube, J. optimal integration of directly combined hydrophobic interaction and ion exchange chromatography purification processes. Chem. Eng. Technol. 2012, 35, 1786–1796. [Google Scholar] [CrossRef]
- Fröhlich, H.; Villian, L.; Melzner, D.; Strube, J. Membrane technology in bioprocess science. Chem. Ing. Tech. 2012, 84, 905–917. [Google Scholar]
- Rosa, P.A.J.; Ferreira, I.F.; Azevedo, A.M.; Aires-Barros, M.R. Aqueous two-phase systems: A viable platform in the manufacturing of biopharmaceuticals. J. Chromatogr. A 2010, 1217, 2296–2305. [Google Scholar] [CrossRef]
- Rosa, P.A.J.; Azevedo, A.M.; Sommerfeld, S.; Bäcker, W.; Aires-Barros, M.R. Continuous aqueous two-phase extraction of human antibodies using a packed column. J. Chromatogr. B 2012, 880, 148–156. [Google Scholar] [CrossRef]
- Eggersgluess, J.; Both, S.; Strube, J. Process development for extraction of biomolecules. Application downstream processing of proteins in aqueous two-phase systems. Chem. Today 2012, 30, 32–36. [Google Scholar]
- Zang, Y.; Kammerer, B.; Eisenkolb, M.; Lohr, K.; Kiefer, H. Towards protein crystallization as a process step in downstream processing of therapeutic antibodies: Screening and optimization at microbatch scale. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Smejkal, B.; Agrawal, N.; Helk, B.; Schulz, H.; Giffard, M.; Mechelke, M.; Ortner, F.; Heckmeier, P.; Trout, B.; Hekmat, D. Fast and scalable purification of a therapeutic full-length antibody based on process crystallization. Biotechnol. Bioeng. 2013, 110, 2452–2461. [Google Scholar] [CrossRef]
- Cramer, S.M.; Holstein, M.A. Downstream bioprocessing: Recent advances and future promise. Curr. Opin. Chem. Eng. 2011, 1, 27–37. [Google Scholar] [CrossRef]
- Saxena, A.; Tripathi, B.P.; Kumar, M.; Shahi, V.K. Membrane-based techniques for the separation and purification of proteins: An overview. Adv. Colloid Interfac. 2009, 145, 1–22. [Google Scholar] [CrossRef]
- Kumar, M.; Ulbricht, M. Advanced ultrafiltration membranes based on functionalized poly(arylene ether sulfone) block copolymers. RSC Adv. 2013, 3, 12190–12203. [Google Scholar] [CrossRef]
- Kumar, M.; Ulbricht, M. Novel ultrafiltration membranes with adjustable charge density based on sulfonated poly(arylene ether sulfone) block copolymers and their tunable protein separation performance. Polymer 2014, 55, 354–365. [Google Scholar]
- Chenette, H.C.S.; Robinson, J.R.; Hobley, E.; Husson, S.M. Development of high-productivity, strong cation-exchange adsorbers for protein capture by graft polymerization from membranes with different pore sizes. J. Membrane Sci. 2012, 423–424, 43–52. [Google Scholar] [CrossRef]
- Weaver, J.; Husson, S.M.; Murphy, L.; Wickramasinghe, S.R. Anion exchange membrane adsorbers for flow-through polishing steps: Part II. Virus, host cell protein, DNA clearance, and antibody recovery. Biotechnol. Bioeng. 2013, 110, 500–510. [Google Scholar] [CrossRef]
- Drioli, E.; Stankiewicz, A.I.; Macedonio, F. Membrane engineering in process intensification—An overview. J. Membrane Sci. 2011, 380, 1–8. [Google Scholar] [CrossRef]
- Benavides, J.; Aguilar, O.; Lapizco-Encinas, B.H.; Rito-Palomares, M. Extraction and purification of bioproducts and nanoparticles using aqueous two-phase systems strategies. Chem. Eng. Technol. 2008, 31, 838–845. [Google Scholar] [CrossRef]
- Somani, S.; Padmanabhan, S. Process for Purification of Recombinant Human Granulocyte Colony Stimulating Factor. U.S. Patent App. 13/378,973, 2010. [Google Scholar]
- Giese, G.; Myrold, A.; Gorrell, J.; Persson, J. Purification of antibodies by precipitating impurities using Polyethylene Glycol to enable a two chromatography step process. J. Chromatogr. B 2013, 938, 14–21. [Google Scholar] [CrossRef]
- Oelmeier, S.A.; Ladd-Effio, C.; Hubbuch, J. Alternative separation steps for monoclonal antibody purification: Combination of centrifugal partitioning chromatography and precipitation. J. Chromatogr. A 2013, 1319, 118–126. [Google Scholar] [CrossRef]
- Kuczewski, M.; Schirmer, E.; Lain, B.; Zarbis-Papastoitsis, G. A single-use purification process for the production of a monoclonal antibody produced in a PER.C6 human cell line. Biotechnol. J. 2011, 6, 56–65. [Google Scholar] [CrossRef]
- McDonald, P.; Victa, C.; Carter-Franklin, J.N.; Fahrner, R. Selective antibody precipitation using polyelectrolytes: A novel approach to the purification of monoclonal antibodies. Biotechnol. Bioeng. 2009, 102, 1141–1151. [Google Scholar] [CrossRef]
- Kuczewski, M.; Schirmer, E.; Lain, B. PEG precipitation: A powerful tool for Monoclonal Antibody Purification. Available online: http://www.biopharminternational.com (accessed on 1 August 2014).
- Grodzki, A.; Berenstein, E. Antibody purification: Ammonium sulfate fractionation or gel filtration. Methods Mol. Biol. 2010, 588, 15–26. [Google Scholar]
- Ma, J.; Hoang, H.; Myint, T.; Peram, T.; Fahrner, R.; Chou, J. Using precipitation by polyamines as an alternative to chromatographic separation in antibody purification processes. J. Chromatogr. B 2010, 878, 798–806. [Google Scholar] [CrossRef]
- Buyel, J.F.; Fischer, R. Flocculation increases the efficacy of depth filtration during the downstream processing of recombinant pharmaceutical proteins produced in tobacco. Plant. Biotechnol. J. 2014, 12, 240–252. [Google Scholar] [CrossRef]
- Fan, L.; Zhao, L.; Sun, Y.; Kou, T.; Zhou, Y.; Tan, W.-S. A high-yielding, generic fed-batch process for recombinant antibody production of GS-engineered cell lines. J. Microbiol. Biotechnol. 2009, 19, 1695–1702. [Google Scholar] [CrossRef]
- Food and Drug Administration. Federal Register/Vol. 63, No. 110/Tuesday, June 9, 1998/Notices, 1998. Available online: http://www.gpo.gov/fdsys/pkg/FR-1998-06-09/pdf/98-15195.pdf (accessed on 1 August 2014).
- Raijada, D.K.; Prasad, B.; Paudel, A.; Shah, R.P.; Singh, S. Characterization of degradation products of amorphous and polymorphic forms of clopidogrel bisulphate under solid state stress conditions. J. Pharma. Biomed. 2010, 52, 332–344. [Google Scholar] [CrossRef]
- Pan, C.; Liu, F.; Motto, M. Identification of pharmaceutical impurities in formulated dosage forms. J. Pharm Sci. 2011, 100, 1228–1259. [Google Scholar] [CrossRef]
- Mazur, M.; Seipert, R.; Mahon, D.; Zhou, Q.; Liu, T. A platform for characterizing therapeutic monoclonal antibody breakdown products by 2D chromatography and top-down mass spectrometry. AAPS J. 2012, 14, 530–541. [Google Scholar] [CrossRef]
- Horak, J.; Ronacher, A.; Lindner, W. Quantification of immunoglobulin G and characterization of process related impurities using coupled Protein A and size exclusion high performance liquid chromatography. J. Chromatogr. A 2010, 1217, 5092–5102. [Google Scholar] [CrossRef]
- Guiochon, G.; Beaver, L.A. Separation science is the key to successful biopharmaceuticals. J. Chromatogr. A 2011, 1218, 8836–8858. [Google Scholar] [CrossRef]
- Wang, X.; Hunter, A.K.; Mozier, N.M. Host cell proteins in biologics development: Identification, quantitation and risk assessment. Biotechnol. Bioeng. 2009, 103, 446–458. [Google Scholar] [CrossRef]
- Prieto, Y.; Rojas, L.; Pérez, R. Towards the molecular characterization of the stable producer phenotype of recombinant antibody-producing NS0 myeloma cells. Cytotechnology 2011, 63, 351–362. [Google Scholar] [CrossRef]
- Grzeskowiak, J.K.; Tscheliessnig, A.; Wu, M.W.; Toh, P.C.; Chusainow, J.; Lee, Y.Y.; Wong, N.; Jungbauer, A. Two-dimensional difference fluorescence gel electrophoresis to verify the scale-up of a non-affinity-based downstream process for isolation of a therapeutic recombinant antibody. Electrophoresis 2010, 31, 1862–1872. [Google Scholar] [CrossRef]
- Ditz, R. Separation Technologies 2030—Are 100 Years of Chromatography Enough? Chem. Ing. Tech. 2012, 84, 875–879. [Google Scholar] [CrossRef]
- Kontoravdi, C.; Samsatli, N.J.; Shah, N. Development and design of bio-pharmaceutical processes. Curr. Opin. Chem. Eng. 2013, 2, 435–441. [Google Scholar]
- Kruhlak, N.L.; Benz, R.D.; Zhou, H.; Colatsky, T.J. (Q)SAR modeling and safety assessment in regulatory review. Clin. Pharmacol. Ther. 2012, 91, 529–534. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inf. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Buyel, J.F.; Woo, J.A.; Cramer, S.M.; Fischer, R. The use of quantitative structure–activity relationship models to develop optimized processes for the removal of tobacco host cell proteins during biopharmaceutical production. J. Chromatogr. A 2013, 1322, 18–28. [Google Scholar] [CrossRef]
- Konstantinov, K. Continous bioprocessing: An interview with Konstantin Konstantinov from Genzyme. Interviewed by Prof. Alois Jungbauer and Dr. Judy Peng. Biotechnol. J. 2011, 6, 1431–1433. [Google Scholar] [CrossRef]
- Warikoo, V.; Godawat, R.; Brower, K.; Jain, S.; Cummings, D.; Simons, E.; Johnson, T.; Walther, J.; Yu, M.; Wright, B.; McLarty, J.; Karey, K.P.; Hwang, C.; Zhou, W.; Riske, F.; Konstantinov, K. Integrated continuous production of recombinant therapeutic proteins. Biotechnol. Bioeng. 2012, 109, 3018–3029. [Google Scholar] [CrossRef]
- Strube, J.; Ditz, R.; Fröhlich, H.; Köster, D.; Grützner, T.; Koch, J.; Schütte, R. Efficient engineering and production concepts for products in regulated environments—Dream or nightmare? Chem. Ing. Tech. 2014, 86, 687–694. [Google Scholar] [CrossRef]
- Asenjo, J.A.; Andrews, B.A. Aqueous two-phase systems for protein separation: Phase separation and applications. J. Chromatogr. A 2012, 1238, 1–10. [Google Scholar] [CrossRef]
- Rosa, P.A.J.; Azevedo, A.M.; Sommerfeld, S.; Mutter, M.; Bäcker, W.; Aires-Barros, M.R. Continuous purification of antibodies from cell culture supernatant with aqueous two-phase systems: From concept to process. Biotechnol. J. 2013, 8, 352–362. [Google Scholar] [CrossRef]
- Chatel, A.; Kumpalume, P.; Hoare, M. Ultra scale-down characterization of the impact of conditioning methods for harvested cell broths on clarification by continuous centrifugation-recovery of domain antibodies from rec E. coli. Biotechnol. Bioeng. 2014, 111, 913–924. [Google Scholar] [CrossRef]
- Müller-Späth, T.; Krättli, M.; Aumann, L.; Ströhlein, G.; Morbidelli, M. Increasing the activity of monoclonal antibody therapeutics by continuous chromatography (MCSGP). Biotechnol. Bioeng. 2010, 107, 652–662. [Google Scholar] [CrossRef]
- Zobel, S.; Helling, C.; Ditz, R.; Strube, J. Design and operation of continuous countercurrent chromatography in biotechnological production. Ind. Eng. Chem. Res. 2014, 53, 9169–9185. [Google Scholar] [CrossRef]
- Liu, Z.; Bartlow, P.; Varakala, R.; Beitle, R.; Koepsel, R.; Ataai, M.M. Use of proteomics for design of a tailored host cell for highly efficient protein purification. J. Chromatogr. A 2009, 1216, 2433–2438. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gronemeyer, P.; Ditz, R.; Strube, J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering 2014, 1, 188-212. https://doi.org/10.3390/bioengineering1040188
Gronemeyer P, Ditz R, Strube J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering. 2014; 1(4):188-212. https://doi.org/10.3390/bioengineering1040188
Chicago/Turabian StyleGronemeyer, Petra, Reinhard Ditz, and Jochen Strube. 2014. "Trends in Upstream and Downstream Process Development for Antibody Manufacturing" Bioengineering 1, no. 4: 188-212. https://doi.org/10.3390/bioengineering1040188
APA StyleGronemeyer, P., Ditz, R., & Strube, J. (2014). Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering, 1(4), 188-212. https://doi.org/10.3390/bioengineering1040188