Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues


1
Faculty of Industrial Chemistry and Environmental Engineering, Politehnica University Timisoara, Bd. V. Parvan Nr. 6, 300223 Timisoara, Romania
2
National Institute for Research and Development in Electrochemistry and Condensed Matter, Str. Dr. Aurel Paunescu Podeanu Nr. 144, 300587 Timisoara, Romania
*
Author to whom correspondence should be addressed.
ChemEngineering 2017, 1(2), 9; https://doi.org/10.3390/chemengineering1020009
Submission received: 9 September 2017
/
Revised: 6 October 2017
/
Accepted: 8 October 2017
/
Published: 11 October 2017
(This article belongs to the Special Issue Selected Papers from ICEER 2017: 2017 the 4th International Conference on Energy and Environment Research)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Reducing the levels of heavy metals in wastewaters below the permissible limits is imperative before they are discharged into the environment. At the same time, water treatment technologies should be not only efficient, but also affordable. In accordance with these principles, this study assessed the possibility of recovering iron-contaminated residues, resulting from the treatment of synthetic acid mine drainage, for the subsequent remediation of Cr(VI) polluted aqueous solutions. Bentonite, an inexpensive and available natural material, was used as an adsorbent for the removal of Fe(II) from synthetic acid mine drainage (AMD). Then, Fe(II)-contaminated bentonite, the residue generated during the treatment of AMD, was recovered and activated with sodium borohydride in order to convert the adsorbed Fe(II) to Fe(0). Subsequently, the Fe(0)-containing bentonite (Be-Fe(0)) was further used for the treatment of Cr(VI) contaminated aqueous solutions. Reactive materials investigated in this work were characterized by means of scanning electron microscopy-energy dispersive angle X-ray spectrometry (SEM-EDX), X-ray diffraction spectroscopy (XRD), point of zero charge, and image photographs. The effect of several important parameters (pH, temperature, metal concentration, and ionic strength) on both treatment processes was investigated and discussed. It was shown that the efficiency of Cr(VI) removal with Be-Fe(0) was much higher than with bentonite. On the basis of the present study it can be concluded that residues generated during the treatment of AMD with bentonite can be used as a cheap precursor for the production an Fe(0)-based reagent, with good Cr(VI) removal potential.
1. Introduction
Metallic elements are widespread components of the rocks and minerals of the earth’s crust. Heavy metals are extremely toxic, non-biodegradable, persistent, and have a great tendency for bioaccumulation in the food chain [1]. Even though some of them (e.g., Fe, Cu, Mn, Zn) are part of many natural biogeochemical cycles that support life [2], the uncontrolled release of heavy metals into the aquatic ecosystem, as a result of industrial, agricultural, and urban pollution, poses a serious threat to the environmental quality. Chromium is an important heavy metal used in a variety of industrial applications, including wood preservation, the preparation of inorganic chemicals and pigments, textile dying, leather tanning, metallurgy, and metal electroplating; all these industries have released large quantities of chromium into the environment due to accidental events or inadequate precautionary measures [3]. Chromium can exist in oxidation states ranging from (−IV) to (+VI); however, in natural environments, chromium can be found mainly in two of its most stable oxidation states, (+III) and (+VI) [4], which display a markedly different toxicity, chemical behavior, and mobility [5]. Under circumneutral pH conditions, Cr(VI) is present as highly mobile anions, which are toxic to living organisms and well-established human carcinogens [6,7,8]. On the other hand, Cr(III) has a very low mobility in the environment and is 500 to 1000 times less toxic to a living cell than Cr(VI) [9]. Therefore, it is important to reduce the concentrations of Cr(VI) in wastewaters below the permissible limits, before being discharged into the environment. Iron is another important heavy metal with numerous industrial applications, which can also be found in natural aqueous environments, mainly in two oxidation states: Fe(II) and Fe(III) [10]. Because under circumneutral conditions Fe(III) is frequently confined to the particulate phase, Fe(II) is the more soluble and mobile form of iron [11,12]. The two oxidation states of iron are capable of accepting and donating electrons; hence, iron participates in redox reactions which are essential for the diverse metabolic processes of most living cells [13,14,15,16]. However, in the case of an excessive concentration within cells, the same redox property of iron also accounts for its toxicity [10,14,17,18]. Therefore, it is obvious that iron concentration in body tissues must be tightly regulated, as well as its concentration in natural effluents. In recent years, there has been great interest in the use of natural materials, available in large quantities, as inexpensive alternative adsorbents [19,20]. Clay minerals are classified as promising cheap adsorbents for heavy metal removal because they offer several advantages, including that they: (1) are readily available, (2) are inexpensive, (3) have a layered structure and large specific surface area, (4) have a good chemical and mechanical stability, and (5) have a high cation exchange capacity [20,21]. At the same time, using metallic iron (Fe(0)) for the removal of various pollutants, including heavy metals, has also become increasingly popular in the last few decades [3,22,23]. The policy framework for managing solid waste encourages the prevention of their generation and/or waste recycling. Accordingly, the reuse of waste materials resulting from water treatment technologies, as reactive materials for other water treatment processes, can become a crucial component, especially in developing nations, in their efforts to address pollution caused by industrial activities. Therefore, in the first phase of this study, we investigated the use of bentonite as a cheap adsorbent for the removal of Fe(II) from synthetic acid mine drainage (AMD). Then, the exhausted bentonite resulting from this step was recovered and treated with sodium borohydride in order to reduce the adsorbed iron from Fe(II) to Fe(0). Finally, the resultant Fe(0)-doped bentonite, hereinafter referred to as Be-Fe(0), was further used for the remediation of Cr(VI) polluted waters. The effect of several important parameters on both treatment processes was investigated, and the efficiency of Cr(VI) removal with Be-Fe(0) and with normal bentonite was comparatively discussed. In addition, the mechanism of Cr(VI) removal with Be-Fe(0) was also evaluated.
2. Materials and Methods
2.1. Materials
The clay employed in the present study for the retaining of Fe(II) was bentonite (O: 53.99%; Si: 34.09%; Al: 6.27%; K: 2.53%; Ca: 1.76%; Fe: 0.90%; Mg: 0.46%) obtained from a local Romanian deposit. The fresh bentonite was ground and passed through a 0.5 mm sieve, washed with distilled water, dried in oven at 105 °C for 24 h, and further used in treatability experiments. Fe(II) (AMD) and Cr(VI) stock solutions (1000 mg/L) were prepared by dissolving the required amount of AR grade FeSO4·7H2O and K2Cr2O7, respectively, in distilled water. Then, synthetic AMD and Cr(VI) working solutions with desired concentrations were prepared by the appropriate dilution of stock solution. The synthetic AMD solution contained only Fe2+ because this is usually the major metal cation in real AMD generated as a result of iron-based sulfide mineral oxidation (e.g., pyrite) in the presence of water and atmospheric oxygen [24,25]. The pH of synthetic solutions was adjusted before experiments to the required value by the addition of small amounts of concentrated H2SO4.
2.2. Experimental Procedure
2.2.1. Fe(II) Treatability Experiments
Batch Fe(II) treatability experiments were conducted in 800 mL Berzelius flasks, by introducing 15 g of bentonite into 500 mL synthetic AMD solution. All experiments were conducted at an initial Fe(II) concentration of 100 mg/L, except those investigating the impact of Fe(II) initial concentration. The mixture was stirred using an Ovan agitator (200 rpm) and, at timed intervals, samples were withdrawn, filtered using a 0.45 μm filter, and analyzed for Fe(total).
2.2.2. Recovery and Activation of Exhausted Bentonite
At the end of the Fe(II) treatability experiments, about 200 g of spent bentonite was recovered. Because Fe(II) treatability experiments were conducted with variations of several important experimental parameters (pH, temperature, Fe(II) concentration, ionic strength), they resulted in different Fe(II) removal efficiencies of bentonite; therefore, samples of exhausted bentonite from different experiments were characterized by different amounts of Fe(II) adsorbed. In order to obtain a homogeneous concentration of adsorbed Fe(II), the 200 g of exhausted bentonite was further reacted with 1000 mL solution Fe(II) 100 mg/L, at room temperature and pH 2.9. After 24 h of mixing, the spent bentonite was separated by filtration, washed thoroughly with distilled water to drain out the un-exchanged iron, and, finally, dried at 105 °C for another 24 h. By means of a mass balance calculation, the concentration of adsorbed iron was determined to be about 1.5 mg Fe/g bentonite. Then, 300 mL distilled water was added over the recovered bentonite, and the obtained slurry was stirred at a rate of 200 rpm, in order to keep the bentonite particles in suspension. Sodium borohydride (NaBH4) was used as a reducing agent to convert the adsorbed iron from Fe(II) to Fe(0) via the liquid-phase reduction method [26]:
Fe(H2O)62+(aq) + 2BH4−(aq) → Fe0(s)↓+ 2B(OH)3(aq) + 7H2(g)↑
This process was carried out because Fe(0) is a stronger reductant than Fe(II) (E° = −0.44 V and +0.77 V for Fe2+/Fe0 and Fe3+/Fe2+, respectively, [3]). A 200% excess of borohydride was applied in order to accelerate the reaction and to compensate for any NaBH4 that reacts with the water. After the complete addition of NaBH4, the mixture was further stirred for 1 h. The resultant Be-Fe(0) was separated from the solution, washed twice with ethanol, dried at 50 °C overnight in an oven, and kept in a vacuum desiccator prior to use.
2.2.3. Cr(VI) Treatability Experiments
Cr(VI) treatability experiments were conducted in 800 mL Berzelius flasks by introducing 10 g of Be-Fe(0) into a 500 mL Cr(VI) solution. All experiments were conducted at an initial Cr(VI) concentration of 2 mg/L, except those investigating the impact of Cr(VI) initial concentration. The mixture was stirred (200 rpm) and, periodically, aliquots were extracted, filtered, and analyzed for Cr(VI). In addition, control experiments with fresh (unmodified) bentonite were also carried out, under similar experimental conditions; however, because preliminary tests showed a very low affinity of bentonite towards Cr(VI) even at a very low pH, the dose of bentonite used in the control experiments was increased to 60 g/L.
2.2.4. Analytical Procedure
Cr(VI) concentration in the filtrate was analyzed by the 1,5-diphenylcarbazide colorimetric method at 540 nm [27]. In addition, for the investigation of the mechanism of Cr(VI) removal with Be-Fe(0), Cr(total) was also determined, by oxidizing any Cr(III) with KMnO4, followed by analysis as Cr(VI) [27]; then, Cr(III), if any, was evaluated as the difference between Cr(total) and Cr(VI). Fe(total) concentration was determined via the 1,10-ortophenantroline method, at 510 nm, by the reduction of any Fe(III) to Fe(II) with hydroxylamine hydrochloride and subsequent analysis as Fe(II) [27]. After the color (purple and orange, for Cr(VI) and Fe(total), respectively) was fully developed, samples were transferred to a Specord 200 PLUS spectrophotometer, where the absorbance was measured in 10 mm pathlength glass cells. The pH of the samples was measured using an Inolab 7320 pH-meter calibrated with three standards. The point of zero charge (pHpzc) of the bentonite surface was determined using the pH drift method [28]. Scanning electron microscopy (SEM)-energy dispersive angle X-ray spectrometry (EDX) and X-ray diffraction (XRD) were employed to investigate the surface morphology and chemical composition of bentonite. The SEM-EDX analysis was performed on an Inspect S scanning electron microscope (FEI, Eindhoven, Holland) coupled with a GENESIS XM 2i energy dispersive angle X-ray spectrometer. The XRD measurements were performed at 40 kV and 30 mA on a X’Pert PRO MPD Diffractometer (FEI, Eindhoven, Holland) equipped with a Cu anode X-ray tube and PixCEL detector (Cu Kα radiation, λ = 1.54056 Å). The ionic strength of the solutions was estimated using the empirical relation of Russel [29]:
where I is the ionic strength (M) and is the conductivity (μS/cm).
3. Results and Discussion
3.1. Mitigation of Fe(II) Pollution
3.1.1. Effect of pH
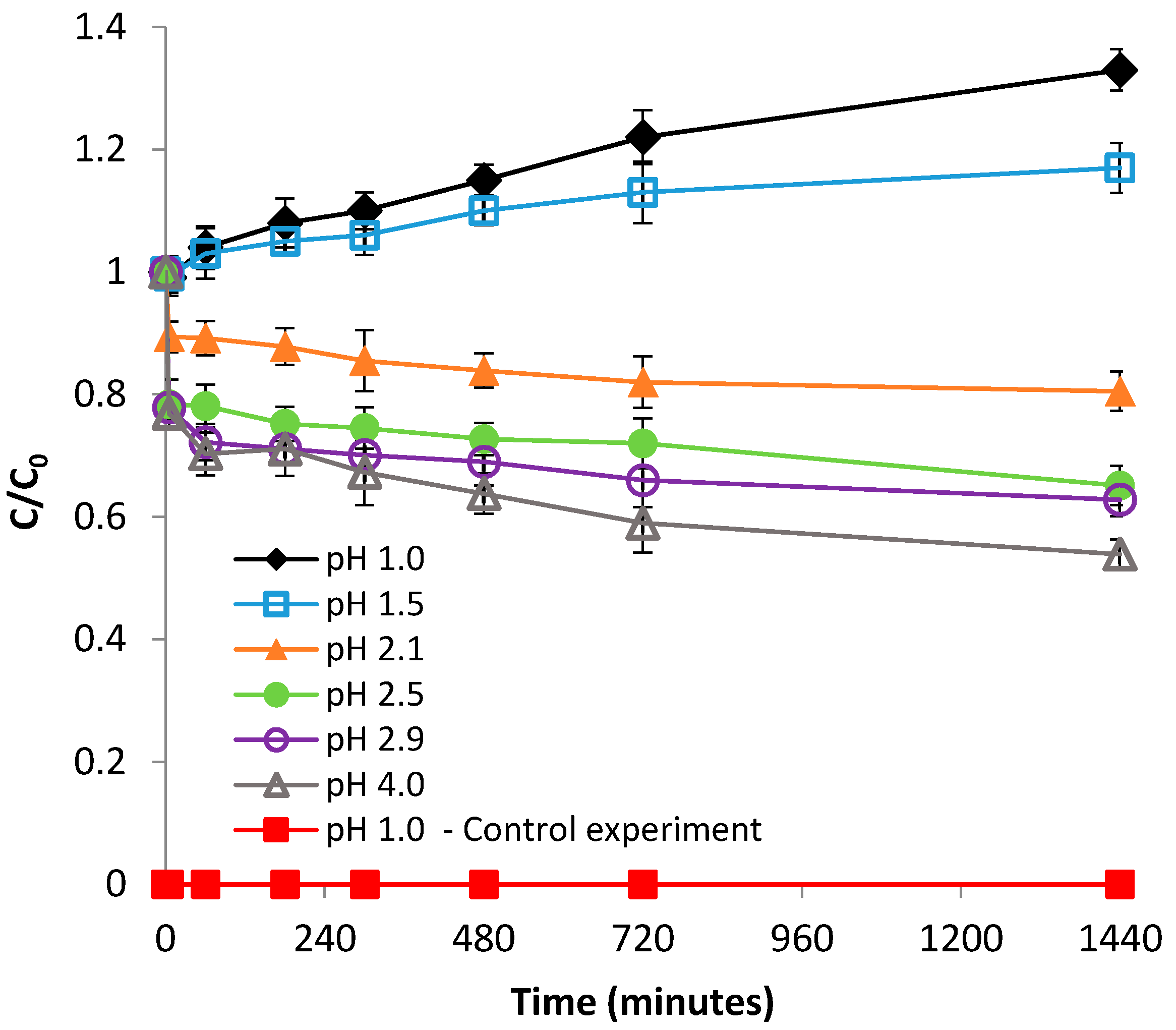
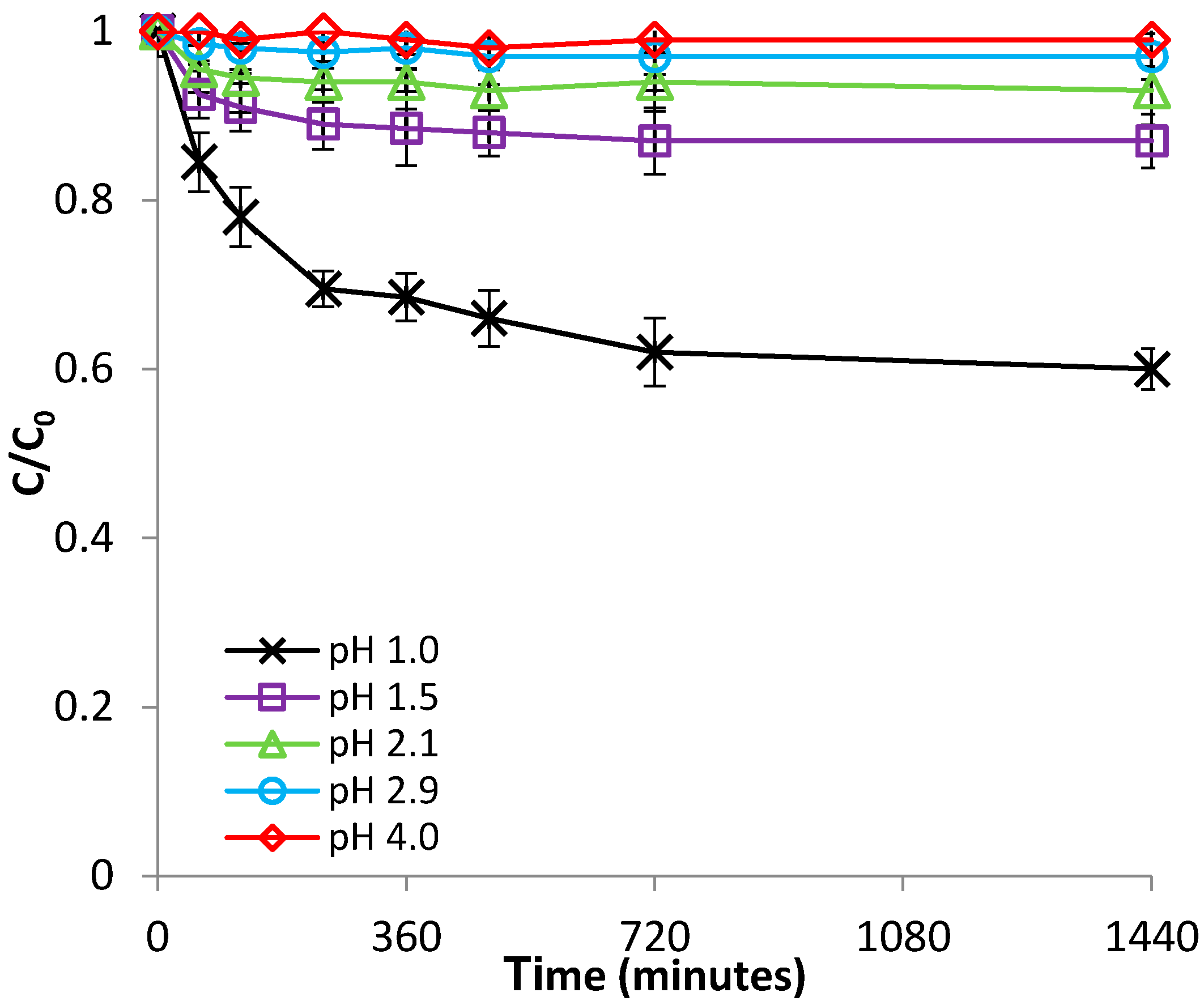
In this study, the impact of initial pH was studied within the pH range of 1.0–4.0, at room temperature (22 °C). These pH values were selected because they are within the range of levels reported for pH in AMD environments [30,31]. It can be seen from Figure 1 and Figure S1 (supplementary material) that both the rate of Fe(II) adsorption and adsorption capacity of bentonite decreased with decreasing pH from 4.0 to 2.1.
On the one hand, this observation can be related to electrostatic interactions between cationic Fe2+ species and anionic charged centers on the surface of bentonite. Acid-base equilibria of bentonite surface groups in aqueous solutions can be represented as follows:
Be-OH + H+ ⇔ Be-OH2+
Be-OH + HO− ⇔ Be-O− + H2O
The value of bentonite pHpzc was found to be 7.2 (Figure S2); therefore, over the entire pH range that was studied for the adsorption of Fe(II), the number of positive charges existent at the surface of bentonite was greater than that of negative charges; hence, the net charge of the surface of bentonite was positive. However, by decreasing the solution pH from 4.0 to 2.1, Equation (3) becomes favored, leading to a gradual increase of the net positive charge. Accordingly, the hindering effect of decreasing the pH from 4.0 to 2.1 can be attributed to the increase in electrostatic repulsion forces between Fe2+ cations and the positively charged bentonite surface. On the other hand, the observed pH dependence may be attributed to a higher competition for the negatively charged sites between Fe2+ and H3O+ cations, as a result of the increasing H3O+ concentration with decreasing solution pH. Figure 1 also reveals that, by further decreasing the pH to even lower values (1.5 and 1.0), the concentration of Fe(II) in solution increased over the initial value of 100 mg/L, up to about 133 mg/L. Since the control experiments carried out in the absence of Fe(II), at pH 1.0 (just distilled water with pH 1.0), over a 24 h period, showed no release of dissolved iron from the stainless steel stirring rods of the agitator, the phenomenon noticed at pH 1.0 and 1.5 can be attributed to the solubilization of iron existent in the bentonite structure.
3.1.2. Effect of Temperature
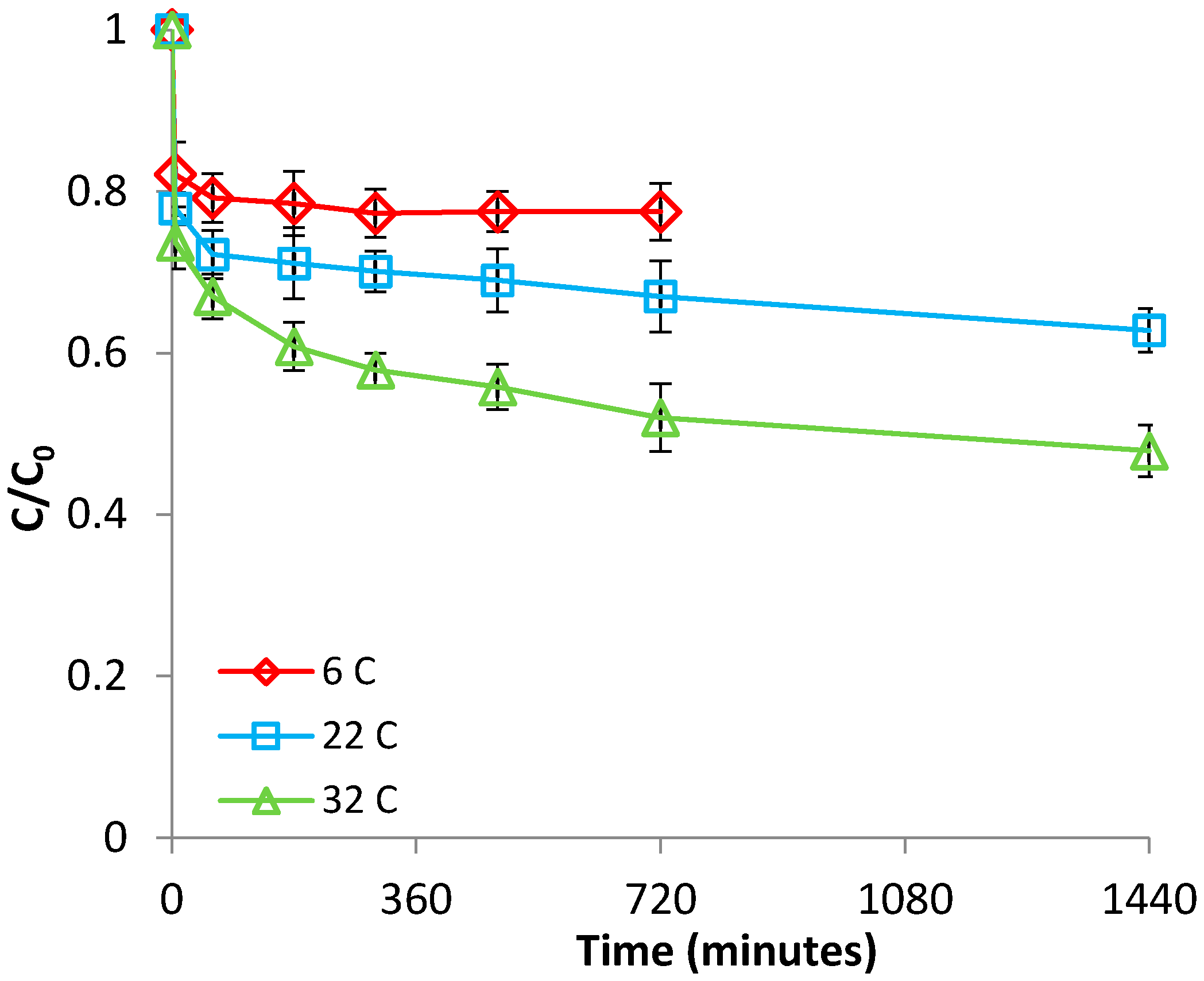
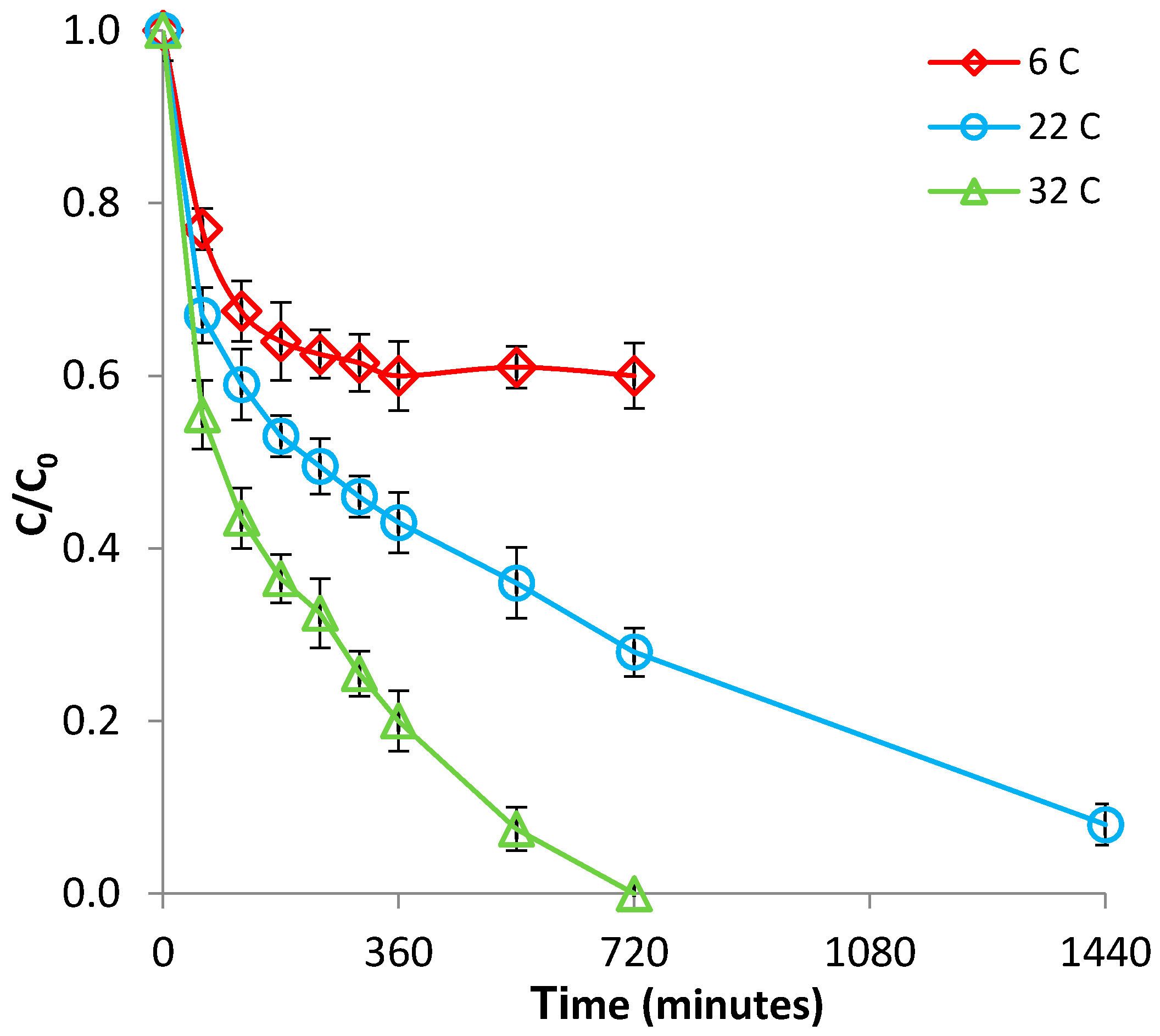
The effect of temperature was investigated over the range of 6–32 °C, at pH 2.9. The results presented in Figure 2 and Figure S3 (supplementary material) show that the adsorption of Fe(II) on bentonite was significantly affected by this parameter, increasing with increasing temperature; this behavior indicates that the adsorption process was endothermic in nature. Furthermore, from Figure 2, it is apparent that adsorption reached saturation faster at low temperatures than at high temperatures. The enhancement of adsorption efficacy with increasing temperature may be attributed to better interactions between Fe(II) and bentonite, as a result of the creation of new adsorption sites, or of increased rates of intraparticle diffusion of Fe(II) ions into the pores of bentonite at higher temperatures [32]. The positive value of ΔH (Table S1, supplementary material) supports the endothermic nature of adsorption, while the positive value of ΔS (Table S1, supplementary material) reflects an increased randomness at the solid/solution interface during the adsorption process [33]. In addition, the positive value of the ΔG at 6 °C suggests that, at low temperatures, the adsorption of Fe(II) on bentonite is not spontaneous; however, the process becomes spontaneous at temperatures ≥ 22 °C, when negative ΔG values were obtained (Table S1, supplementary material). All this supports the fact that Fe(II) adsorption is favored by an increase in temperature.
3.1.3. Effect of Fe(II) Concentration
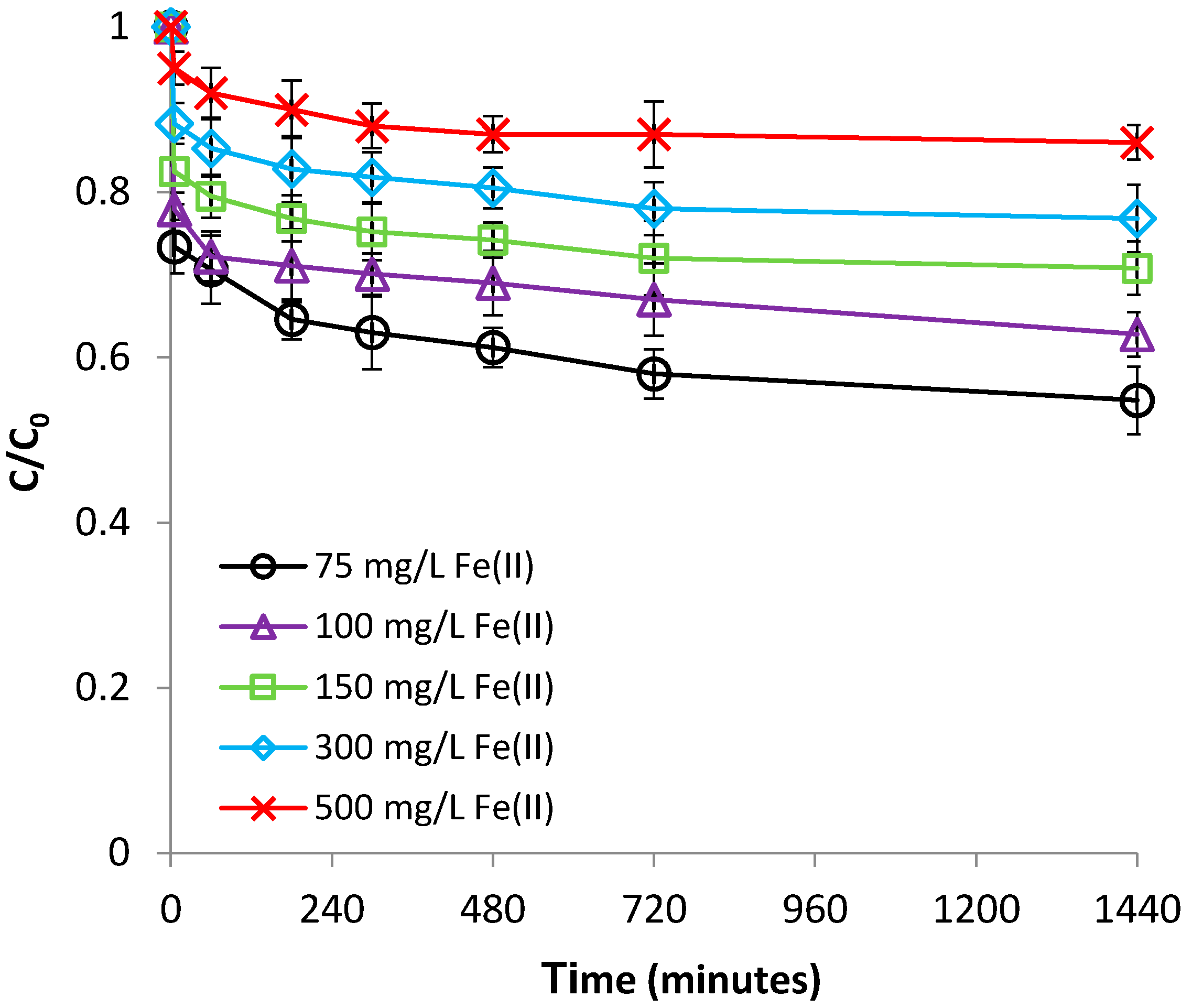
The influence of Fe(II) concentration was researched by varying the initial Fe(II) concentration over the range of 75–500 mg/L, at pH 2.9 and room temperature (22 °C). These concentrations were selected because they are within the range of the levels of reported Fe(II) in AMD environments [30,31]. As revealed in Figure 3, the efficiency of adsorption was found to increase proportionally with the decrease of Fe(II) concentration. This phenomenon may be ascribed to increasing the bentonite:Fe(II) mass ratio with decreasing Fe(II) concentration, while maintaining the bentonite dose. As a result, a greater surface area, and thus a higher number of adsorption sites, will be available for the retaining of low concentrations of Fe(II). Instead, at higher Fe(II) concentrations, no sufficient adsorption sites were available at the bentonite surface and, therefore, Fe(II) was much more slowly retained from the solution, due the rapid saturation of the adsorption centers. On the other hand, Figure S4 (supplementary material) indicates that the adsorption capacity of bentonite increased from 1.13 to 2.33 mg/g by increasing the initial concentration of Cr(VI) from 75 to 500 mg/L. One can see from this figure that the adsorption capacity of bentonite constantly increased with increasing Fe(II) concentration, until a steady state value was reached, corresponding to the maximal adsorption capacity. The improved bentonite adsorption capacities observed at higher Fe(II) concentrations are attributable to the increasing of the Fe(II) concentration gradient at the solution-bentonite interface, leading to more frequent collisions between Fe(II) and bentonite, and therefore, to increased mass transfer driving forces [34].
3.1.4. Effect of Ionic Strength
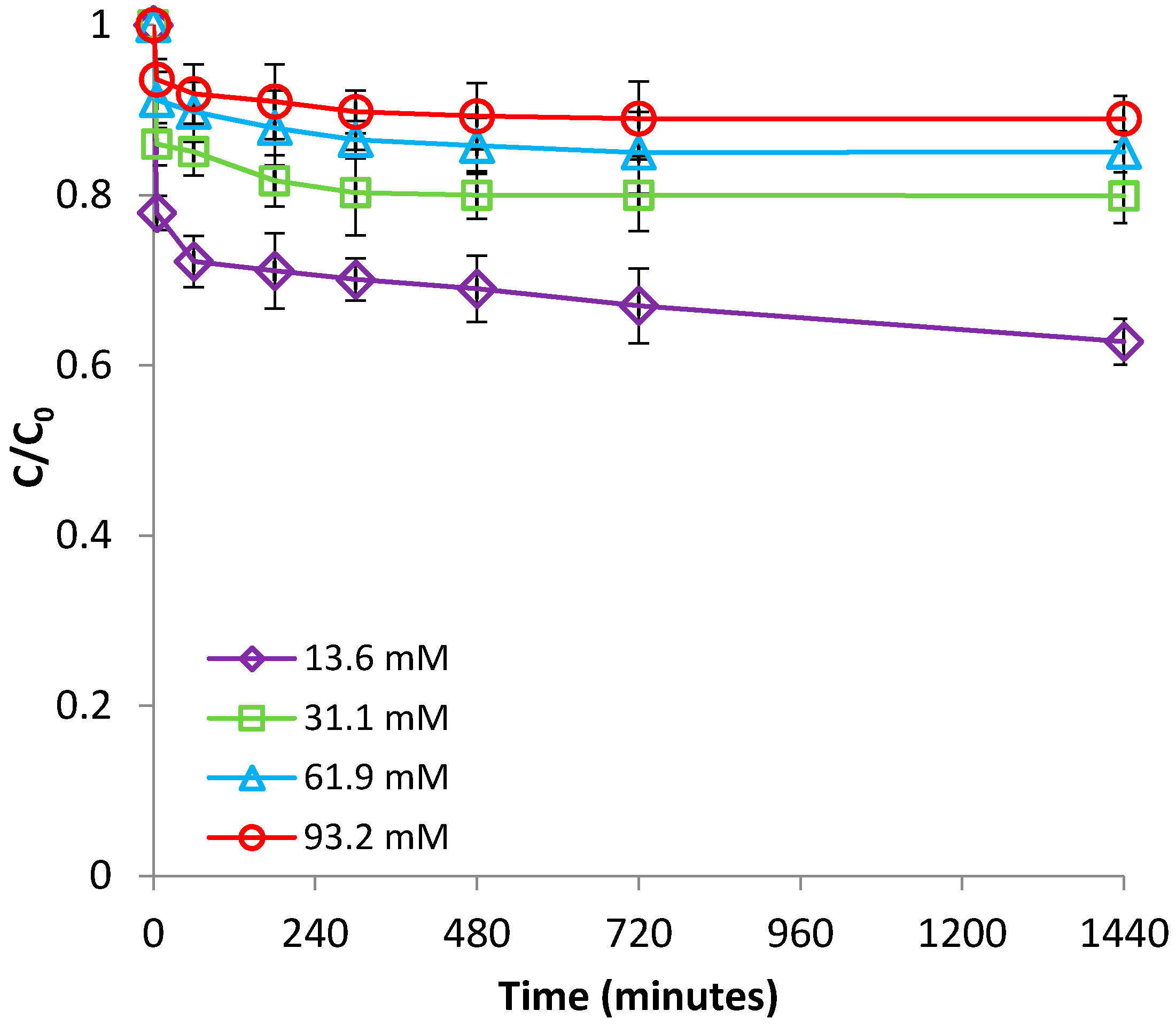
The ionic strength effect was tested at room temperature (22 °C) and pH 2.9, by using NaCl with concentrations of 0, 0.01, 0.03, and 0.05 M as a background electrolyte. Figure 4 and Figure S5 (supplementary material) portray the impact of ionic strength on the adsorption process. These figures clearly reveal that less Fe(II) was adsorbed by bentonite in the presence of competing cations (Na+) than when no cations were added; in addition, the efficiency of Fe(II) adsorption was progressively hindered with increasing ionic strength. This effect is usually interpreted as indicating a non-specific adsorption mechanism (physisorption) [35].
3.2. Mitigation of Cr(VI) Pollution
3.2.1. Effect of pH
The effect of solution pH was studied within the initial pH range of 1.0–4.0, at room temperature. The present experiments showed that both the rate of Cr(VI) removal (Figure 5) and Cr(VI) removal capacity of Be-Fe(0) (Figure S6, supplementary material) were enhanced by decreasing the pH from 4.0 to 1.0. Nevertheless, it is important to point out that the most notable improvement in Cr(VI) removal was achieved by decreasing the pH from 4.0 to 1.5; a further decrease of the pH to 1.0 led only to a minor enhancement of Cr(VI) removal efficacy. By comparing these findings with the results of control experiments carried out with bentonite under the same experimental conditions, but with a three times higher bentonite dose, it clearly shows that the efficacy of Be-Fe(0) to remove Cr(V) was much higher than that of bentonite (Figure 6 and Figure S7, supplementary material).
This behavior can only be ascribed to the existence of Fe(0) centers at the surface of Be-Fe(0), which are responsible for a much more efficient mechanism of Cr(VI) removal, as discussed later in Section 3.4. From Equations (6)–(12), it is obvious that for both mechanisms of Cr(VI) removal (adsorption and chemical reduction), a decrease of pH (thus, an increase in H+ concentration) will lead to an increase in the efficiency of Cr(VI) removal. On the one hand, by decreasing the solution pH from 4.0 to 1.0, Equation (3) becomes favored, leading to a gradual increase of the Be-Fe(0) net positive charge. As a result, the attracting electrostatic forces between Cr(VI) anions and the positively charged Be-Fe(0) surface will also increase, leading to a better retaining of Cr(VI). At the same time, the observed effect of decreasing the pH may be attributed to lower competition for the positively charged sites between HCrO4− and HO− anions, as a result of decreasing the HO− concentration with decreasing the solution pH. On the other hand, the stoichiometry of Equations (8)–(12) clearly shows that all these reactions are highly dependent on the H+ concentration in the solution; for most of them, the chemical reduction of Cr(VI) will be favored when the concentration of H+ in the solution increases (LeChatelier’s principle).
3.2.2. Effect of Temperature
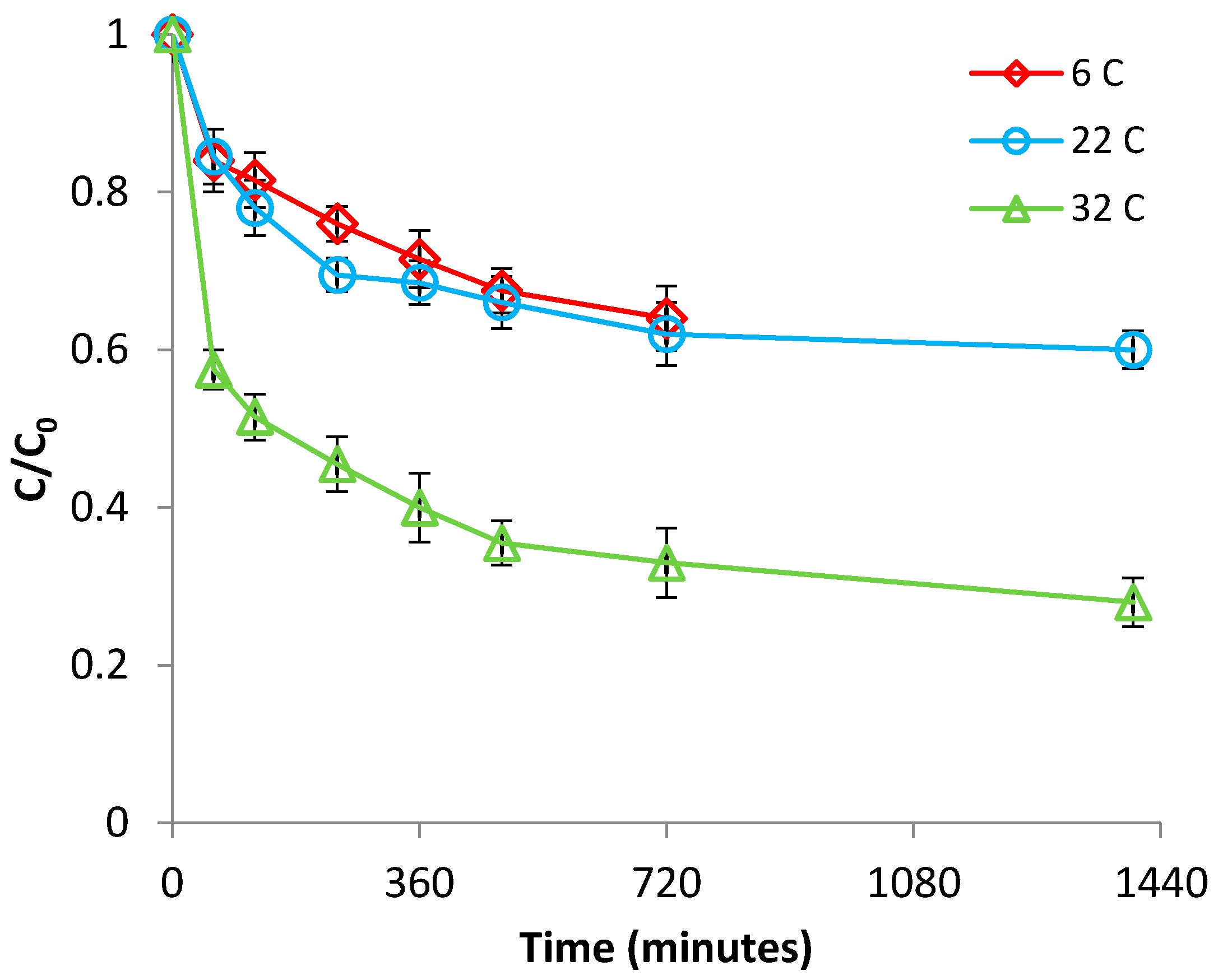
The impact of temperature was investigated over the range of 6–32 °C, at pH 1.0. From Figure 7 and Figure S8 (supplementary material), it can be seen that the higher the solution temperature, the better the efficiency of Cr(VI) removal and the Cr(VI) removal capacity of Be-Fe(0). The same effect was observed for the control experiments, conducted with bentonite under similar experimental conditions (Figure 8 and Figure S9, supplementary material). This result indicates that the removal of Cr(VI) with both Be-Fe(0) and bentonite is endothermic in nature. The endothermic nature of Cr(VI) removal process with both materials is confirmed by the positive value of ΔH (Table S1, supplementary material), while the positive value of ΔS (Table S1, supplementary material) reflects an increased randomness at the solid/solution interface during the adsorption process [33]. Figure 8 and Figure S9 (supplementary material) also reveal that the very low efficiency of Cr(VI) removal with bentonite was only slightly improved when the temperature was raised from 6 to 22 °C, and then significantly enhanced by further increasing the temperature from 22 to 32 °C; this is confirmed by the positive values of the ΔG observed for the process of Cr(VI) removal with bentonite at 6 °C and 22 °C, suggesting that, at these temperatures, the process is not spontaneous; instead, the Cr(VI) removal with bentonite was much more efficient at high temperatures (32 °C), when it was found to be spontaneous (Table S1, supplementary material). On the other hand, important improvements in Cr(VI) removal efficacy were achieved with Be-Fe(0) by increasing the temperature not only from 22 to 32 °C, but also from 6 to 22 °C (Figure 7 and Figure S8, supplementary material). This observation is endorsed by the negative values of the ΔG observed for the process of Cr(VI) removal with Be-Fe(0) (Table S1, supplementary material), indicating the spontaneous nature and feasibility of this process over the entire studied temperature range; moreover, since the values of ΔG become more negative at a higher temperature, it means that Cr(VI) removal with Be-Fe(0) is thermodynamically favored by increasing the temperature. All these discrepancies between the influence of temperature on Cr(VI) removal with Be-Fe(0) and bentonite strongly suggest that the mechanisms responsible for the removal of Cr(VI) with the two materials are also different, confirming thus the existence of Fe(0) centers at the surface of Be-Fe(0).
3.2.3. Effect of Cr(VI) Concentration
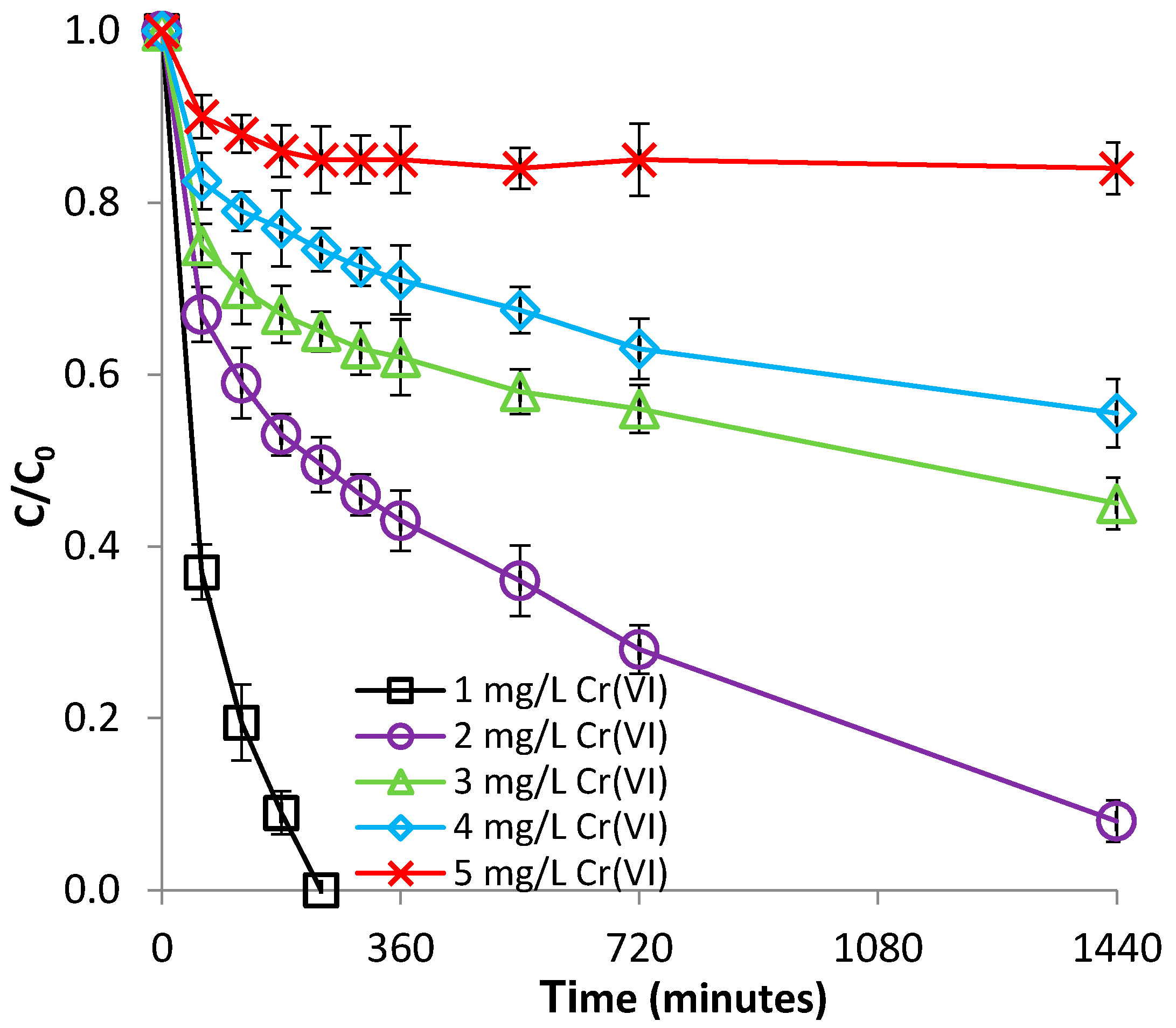
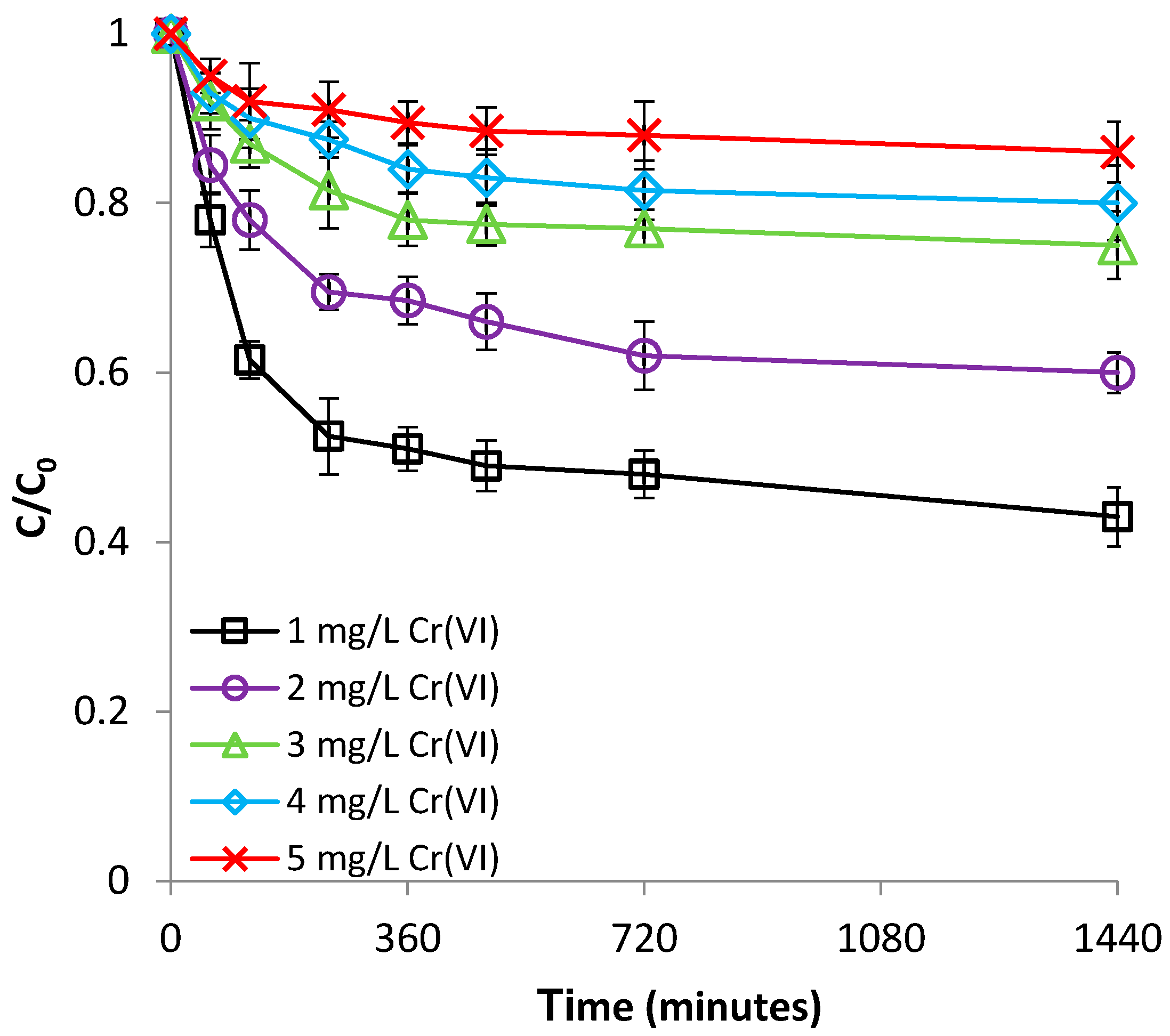
The influence of the Cr(VI) concentration was researched by varying the Cr(VI) concentration over the range of 1–5 mg/L, at room temperature (22 °C) and pH 1.0. These concentrations were chosen because they are within the common levels in subsurface Cr(VI)-contaminated environments [36]. As presented in Figure 9, the removal of Cr(VI) with Be-Fe(0) is significantly influenced by the initial Cr(VI) concentration: the higher the initial Cr(VI) concentration, the lower the efficiency of the removal process. This phenomenon is attributable to the fact that, by maintaining the Be-Fe(0) dose and increasing the Cr(VI) concentration, the number of reactive sites (available for the interaction with Cr(VI)) of each gram of Be-Fe(0) will decrease, and saturation of the adsorption centers will occur more rapidly.
On the other hand, Figure S10 (supplementary material) indicates that the highest Cr(VI) removal capacity of Be-Fe(0) was observed over the Cr(VI) concentration range of 2–4 mg/L. The Cr(VI) removal capacity of Be-Fe(0) increased from 0.05 to about 0.9 mg/g, by increasing the initial concentration of Cr(VI) from 1 to 4 mg/L; subsequently, the Cr(VI) removal capacity of Be-Fe(0) decreased to 0.04 mg/g when the Cr(VI) initial concentration was further increased to 5 mg/L (Figure S10, supplementary material). As reported for the retaining of Fe(II) on bentonite at Section 3.1.3, normally, the retaining capacity should constantly increase with an increasing adsorbate concentration, until a steady state value is reached, corresponding to the maximal adsorption capacity of that adsorbent. The initial increase of the Be-Fe(0) removal capacity with increasing Cr(VI) concentration is attributable to the increasing Cr(VI) concentration gradient at the solution-Be-Fe(0) interface, which leads to more frequent collisions between Cr(VI) and Be-Fe(0), hence, to increased mass transfer driving forces. The subsequent decline of the Be-Fe(0) Cr(VI) removal capacity at a Cr(VI) concentration of 5 mg/L can be related to the ability of HCrO4− to dimerize [3]:
HCrO4− + HCrO4− ⇔ Cr2O72− + H2O
With increasing HCrO4− concentration, Equilibrium (12) shifts to the right, leading to an increase in the Cr2O72− concentration (Le Châtelier’s principle). Because Cr2O72− anions are species with larger volume than HCrO4−, they cannot access all reactive centers located inside Be-Fe(0) pores, which led to a lower removal capacity of Be-Fe(0). The same effect of increasing the Cr(VI) concentration was also observed for the control experiments conducted with bentonite under similar experimental conditions (Figure 10 and Figure S11, supplementary material).
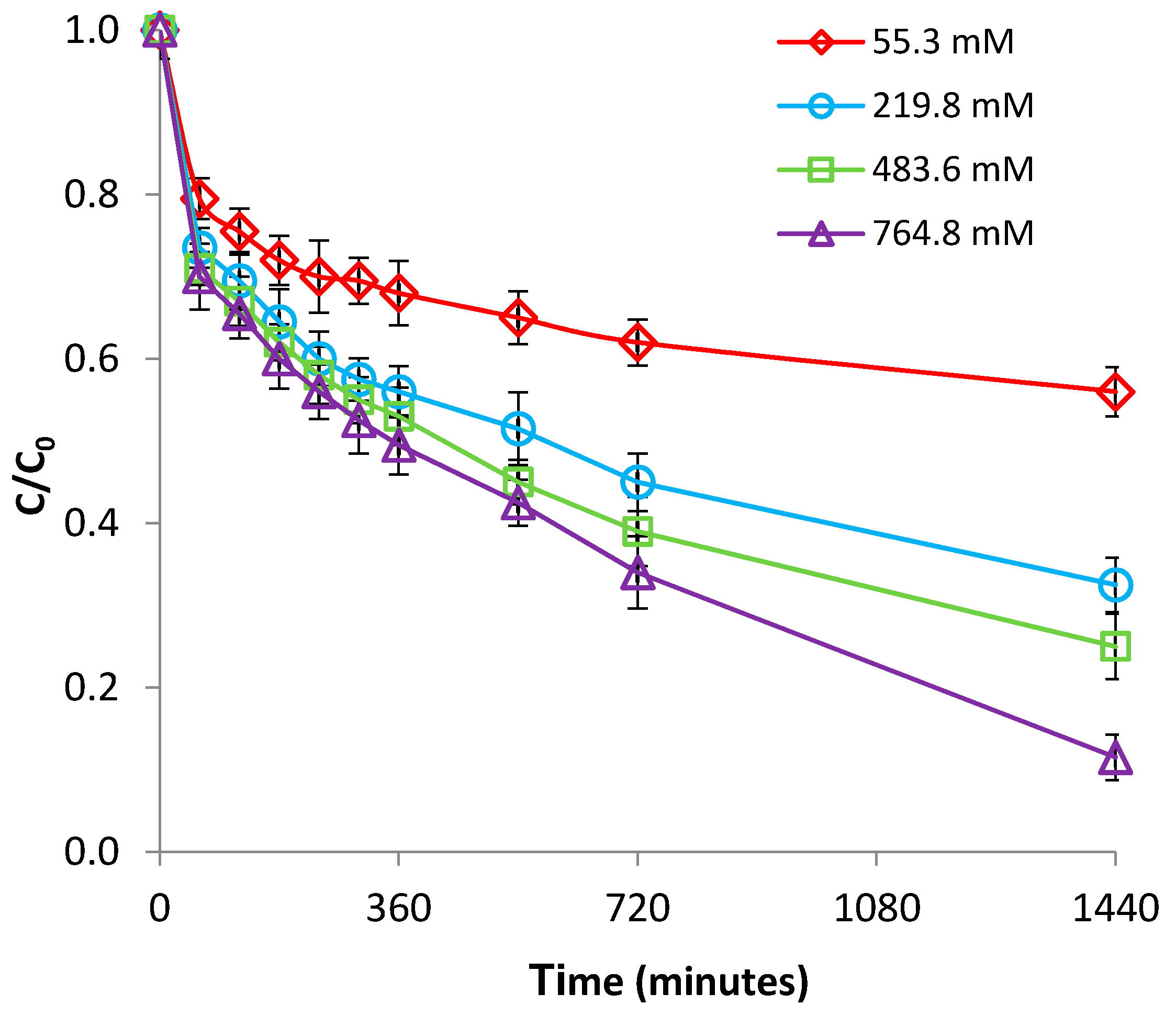
3.2.4. Effect of Ionic Strength
The ionic strength effect was tested at room temperature (22 °C) and pH 2.1 by using NaCl with concentrations of 0, 0.1, 0.3, and 0.5 M as a background electrolyte. As illustrated in Figure 11 and in Figure S12 (supplementary material), the removal of Cr(VI) with Be-Fe(0) was considerably enhanced by increasing the concentration of NaCl. On the contrary, control experiments conducted with bentonite revealed that adsorption of Cr(VI) on bentonite was not influenced by the increase of background ionic strength (Figure 12 and Figure S13, supplementary material). The contrasting effect of ionic strength on the removal of Cr(VI) with bentonite and with Be-Fe(0) clearly indicates that two different mechanisms are involved in the removal of Cr(VI) with the two materials. While the effect of ionic strength on Cr(VI) removal with bentonite can be interpreted as indicating a strong specific adsorption mechanism [35], the results obtained with Be-Fe(0) can be ascribed solely to the existence of Fe(0) particles at the surface of Be-Fe(0).
This is in accordance with previous works which demonstrated that Cl− may accelerate Fe(0) corrosion by forming soluble complexes with Fe(II), which are carried away from the metal surface [37]. Therefore, the enhanced removal of Cr(VI) with Be-Fe(0) in the presence of Cl− can be ascribed to: (1) higher amounts of soluble Fe(II) in solution, which are available for the reduction of Cr(VI) to Cr(III) (Equation (10)); and (2) delaying of the formation of oxide layers on the Fe(0) surface [38].
3.3. Solid Phase Characterization
Figure S13 presents the images of fresh bentonite (A) and Be-Fe(0) (B). It can be seen that the color of Be-Fe(0) is slightly darker than that of bentonite. This confirms the occurrence of black Fe(0) particles at the surface of Be-Fe(0), as a result of reacting exhausted bentonite with borohydride. SEM-EDX and XRD analysis of exhausted bentonite resulting from acid mine drainage treatment gave similar results to those observed for the fresh bentonite (Figures S15–S18, supplementary material). This is attributable to the fact that only small amounts of Fe(II) were adsorbed at the surface of bentonite, and, therefore, differences between fresh and exhausted bentonite are not discernible by this type of instrumental analysis. Similar XRD patterns and SEM images were also obtained for the materials (Be-Fe(0) and bentonite) used in the Cr(VI) treatability experiments (Figures S19–S24, supplementary material). In addition, no supplementary peaks of Fe(0) were noticed in the XRD spectra of fresh Be-Fe(0) (Figure S19, supplementary material), in comparison to the XRD spectra of exhausted bentonite from acid mine drainage treatment (Figure S16, supplementary material), indicating that only a very small amount of Fe(0) was formed at the surface of bentonite after the treatment with sodium borohydride. However, additional Cr peaks were observed in the EDX spectra of exhausted Be-Fe(0) recovered after the treatment of Cr(VI) solution (Figure S21, supplementary material), indicating that the removed Cr(VI) was retained at the surface of Be-Fe(0); in contrast, no Cr was identified at the surface of exhausted bentonite resulting from control experiments with Cr(VI) solution (Figure S23, supplementary material), which confirms the very low affinity of unmodified bentonite for Cr(VI).
3.4. Mechanism of Cr(VI) Removal with Be-Fe(0)
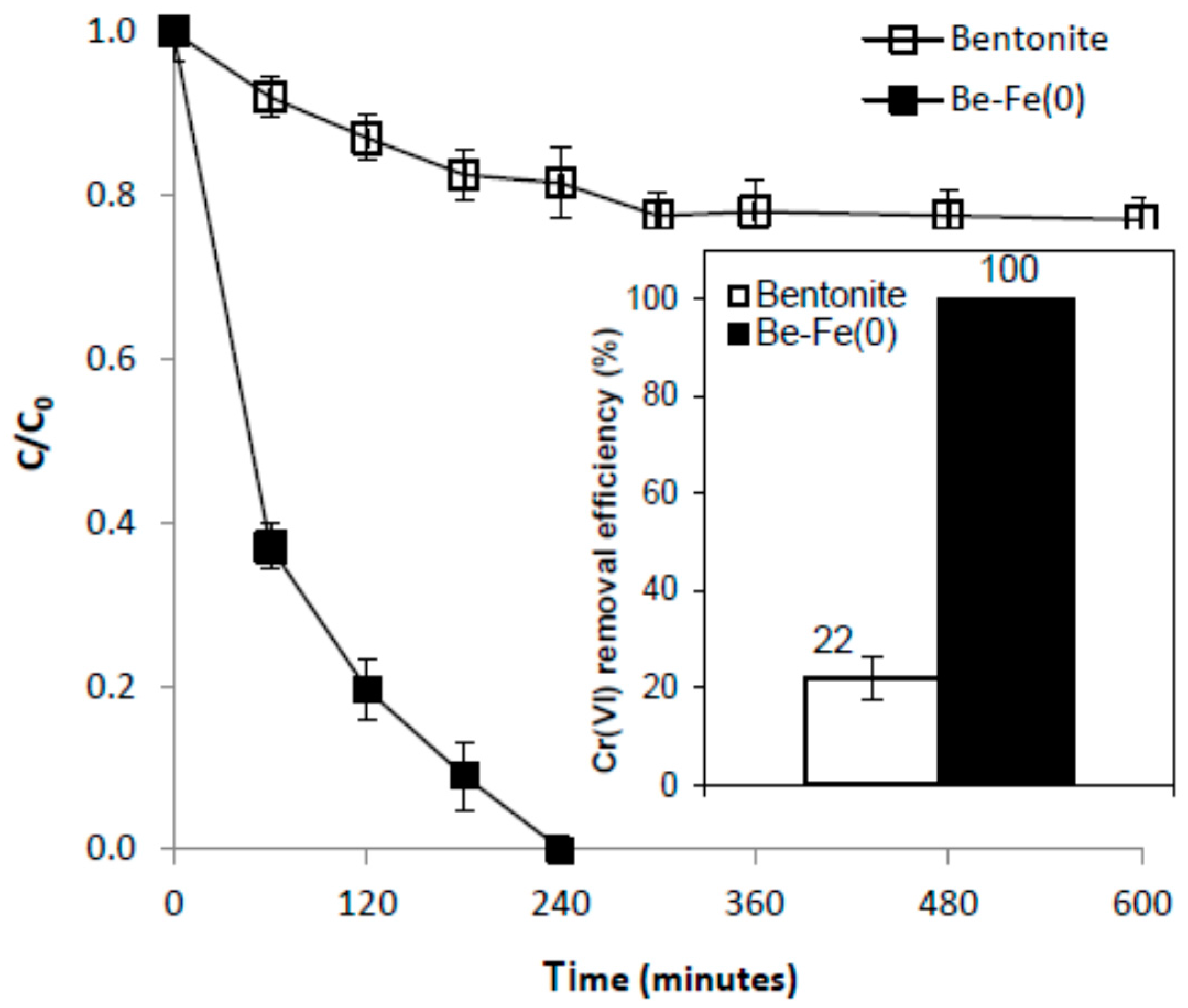
In order to assess the mechanism of Cr(VI) removal with Be-Fe(0), we compared the results of Cr(VI) removal experiments with those of Be-Fe(0) and with unmodified bentonite, carried out at room temperature (22 °C) and pH 1.0, by introducing a dose of 40 g/L solid material into a 2 mg/L Cr(VI) solution. Figure 13 reveals that the highest removal efficiency of normal bentonite was only 22%, obtained after 300 min, while for Be-Fe(0), the integral removal of Cr(VI) was achieved after only 240 min.
These different removal efficacies can be ascribed to the existence of Fe(0) centers at the surface of Be-Fe(0). Accordingly, while the removal of Cr(VI) with bentonite occurred through a pure adsorption mechanism, the removal of Cr(VI) with Be-Fe(0) may take place via a complex mechanism comprising: (1) adsorption on the ”Fe(0)-free” surface of Be-Fe(0); (2) adsorption on Fe(0) centers, or onto (hydr)oxide layers existent at surface of Fe(0) centers; (3) heterogeneous chemical reduction with Fe(0); (4) heterogeneous chemical reduction with solid corrosion products containing Fe(II), existing at the surface of Fe(0); and (5) homogenous Cr(VI) reduction with dissolved indirect reductants generated by Fe(0) corrosion, according to [3]:
Be-OH2+ + HCrO4− ⇔ Be-CrO4 + H2O
Be-Fe-OH2+ + HCrO4− ⇔ Be-Fe-CrO4 + H2O
3FeIIFeIII2O4 + HCrO4− + 14H2O + H+ → 4[Fe0.75Cr0.25](OH)3 + 6Fe(OH)3
3[FeII4FeIII2(OH)12][SO4.3H2O] + 4HCrO4− + 5H2O → 16[Fe0.75Cr0.25](OH)3 + 2H+ +
3SO42− + 6Fe(OH)3
3SO42− + 6Fe(OH)3
2HCrO4− + 3Fe0 + 14H+ → 3Fe2+ + 2Cr3+ + 8H2O
HCrO4− + 3Fe2+ + 7H+ → 3Fe3+ + Cr3+ + 4H2O
2HCrO4− + 3H2 + 8H+ → 2Cr3+ + 8H2O
Figure 13 clearly suggests that, even if Cr(VI) adsorption on the “Fe(0)-free” surface of Be-Fe(0) (Mechanism 1) may take place, its contribution to Cr(VI) removal with Be-Fe(0) is less important. At the same time, since no Cr(III) was detected in the solution after 240 min (when all Cr(VI) was already removed), and the working environment was highly acidic (i.e., adsorption and/or precipitation of dissolved Cr(III) is not significant), Cr(VI) removal via homogenous reduction with dissolved indirect reductants (Mechanism 5) can also be ruled out. Therefore, it can be reasonably presumed that the removal of Cr(VI) with Be-Fe(0) was mainly the result of Cr(VI) adsorption on Fe(0) centers existent at the Be-Fe(0) surface, or, onto (hydr)oxide layers existent at the surface of Fe(0) centers (depending on working pH), followed by possible Cr(VI) heterogeneous chemical reduction to Cr(III) with Fe(0) [3] or with solid structural Fe(II) [39], with both processes being favored by the low pH values involved in this study.
4. Conclusions
Water and wastewater treatment technologies are known to generate important amounts of waste residues, raising concerns about their associated management and environmental costs. In order to reduce the environmental impact induced by classic disposal methods (e.g., landfill), the prevention/minimization of waste generation and recycling/reuse of water treatment process residues should take precedence over disposal. This work presents investigations on two different water treatment technologies which, apparently, have nothing in common: the removal of Fe(II) from contaminated AMD by adsorption on bentonite, and the treatment of Cr(VI) contaminated solutions by use of metallic iron. The present study suggests that the two treatment technologies can be connected if residues resulting from the first one are further used as reactive materials for the second one. Before being reused for the abatement of Cr(VI) pollution, the exhausted bentonite recovered from the treatment of AMD was chemically activated with sodium borohydride, in order to achieve the reduction of adsorbed Fe(II) to Fe(0). The experimental results from this study clearly demonstrated that exhausted bentonite activated with sodium borohydride has a much better capacity to remove Cr(VI) from aqueous solutions than fresh unmodified bentonite, which is attributable to the existence of Fe(0) centers at the surface of Be-Fe(0). The overall data presented in this study strongly suggests that the removal of Cr(VI) with Be-Fe(0) was mainly the result of Cr(VI) adsorption on Fe(0) or onto (hydr)oxide layers existent at the surface of Fe(0), probably followed by its heterogeneous chemical reduction to Cr(III).
Supplementary Materials
The following are available online at www.mdpi.com/2305-7084/1/2/9/s1.
Acknowledgments
This work was supported by a grant of the Romanian National Authority for Scientific Research and Innovation, CNCS—UEFISCDI, project number PN-II-RU-TE-2014-4-0508. The present article is an extension of the work presented at ICEER2017 and published in Energy Procedia. Authors appreciate the efforts of anonymous reviewers and editors for constructive criticisms and valuable suggestions which improved the quality of this manuscript.
Author Contributions
Marius Gheju and Ionel Balcu conceived and designed the experiments; Marius Gheju contributed with reagents/materials, performed the experiments and wrote the paper; Ionel Balcu contributed with analysis tools and analyzed the data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shrivastava, R.; Upreti, R.K.; Chaturvedi, U.C. Various cells of the immune system and intestine differ in their capacity to reduce hexavalent chromium. FEMS Immunol. Med. Microbiol. 2003, 1555, 1–6. [Google Scholar] [CrossRef]
- Keepax, R.E.; Moyes, L.; Livens, F.R. Speciation of heavy metals and radioisotopes. In Environmental and Ecological Chemistry; Sabljic, A., Ed.; Eolss Publishers Co.: Oxford, UK, 2009; Volume 2, pp. 165–199. [Google Scholar]
- Gheju, M. Hexavalent chromium reduction with zero-valent iron (ZVI) in aquatic systems. Water Air Soil Pollut. 2011, 222, 103–148. [Google Scholar]
- Vaiopoulou, E.; Gikas, P. Effects of chromium on activated sludge and on the performance of wastewater treatment plants: A review. Water Res. 2012, 46, 549–570. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Orvig, C. Biosorbents for hexavalent chromium elimination from industrial and municipal effluents. Coord. Chem. Rev. 2010, 254, 2959–2972. [Google Scholar] [CrossRef]
- Saha, R.; Nandi, R.; Saha, B. Sources and toxicity of hexavalent chromium. J. Coord. Chem. 2011, 64, 1782–1806. [Google Scholar] [CrossRef]
- Wise, S.S.; Holmes, A.L.; Liou, L.; Adam, R.M.; Wise, J.P. Hexavalent chromium induces chromosome instability in human urothelial cells. Toxicol. Appl. Pharmacol. 2016, 296, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Choppala, G.; Bolan, N.; Park, J.H. Chromium contamination and its risk management in complex environmental settings. Adv. Agron. 2013, 120, 129–172. [Google Scholar]
- Costa, M. Potential hazards of hexavalent chromate in our drinking water. Toxicol. Appl. Pharmacol. 2003, 188, 1–5. [Google Scholar] [CrossRef]
- Stefansson, A.; Seward, T. A spectrophotometric study of iron (III) hydrolysis in aqueous solutions to 200 °C. Chem. Geol. 2008, 249, 227–235. [Google Scholar] [CrossRef]
- Davison, W.; Seed, G. The kinetics of the oxidation of ferrous iron in synthetic and natural waters. Geochim. Cosmochim. Acta 1983, 47, 67–79. [Google Scholar] [CrossRef]
- Davison, W. Iron and manganese in lakes. Earth Sci. Rev. 1993, 34, 119–163. [Google Scholar] [CrossRef]
- Lieu, P.; Heiskala, M.; Peterson, P.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar] [CrossRef]
- Nair, M.; Iyengar, V. Iron content, bioavailability & factors affecting iron status of Indians. Indian J. Med. Res. 2009, 130, 634–645. [Google Scholar] [PubMed]
- Fontecave, M.; Pierre, J.L. Iron: Metabolism, toxicity and therapy. Biochimie 1993, 75, 767–773. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Andrews, N.C. Iron metabolism: Iron deficiency and iron overload. Annu. Rev. Genom. Hum. Genet. 2000, 1, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Rocha, S.; Fernandes, R. Iron metabolism: From health to disease. J. Clin. Lab. Anal. 2014, 28, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yingxin, Z.; Wenfang, Q.; Guanyi, C.; Min, J.; Zhenya, Z. Behavior of Cr(VI) removal from wastewater by adsorption onto HCl activated Akadama clay. J. Taiwan Inst. Chem. Eng. 2015, 50, 190–197. [Google Scholar]
- Te, B.; Wichitsathian, B.; Yossapol, C. Modification of natural common clays as low cost adsorbents for arsenate adsorption. Int. J. Environ. Sci. Dev. 2015, 6, 799–804. [Google Scholar] [CrossRef]
- Potgieter, J.H.; Potgieter-Vermaak, S.S.; Kalibantonga, P.D. Heavy metals removal from solution by palygorskite clay. Miner. Eng. 2006, 19, 463–470. [Google Scholar] [CrossRef]
- Noubactep, C. Metallic iron for environmental remediation: A review of reviews. Water Res. 2015, 85, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Mwakabona, H.; Ndé-Tchoupé, A.; Njau, K.; Noubactep, C.; Wydra, K. Metallic iron for safe drinking water provision: Considering a lost knowledge. Water Res. 2017, 117, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Suteerapataranon, S.; Bouby, M.; Geckeis, H.; Fanghanel, T.; Grudpan, K. Interaction of trace elements in acid mine drainage solution with humic acid. Water Res. 2006, 40, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Zanetti, M.C. Preliminary test concerning zero-valent iron efficiency in inorganic pollutants remediation. Am. J. Environ Sci. 2009, 5, 555–560. [Google Scholar] [CrossRef]
- Yunfei, X.; Megharaj, M.; Ravendra, N. Reduction and adsorption of Pb2+ in aqueous solution by nano-zero-valent iron—A SEM, TEM and XPS study. Mater. Res. Bull. 2010, 45, 1361–1367. [Google Scholar]
- Gheju, M.; Iovi, A.; Balcu, I. Hexavalent chromium reduction with scrap iron in continuous-flow system. Part 1: Effect of feed solution pH. J. Hazard. Mater. 2008, 153, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Zach-Maor, A.; Semiat, R.; Shemer, H. Synthesis, performance, and modeling of immobilized nano-sized magnetite layer for phosphate removal. J. Colloid Interface Sci. 2011, 357, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Reckhow, D.A. Analytical Chemistry for Environmental Engineers and Scientists. Lecture Notes to Accompany CEE 572 and CEE 772. Department of Civil Engineering University of Massachusetts: Amherst, MA 01003 Chapter XVIII: Electrochemical Methods. Available online: http://www.ecs.umass.edu/cee/reckhow/courses/572/572BKI.html (accessed on 8 September 2017).
- Espana, J.S.; Pamo, E.L.; Pastor, E.S. The oxidation of ferrous iron in acidic mine effluents from the Iberian Pyrite Belt (Odiel Basin, Huelva, Spain): Field and laboratory rates. J. Geochem. Explor. 2007, 92, 120–132. [Google Scholar] [CrossRef]
- Espana, J.S. Acid mine drainage in the Iberian pyrite belt: An overview with special emphasis on generation mechanisms, aqueous composition and associated mineral phases. Macla 2008, 10, 34–43. [Google Scholar]
- Karthikeyan, T.; Rajgopal, S.; Miranda, L.R. Chromium (VI) adsorption from aqueous solution by Hevea Brasilinesis sawdust activated carbon. J. Hazard. Mater. 2005, B124, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Granados-Correa, F.; Jimenez-Becerril, J. Chromium (VI) adsorption on boehmite. J. Hazard. Mater. 2009, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Baral, S.S.; Das, S.N.; Rath, P. Hexavalent chromium removal from aqueous solution by adsorption on treated sawdust. Biochem. Eng. J. 2006, 31, 216–222. [Google Scholar] [CrossRef]
- Brown, G., Jr.; Parks, G. Sorption of trace elements on mineral surfaces: Modern perspectives from spectroscopic studies, and comments on sorption in the marine environment. Int. Geol. Rev. 2001, 43, 963–1073. [Google Scholar] [CrossRef]
- Flury, B.; Eggenberger, U.; Mader, U. First results of operating and monitoring an innovative design of a permeable reactive barrier for the remediaton of chromate contaminated groundwater. J. Appl. Geochem. 2009, 24, 687–697. [Google Scholar] [CrossRef]
- Traubenberg, S.E.; Foley, R.T. The influence of chloride and sulfate ions on the corrosion of iron in sulfuric acid. J. Electrochem. Soc. 1971, 18, 1066–1070. [Google Scholar] [CrossRef]
- Tepong-Tsindé, R.; Phukan, M.; Nassi, A.; Noubactep, C.; Ruppert, H. Validating the efficiency of the MB discoloration method for the characterization of Fe0/H2O systems using accelerated corrosion by chloride ions. Chem. Eng. J. 2015, 279, 353–362. [Google Scholar] [CrossRef]
- White, A.F.; Paterson, M.L. Reduction of aqueous transition metal species on the surface of Fe(II)-containing oxides. Geochim. Cosmochim. Acta 1996, 60, 3799–3814. [Google Scholar] [CrossRef]
Figure 1.
Effect of pH on Fe(II) removal by bentonite.
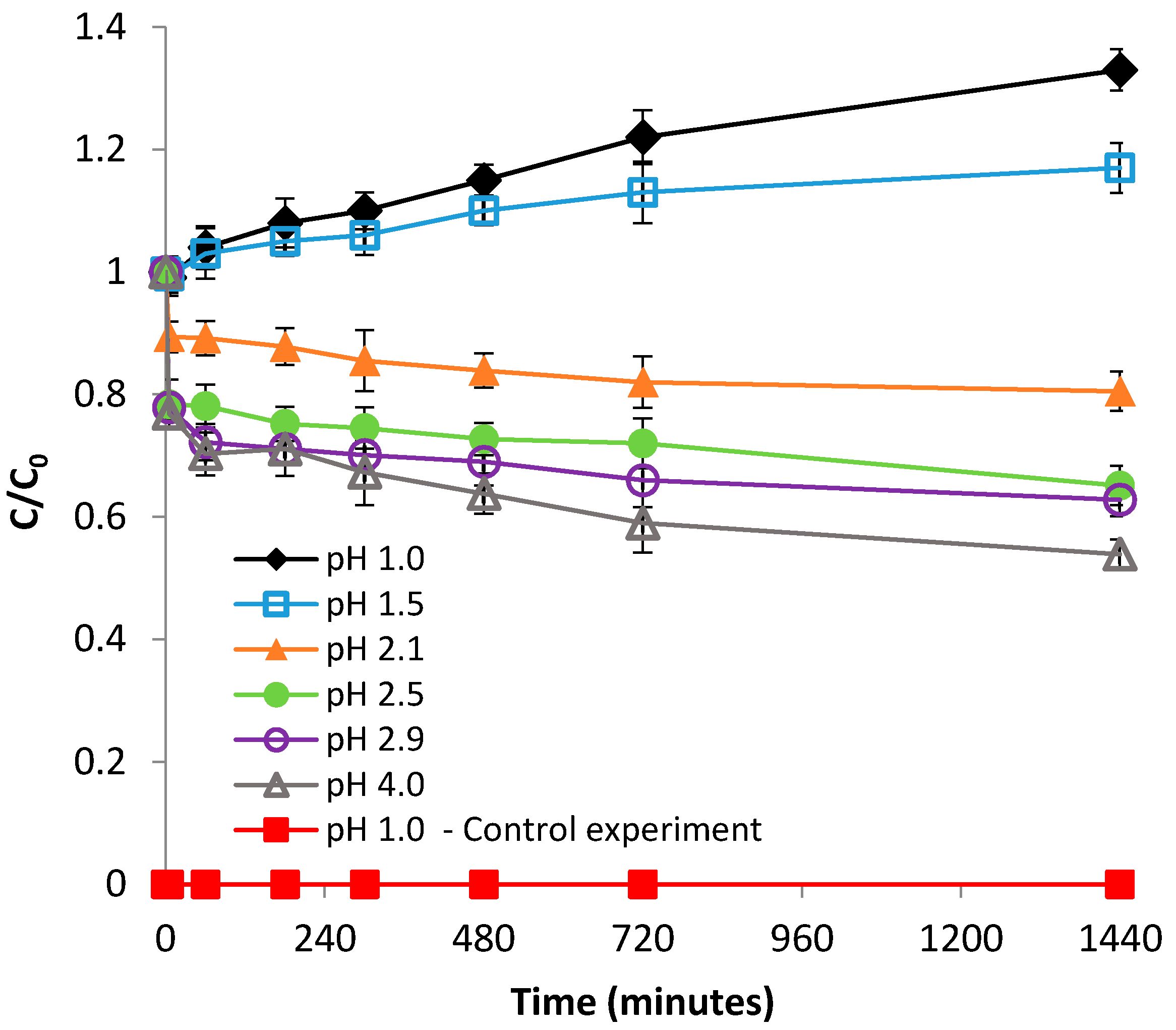
Figure 2.
Effect of temperature on Fe(II) removal by bentonite.
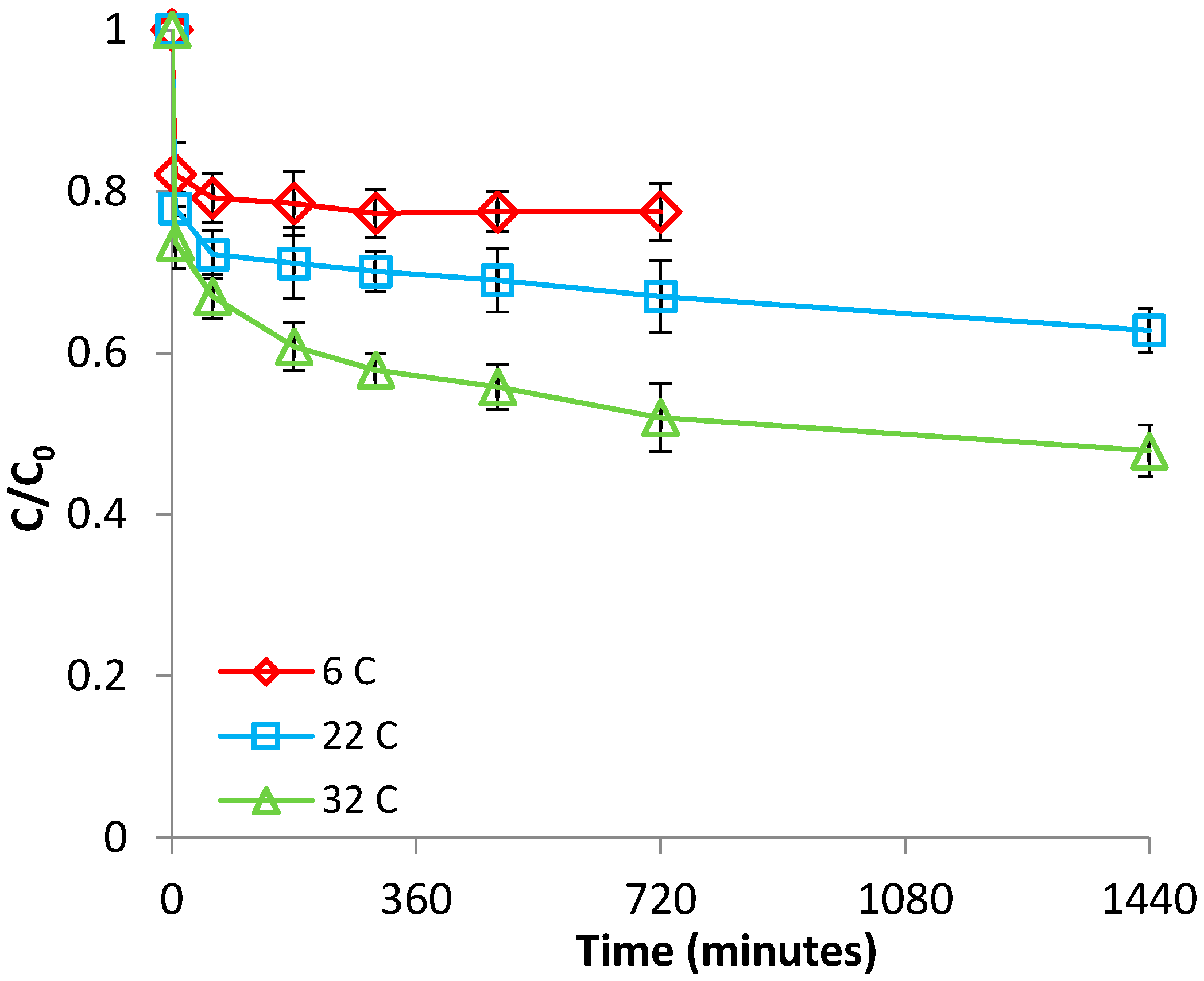
Figure 3.
Effect of Fe(II) concentration on Fe(II) removal by bentonite.
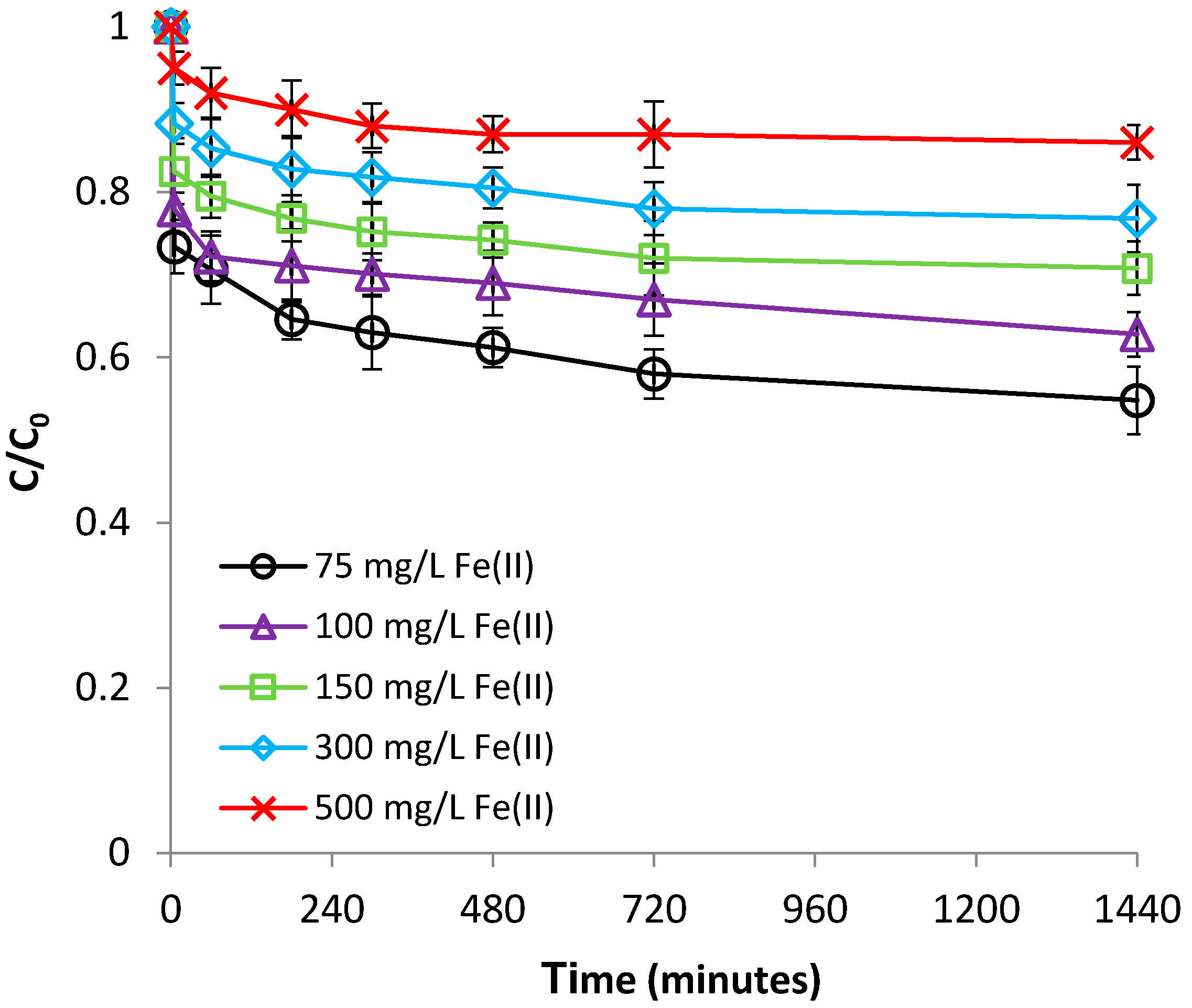
Figure 4.
Effect of ionic strength on Fe(II) removal by bentonite.

Figure 5.
Effect of pH on Cr(VI) removal by Be-Fe(0).
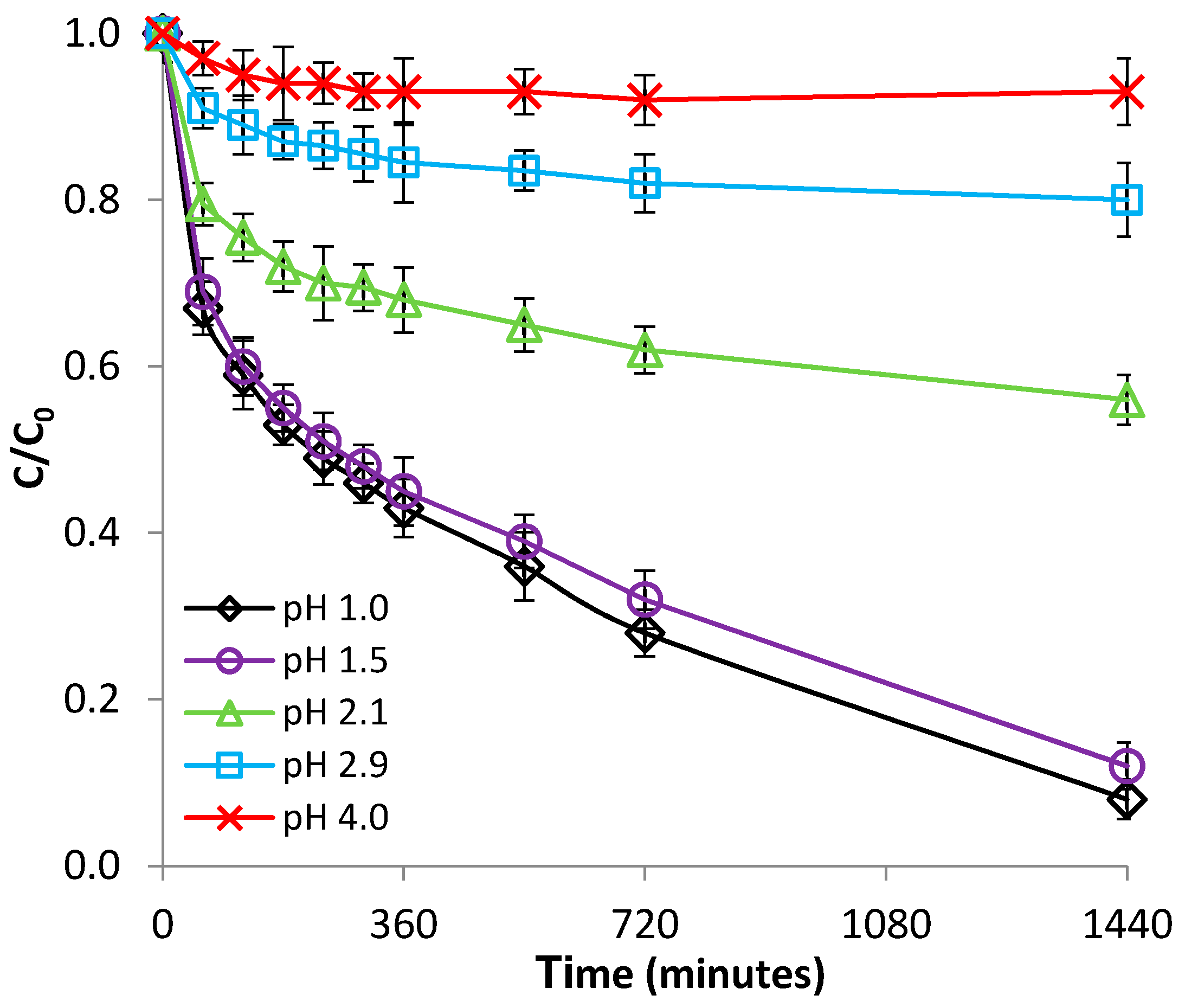
Figure 6.
Effect of pH on Cr(VI) removal by bentonite.
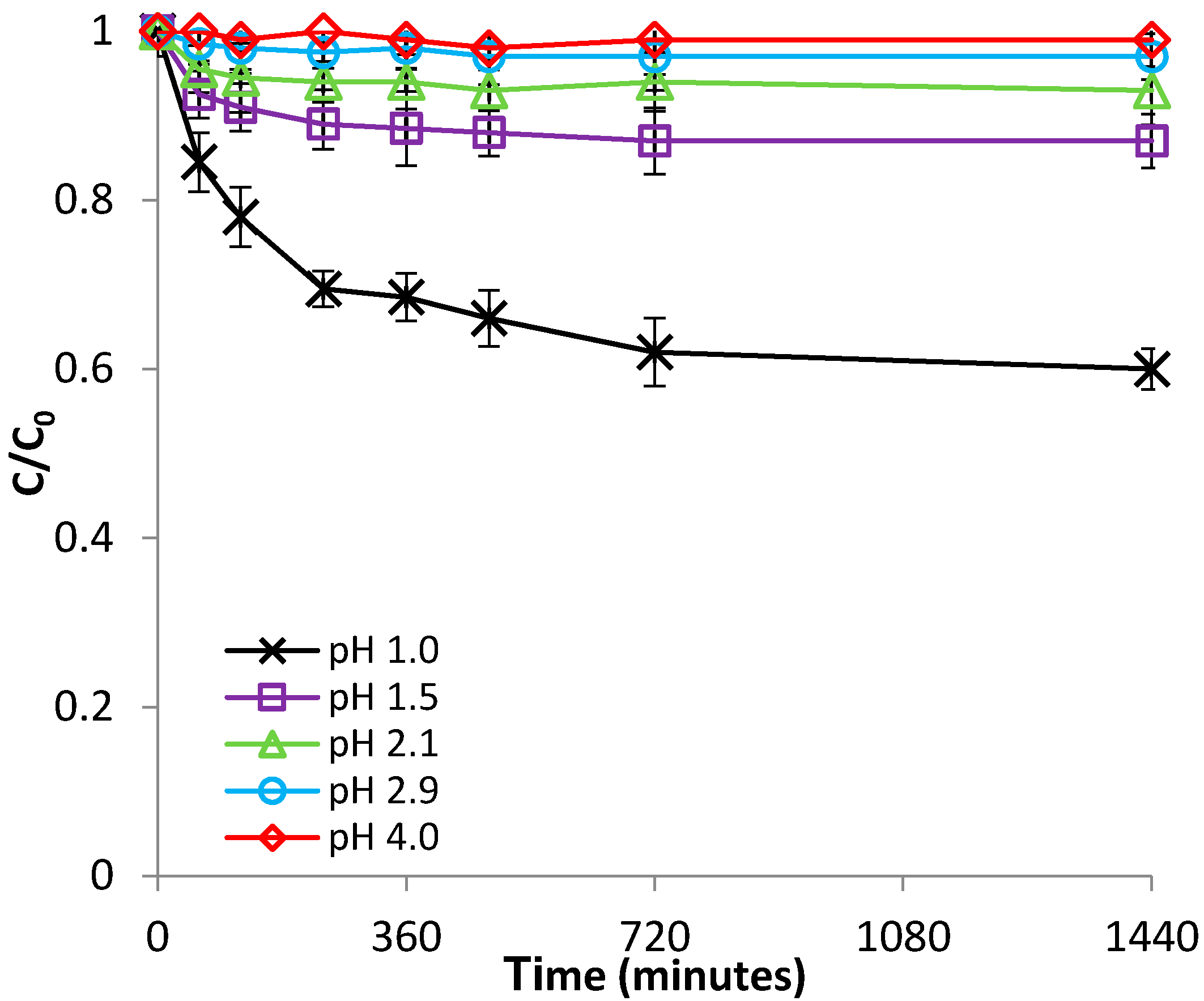
Figure 7.
Effect of temperature on Cr(VI) removal by Be-Fe(0).
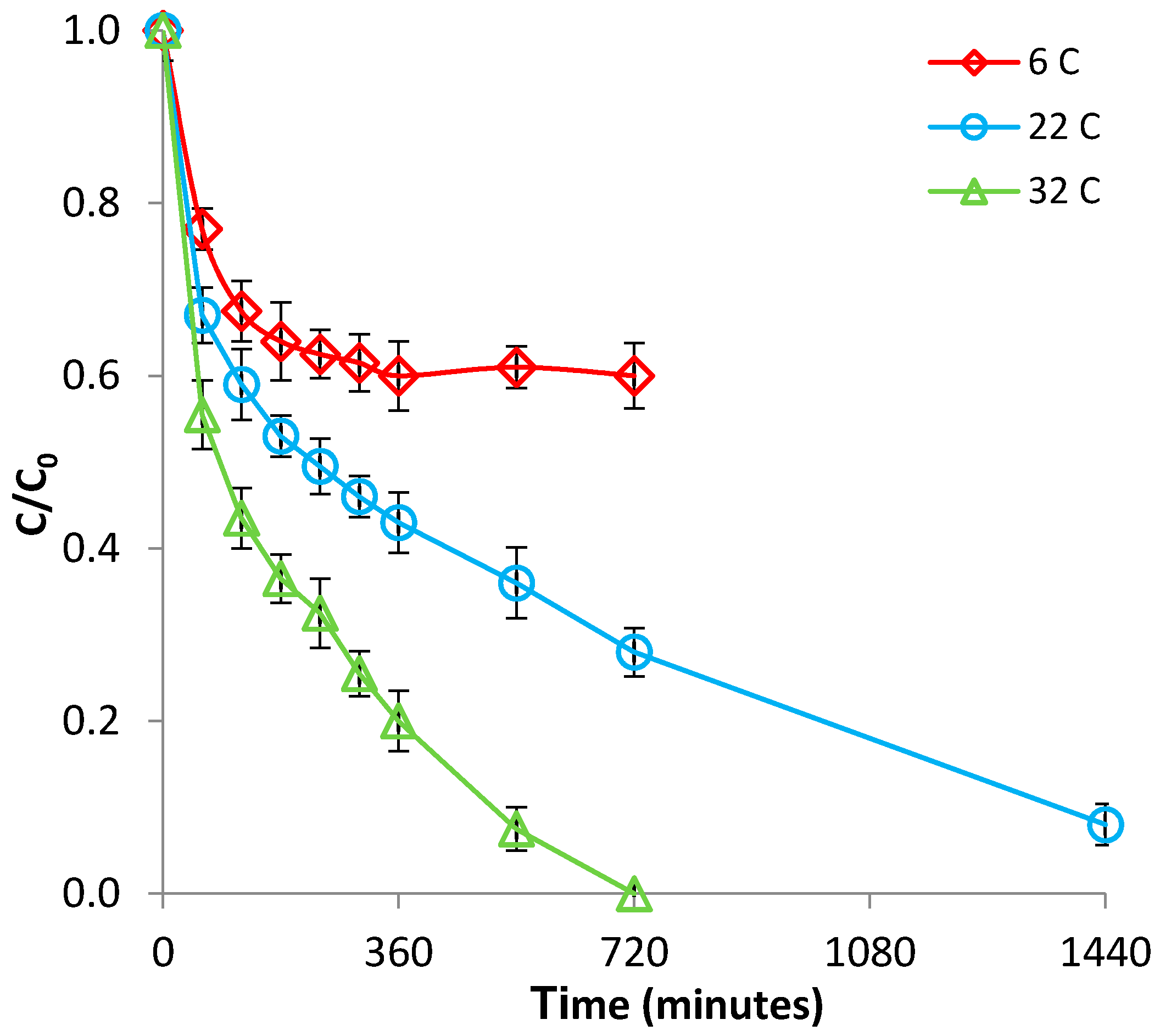
Figure 8.
Effect of temperature on Cr(VI) removal by bentonite.
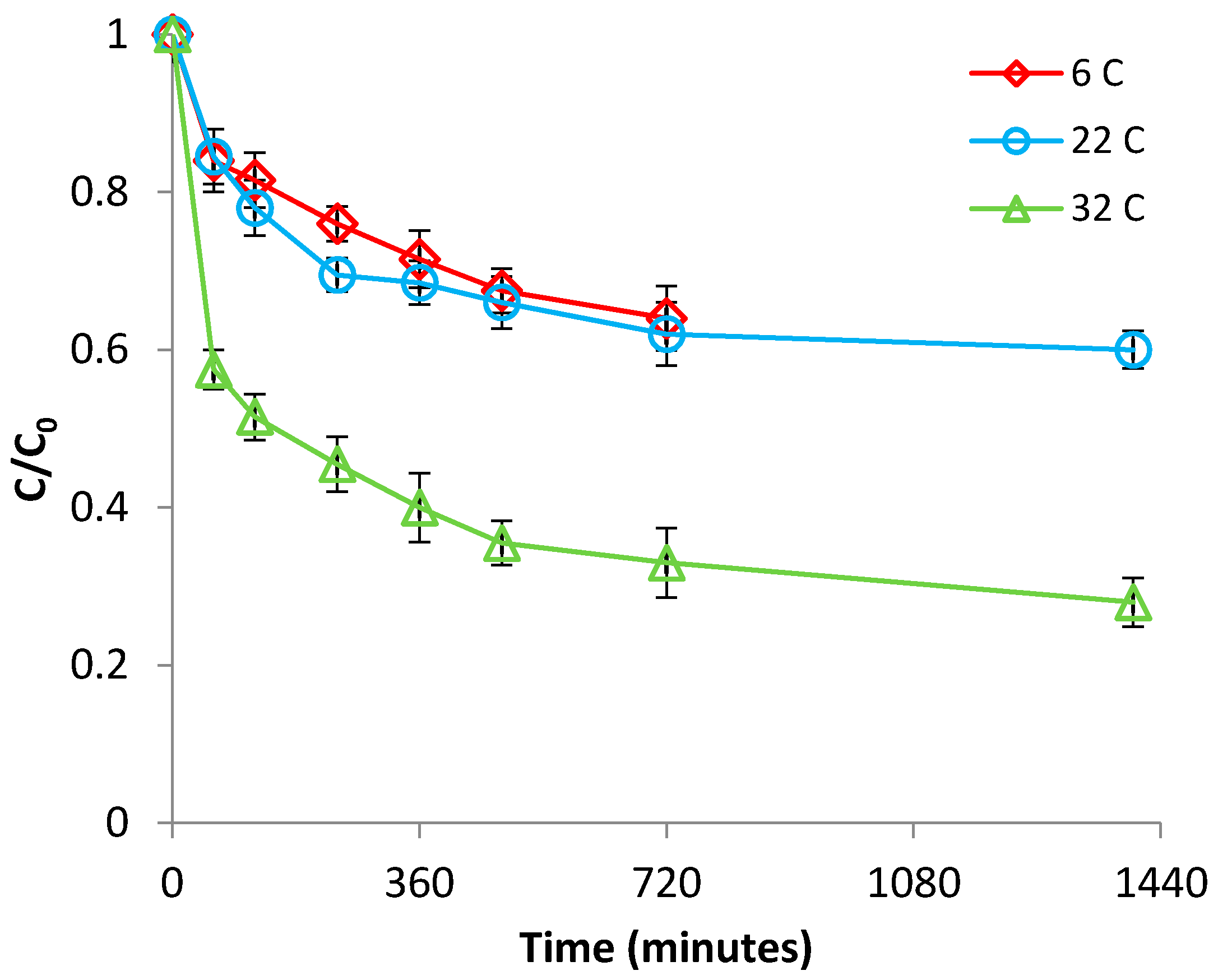
Figure 9.
Effect of Cr(VI) concentration on Cr(VI) removal by Be-Fe(0).
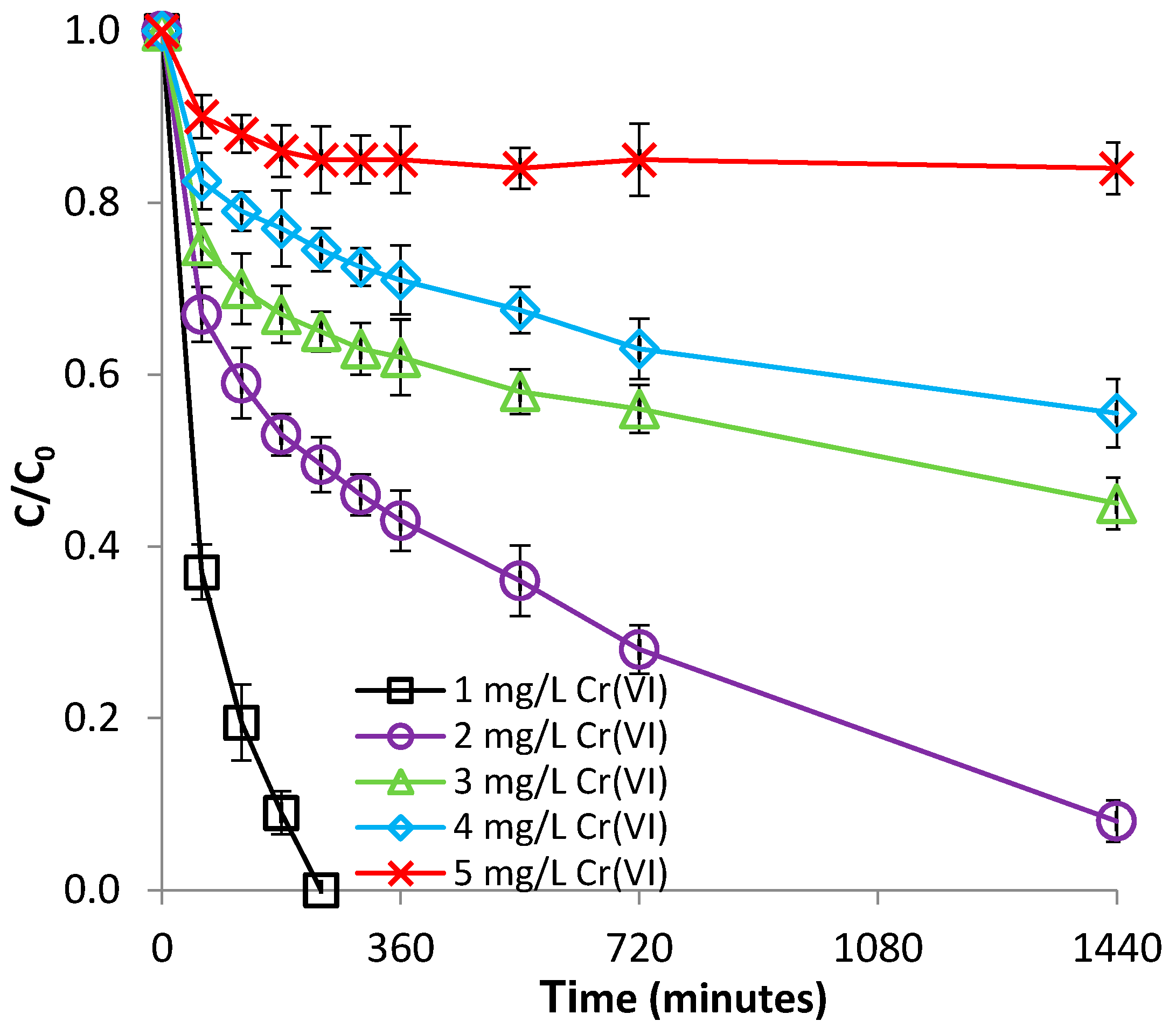
Figure 10.
Effect of Cr(VI) concentration on Cr(VI) removal by bentonite.
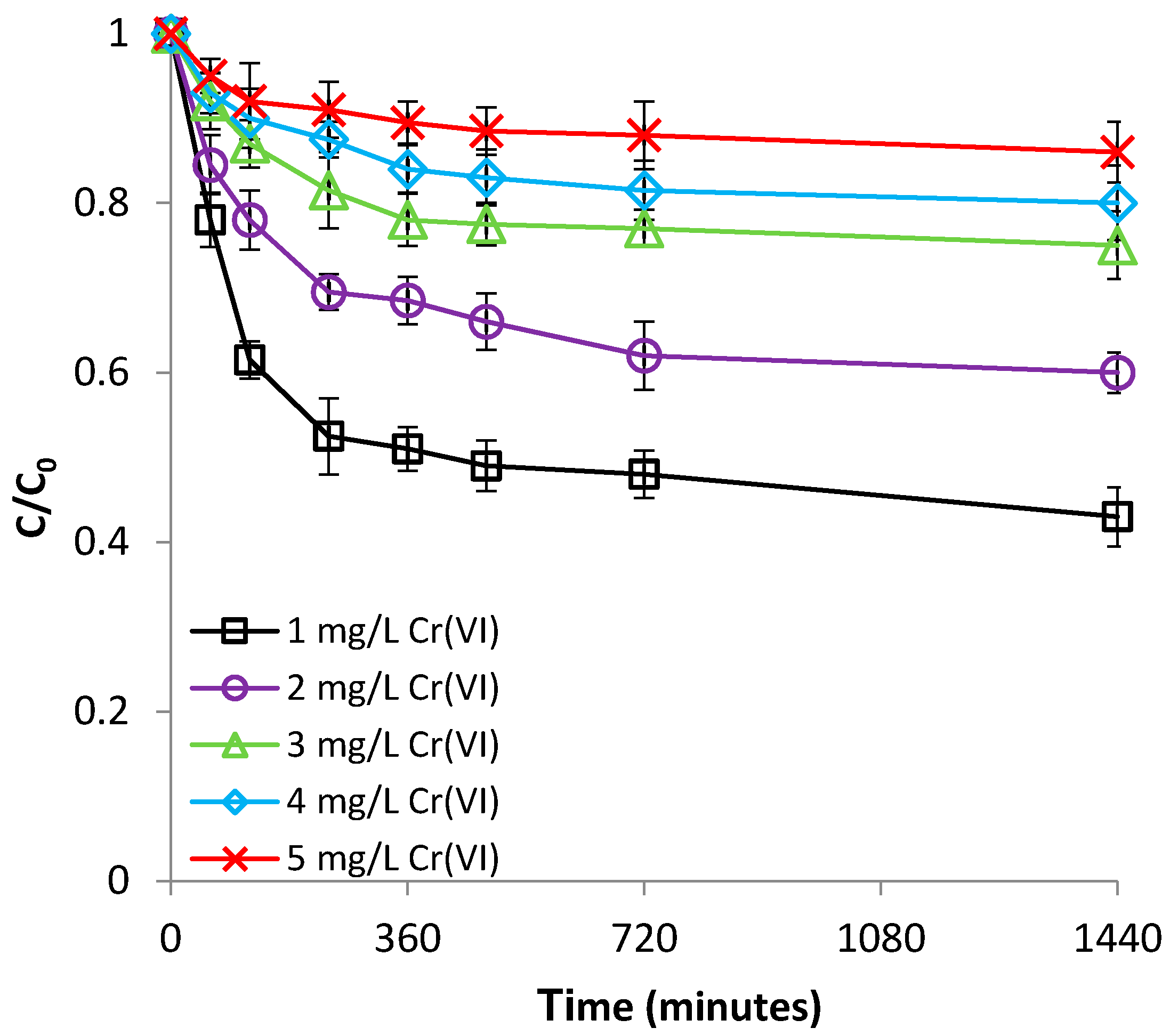
Figure 11.
Effect of ionic strength on Cr(VI) removal by Be-Fe(0).
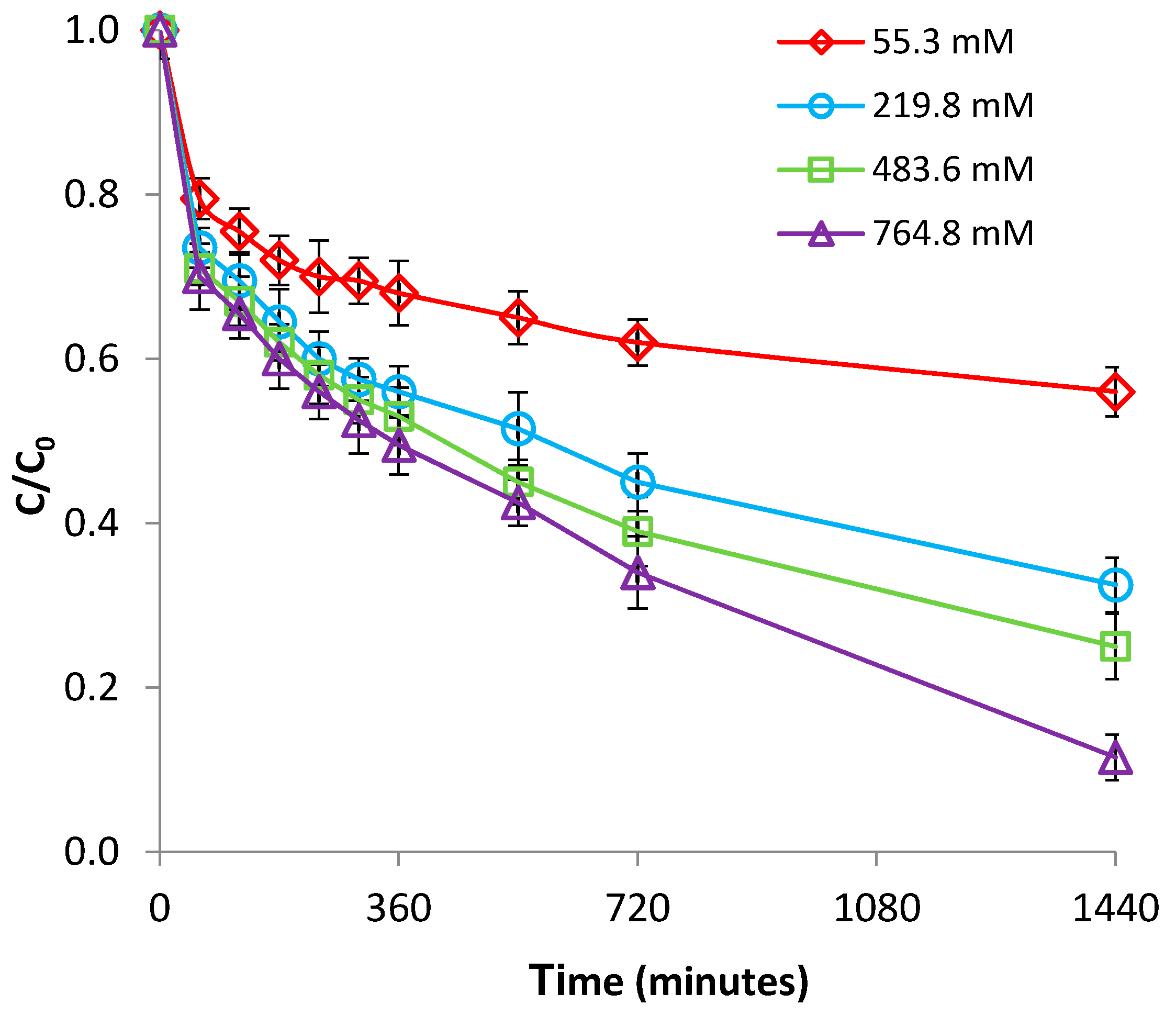
Figure 12.
Effect of ionic strength on Cr(VI) removal by bentonite.
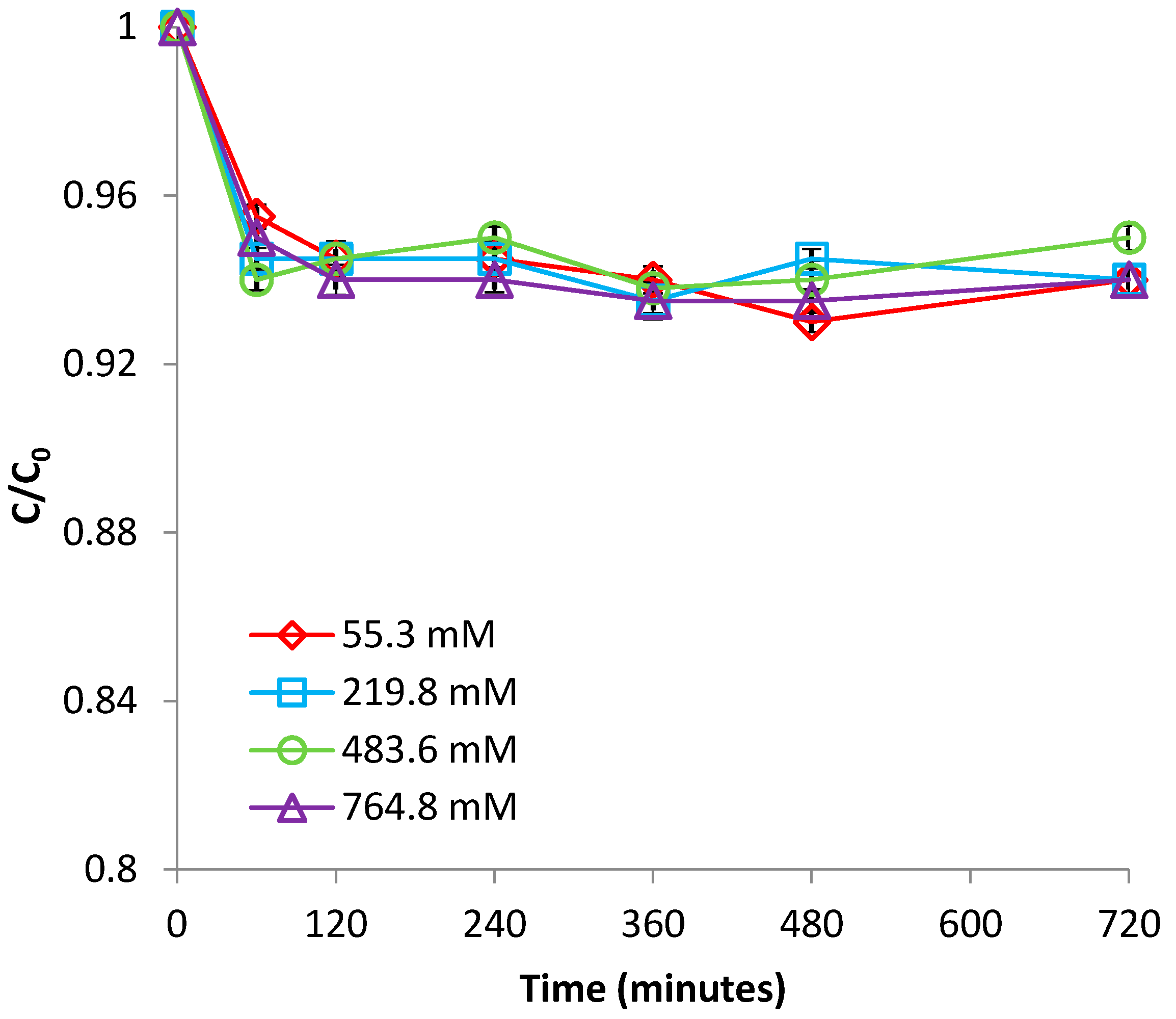
Figure 13.
Comparison of Cr(VI) removal by Be-Fe(0) and bentonite.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gheju, M.; Balcu, I. Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues. ChemEngineering 2017, 1, 9. https://doi.org/10.3390/chemengineering1020009
AMA Style
Gheju M, Balcu I. Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues. ChemEngineering. 2017; 1(2):9. https://doi.org/10.3390/chemengineering1020009
Chicago/Turabian StyleGheju, Marius, and Ionel Balcu. 2017. "Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues" ChemEngineering 1, no. 2: 9. https://doi.org/10.3390/chemengineering1020009