Chemokine CCL4 Induced in Mouse Brain Has a Protective Role against Methylmercury Toxicity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Immunochemistry
2.3. Cell Culture
2.4. siRNA Transfection
2.5. Cell Viability Assay
2.6. Measurement of CCL4 mRNA Levels by Quantitative Real-Time PCR
2.7. Statistical Analysis
3. Results
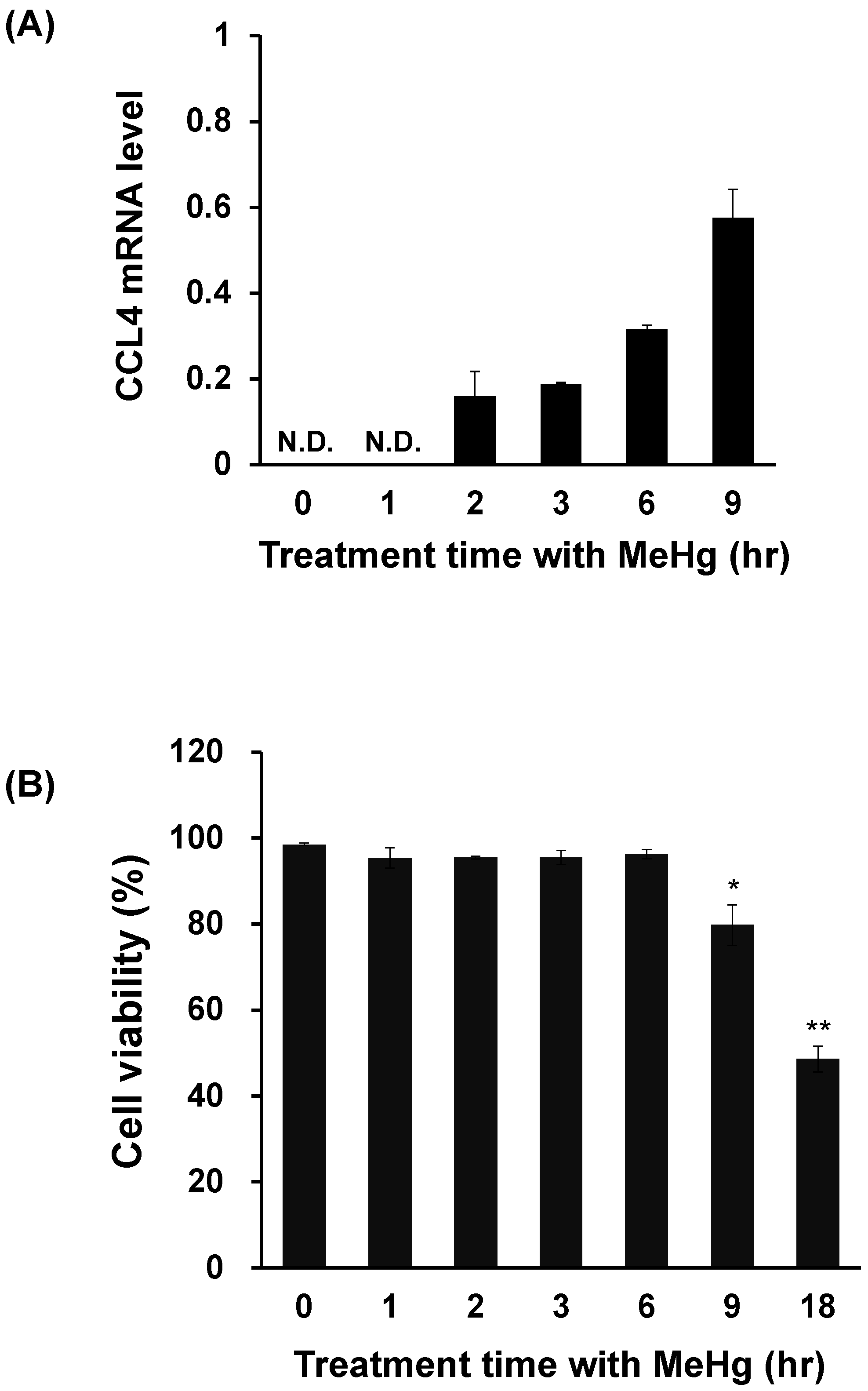
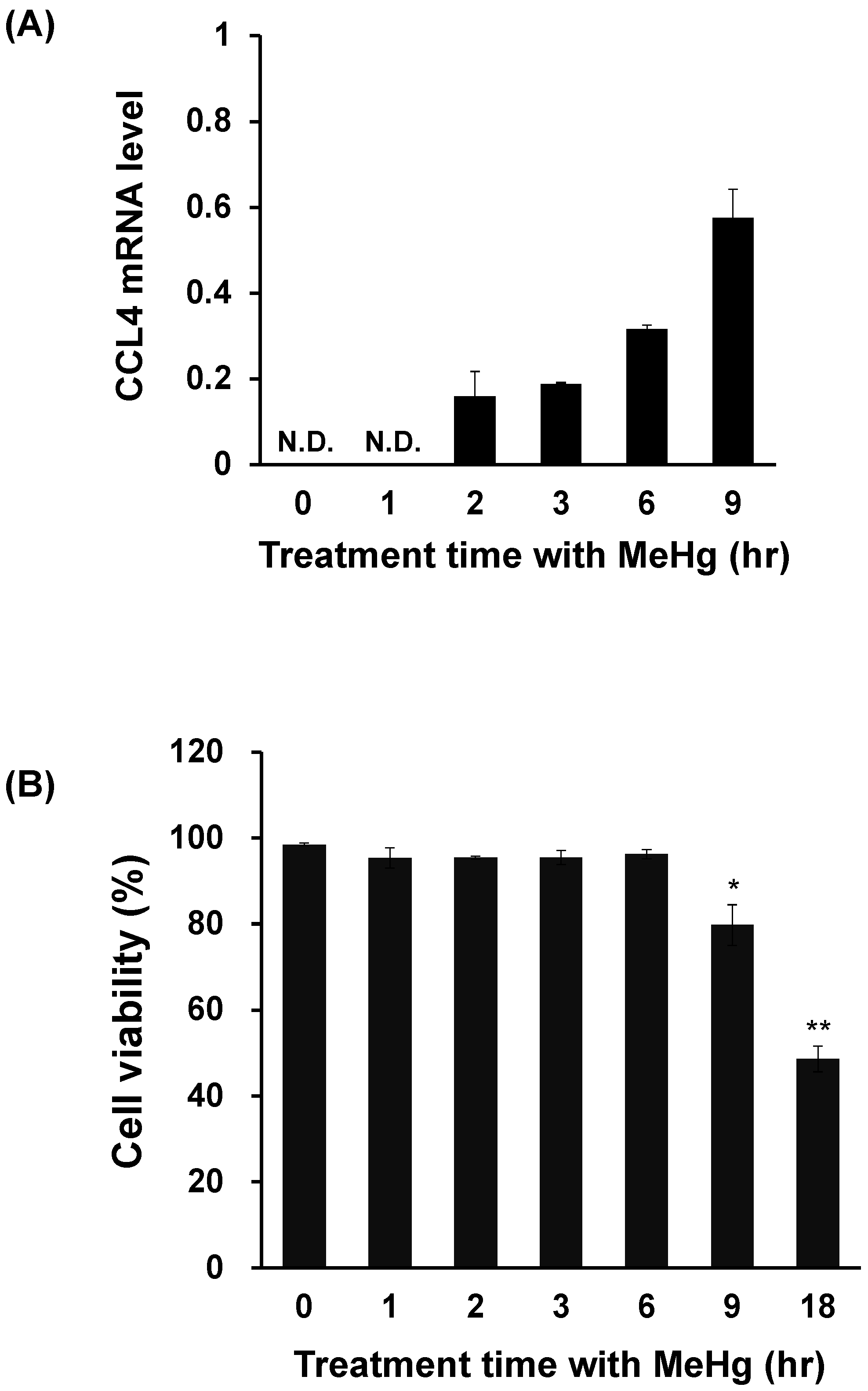
3.1. CCL4 Expression Is Induced Prior to Neuronal Damage Caused by MeHg
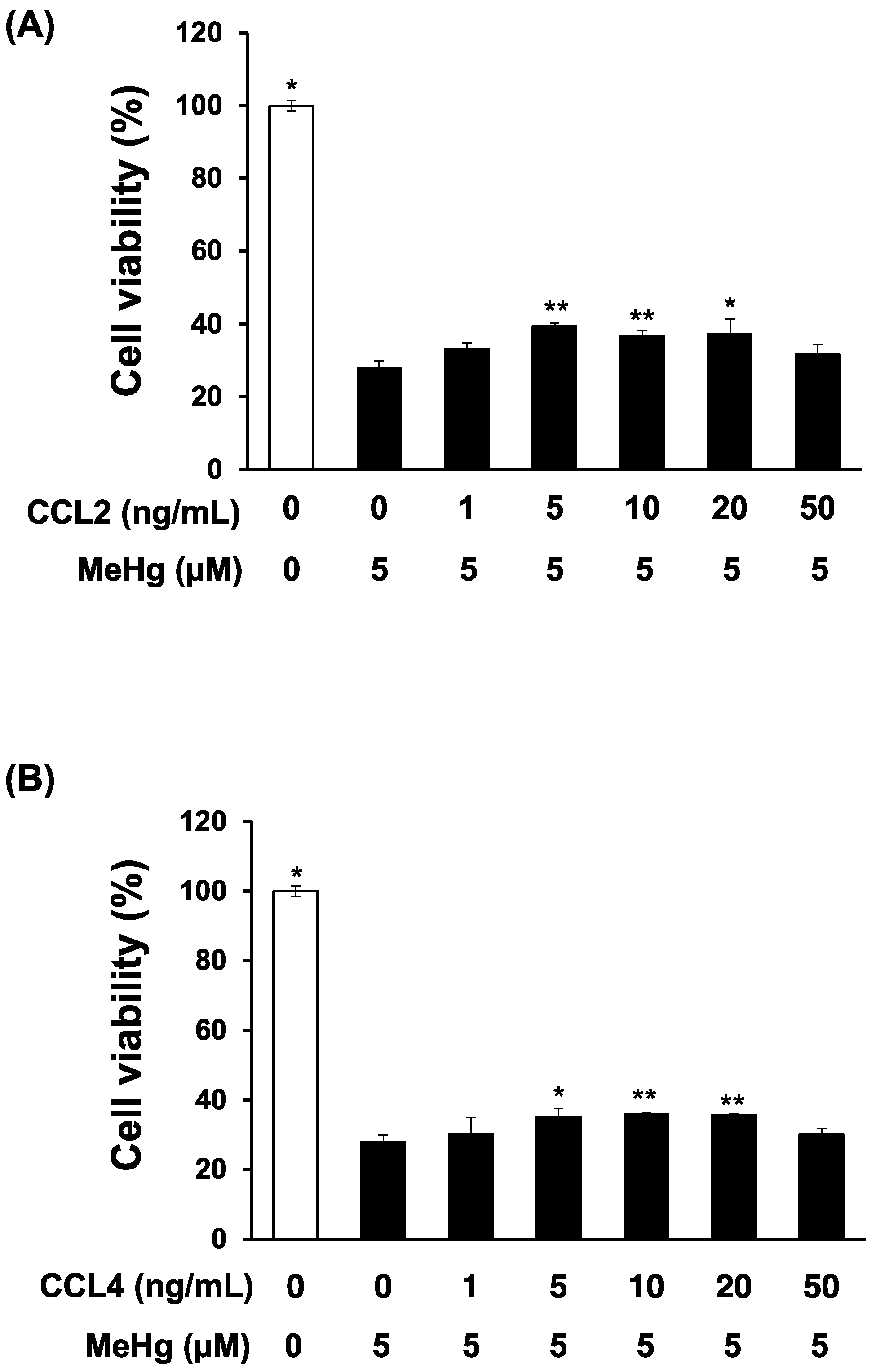
3.2. CCL4 Attenuates MeHg Toxicity in Primary Mouse Neuron Cultures
3.3. CCL4 Expression Is Induced Prior to MeHg-Induced Cytotoxicity in C17.2 Cells
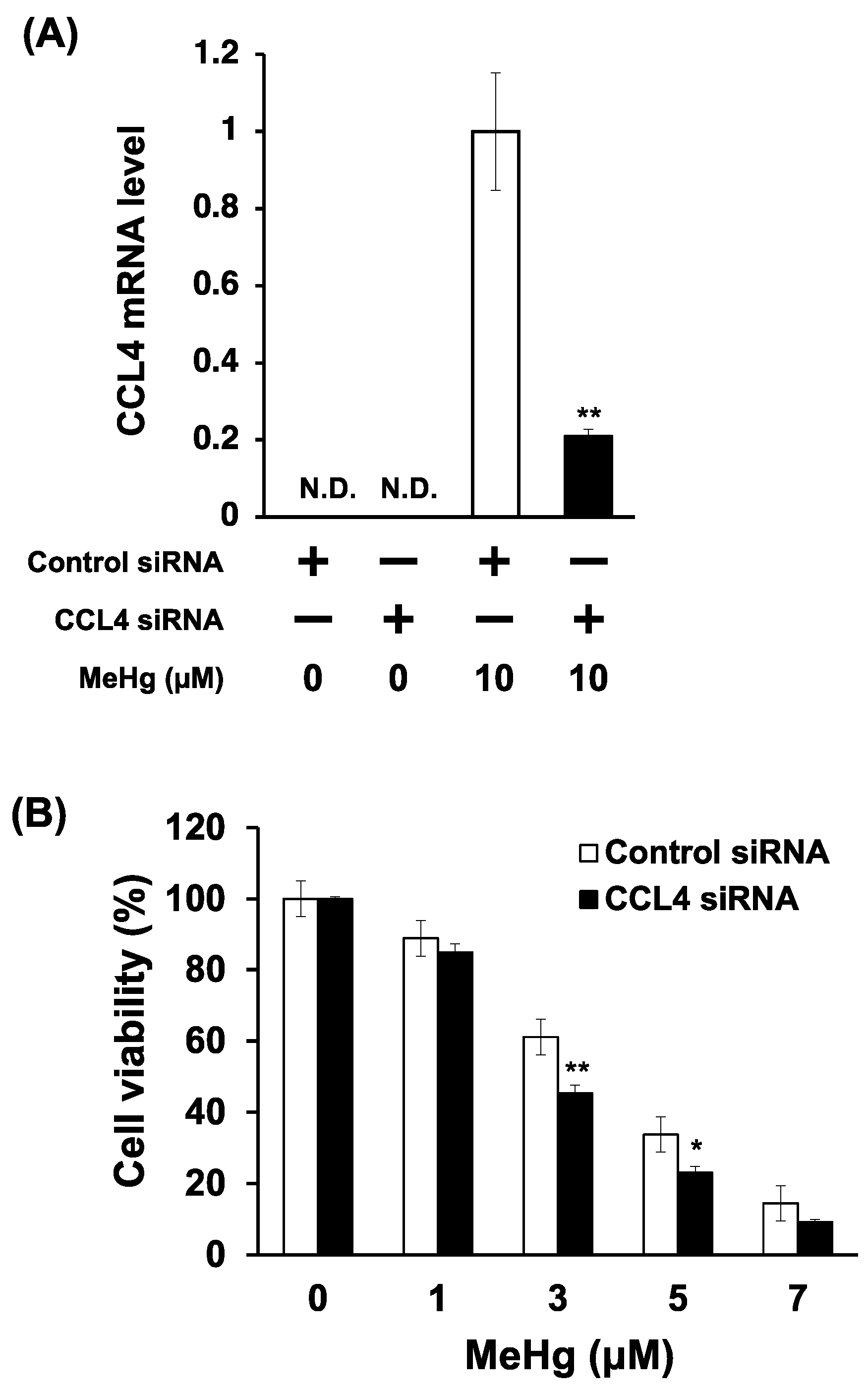
3.4. Knockdown of CCL4 Enhances MeHg Cytotoxicity in C17.2 Cells
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Harada, M. Minamata disease: Methylmercury poisoning in Japan caused by environmental pollution. Crit. Rev. Toxicol. 1995, 25, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Aschner, J.L. Mercury neurotoxicity: Mechanisms of blood-brain barrier transport. Neurosci. Biobehav. Rev. 1990, 14, 169–176. [Google Scholar] [CrossRef]
- Vahter, M.; Akesson, A.; Lind, B.; Bjors, U.; Schutz, A.; Berglund, M. Longitudinal study of methylmercury and inorganic mercury in blood and urine of pregnant and lactating women, as well as in umbilical cord blood. Environ. Res. 2000, 84, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N. Transport of toxic metals by molecular mimicry. Environ. Health. Perspect. 2002, 110, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, A.F.; Johansson, C.; Onishchenko, N.; Coccini, T.; Roda, E.; Vahter, M.; Ceccatelli, S.; Manzo, L. Human developmental neurotoxicity of methylmercury: Impact of variables and risk modifiers. Regul. Toxicol. Pharmacol. 2008, 51, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.W.; Lee, J.Y.; Ryoke, K.; Matsuyama, F.; Kim, J.M.; Takahashi, T.; Naganuma, A. Gene expression profiling using DNA microarray analysis of the cerebellum of mice treated with methylmercury. J. Toxicol. Sci. 2011, 36, 389–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Hwang, G.W.; Kim, M.S.; Takahashi, T.; Naganuma, A. Methylmercury induces a brain-specific increase in chemokine CCL4 expression in mice. J. Toxicol. Sci. 2012, 37, 1279–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai-Shimada, M.; Takahashi, T.; Kim, M.S.; Fujimura, M.; Ito, H.; Toyama, T.; Naganuma, A.; Hwang, G.W. Methylmercury induces the expression of TNF-α selectively in the brain of mice. Sci. Rep. 2016, 6, 38294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Takahashi, T.; Lee, J.Y.; Hwang, G.W.; Naganuma, A. Global chemokine expression in methylmercury-treated mice: Methylmercury induces brain-specific expression of CCL3 and CCL4. J. Toxicol. Sci. 2013, 38, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; von Stebut, E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 2004, 36, 1882–1886. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowell, R.M.; Xu, H.; Galasso, J.M.; Silverstein, F.S. Hypoxic-ischemic injury induces macrophage inflammatory protein-1α expression in immature rat brain. Stroke 2002, 33, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Boven, L.A.; Montagne, L.; Nottet, H.S.; De Groot, C.J. Macrophage inflammatory protein-1α (MIP-1α), MIP-1β, and RANTES mRNA semiquantification and protein expression in active demyelinating multiple sclerosis (MS) lesions. Clin. Exp. Immunol. 2000, 122, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.Q.; Qin, S.X.; Wu, L.J.; Mackay, C.R.; Hyman, B.T. Immunohistochemical study of the β-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am. J. Pathol. 1998, 153, 31–37. [Google Scholar] [CrossRef]
- Szczucinski, A.; Losy, J. Chemokines and chemokine receptors in multiple sclerosis. Potential targets for new therapies. Acta. Neurol. Scand. 2007, 115, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godefroy, D.; Gosselin, R.D.; Yasutake, A.; Fujimura, M.; Combadiere, C.; Maury-Brachet, R.; Laclau, M.; Rakwal, R.; Melik-Parsadaniantz, S.; Bourdineaud, J.P.; et al. The chemokine CCL2 protects against methylmercury neurotoxicity. Toxicol. Sci. 2012, 125, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, M.; Usuki, F. Methylmercury causes neuronal cell death through the suppression of the TrkA pathway: In vitro and in vivo effects of TrkA pathway activators. Toxicol. Appl. Pharmacol. 2015, 282, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. In situ different antioxidative systems contribute to the site-specific methylmercury neurotoxicity in mice. Toxicology 2017, 392, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. Site-specific neural hyperactivity via the activation of MAPK and PKA/CREB pathways triggers neuronal degeneration in methylmercury-intoxicated mice. Toxicol. Lett. 2017, 271, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Van Eldik, L.J. Inflammatory cytokines stimulate the chemokines CCL2/MCP-1 and CCL7/MCP-3 through NFkB and MAPK dependent pathways in rat astrocytes [corrected]. Brain Res. 2009, 1287, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Giraud, S.N.; Caron, C.M.; Pham-Dinh, D.; Kitabgi, P.; Nicot, A.B. Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB-dependent CCL2 expression in reactive astrocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 8416–8421. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, J.; Crabtree, G.; Grove, M.; Daubersies, P.; Bailleul, B.; Wright, E.; Plumb, M. An ATF/CREB-binding site is essential for cell-specific and inducible transcription of the murine MIP-1β cytokine gene. Gene 1995, 152, 173–179. [Google Scholar] [CrossRef]
- Wiesner, P.; Choi, S.H.; Almazan, F.; Benner, C.; Huang, W.; Diehl, C.J.; Gonen, A.; Butler, S.; Witztum, J.L.; Glass, C.K.; et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa B and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 2010, 107, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.M.; Godleski, J.J.; Paulauskis, J.D. Regulation of macrophage inflammatory protein-1α mRNA by oxidative stress. J. Biol. Chem. 1996, 271, 5878–5883. [Google Scholar] [CrossRef] [PubMed]
- Stefini, R.; Catenacci, E.; Piva, S.; Sozzani, S.; Valerio, A.; Bergomi, R.; Cenzato, M.; Mortini, P.; Latronico, N. Chemokine detection in the cerebral tissue of patients with posttraumatic brain contusions. J. Neurosurg. 2008, 108, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Bless, N.M.; Huber-Lang, M.; Guo, R.F.; Warner, R.L.; Schmal, H.; Czermak, B.J.; Shanley, T.P.; Crouch, L.D.; Lentsch, A.B.; Sarma, V.; et al. Role of CC chemokines (macrophage inflammatory protein-1β, monocyte chemoattractant protein-1, RANTES) in acute lung injury in rats. J. Immunol. 2000, 164, 2650–2659. [Google Scholar] [CrossRef] [PubMed]
- Fahey, T.J., 3rd; Tracey, K.J.; Tekamp-Olson, P.; Cousens, L.S.; Jones, W.G.; Shires, G.T.; Cerami, A.; Sherry, B. Macrophage inflammatory protein 1 modulates macrophage function. J. Immunol. 1992, 148, 2764–2769. [Google Scholar] [PubMed]
- Speyer, C.L.; Gao, H.; Rancilio, N.J.; Neff, T.A.; Huffnagle, G.B.; Sarma, J.V.; Ward, P.A. Novel chemokine responsiveness and mobilization of neutrophils during sepsis. Am. J. Pathol. 2004, 165, 2187–2196. [Google Scholar] [CrossRef]
- Noguchi, Y.; Shinozaki, Y.; Fujishita, K.; Shibata, K.; Imura, Y.; Morizawa, Y.; Gachet, C.; Koizumi, S. Astrocytes protect neurons against methylmercury via ATP/P2Y(1) receptor-mediated pathways in astrocytes. PLoS ONE 2013, 8, e57898. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Iwai-Shimada, M.; Syakushi, Y.; Kim, M.S.; Hwang, G.W.; Miura, N.; Naganuma, A. Methylmercury induces expression of interleukin-1β and interleukin-19 in mice brains. Fundam. Toxicol. Sci. 2015, 2, 239–243. [Google Scholar] [CrossRef] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, T.; Kim, M.-S.; Iwai-Shimada, M.; Fujimura, M.; Toyama, T.; Naganuma, A.; Hwang, G.-W. Chemokine CCL4 Induced in Mouse Brain Has a Protective Role against Methylmercury Toxicity. Toxics 2018, 6, 36. https://doi.org/10.3390/toxics6030036
Takahashi T, Kim M-S, Iwai-Shimada M, Fujimura M, Toyama T, Naganuma A, Hwang G-W. Chemokine CCL4 Induced in Mouse Brain Has a Protective Role against Methylmercury Toxicity. Toxics. 2018; 6(3):36. https://doi.org/10.3390/toxics6030036
Chicago/Turabian StyleTakahashi, Tsutomu, Min-Seok Kim, Miyuki Iwai-Shimada, Masatake Fujimura, Takashi Toyama, Akira Naganuma, and Gi-Wook Hwang. 2018. "Chemokine CCL4 Induced in Mouse Brain Has a Protective Role against Methylmercury Toxicity" Toxics 6, no. 3: 36. https://doi.org/10.3390/toxics6030036