Facile and Selective Synthetic Approach for Ruthenium Complexes Utilizing a Molecular Sieve Effect in the Supporting Ligand

Abstract
:1. Introduction

2. Results and Discussion
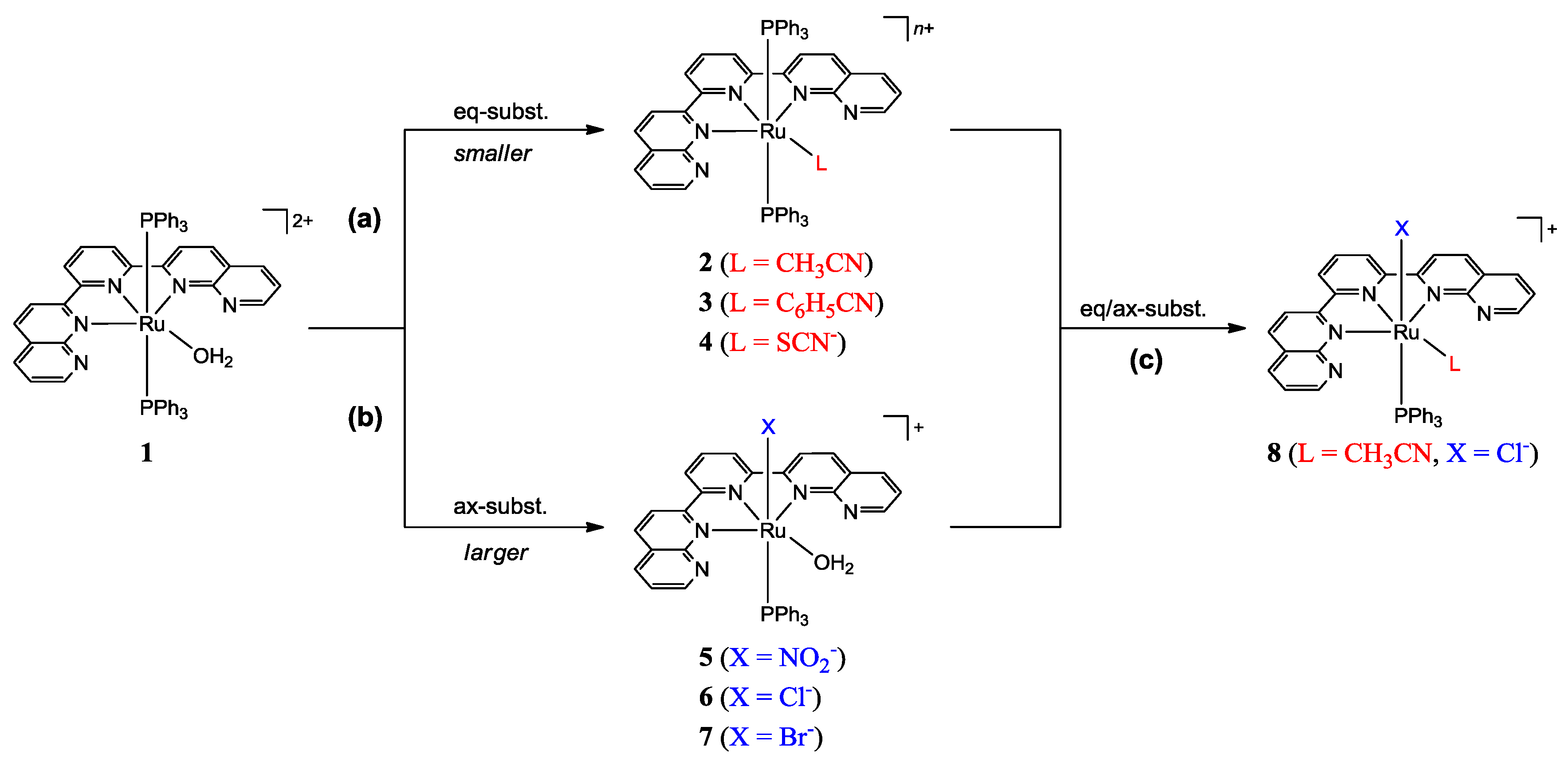
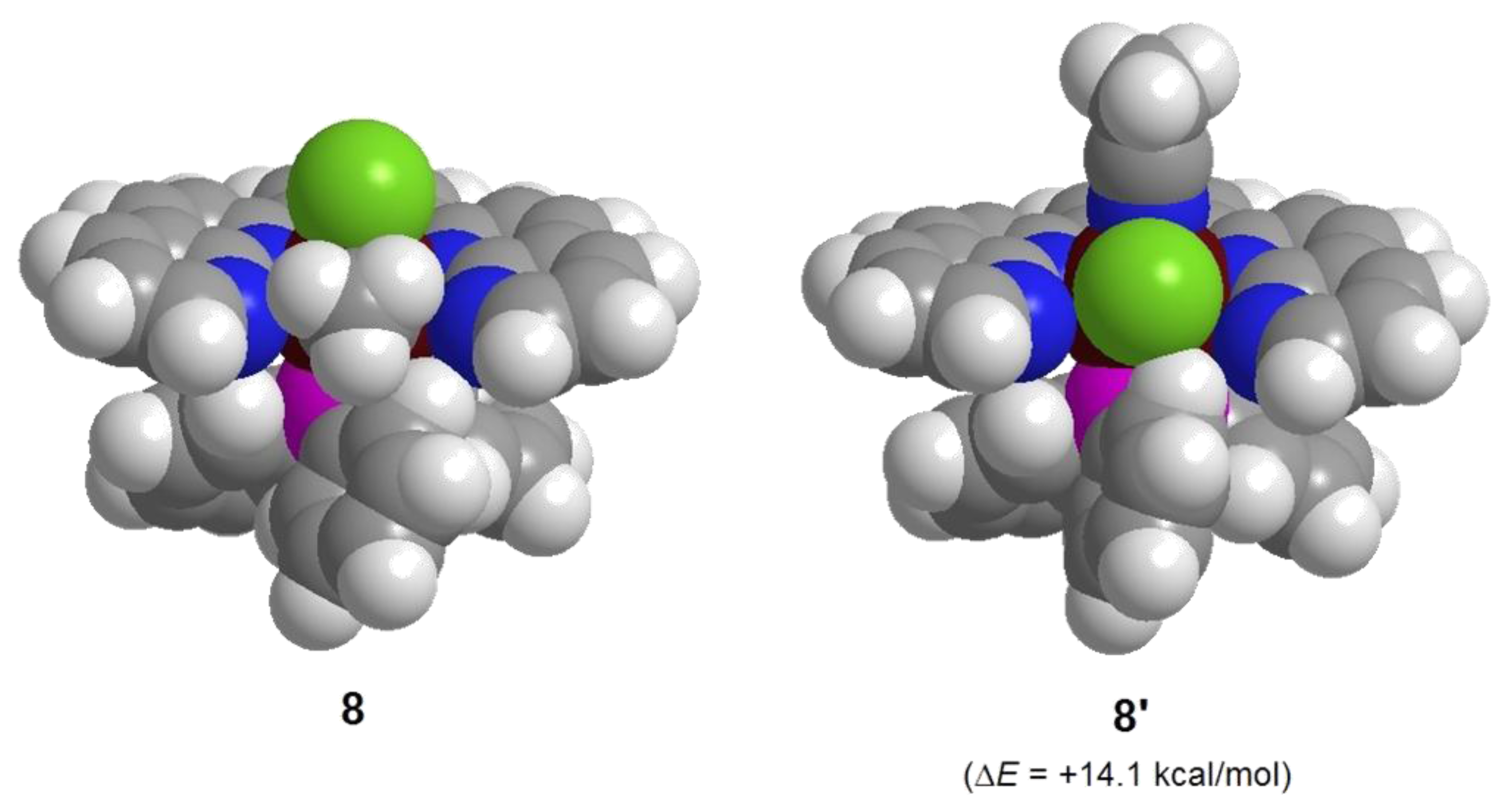
2.1. Synthesis of Ruthenium Complexes Utilizing the Molecular Sieve Effect of the dnp Ligand
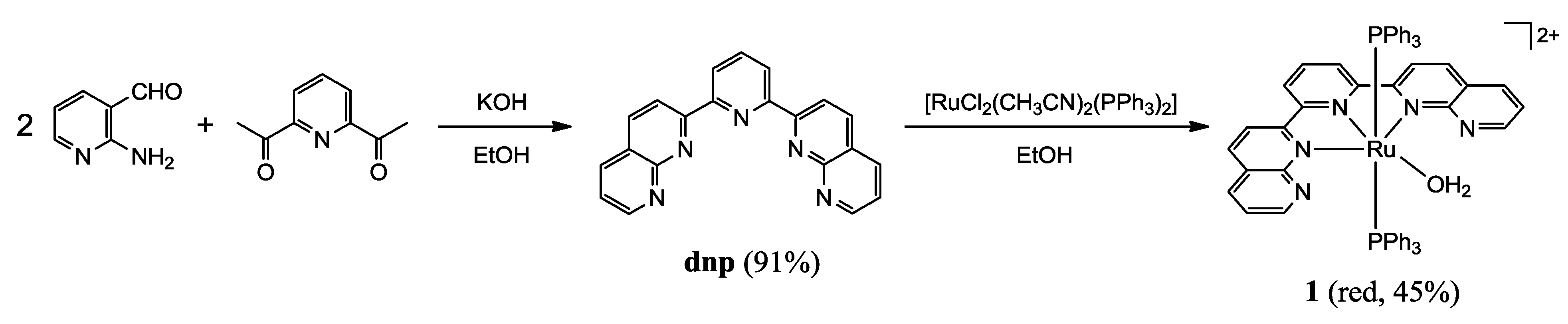
2.1.1. Synthesis of the Precursor

2.1.2. Synthesis of Complexes
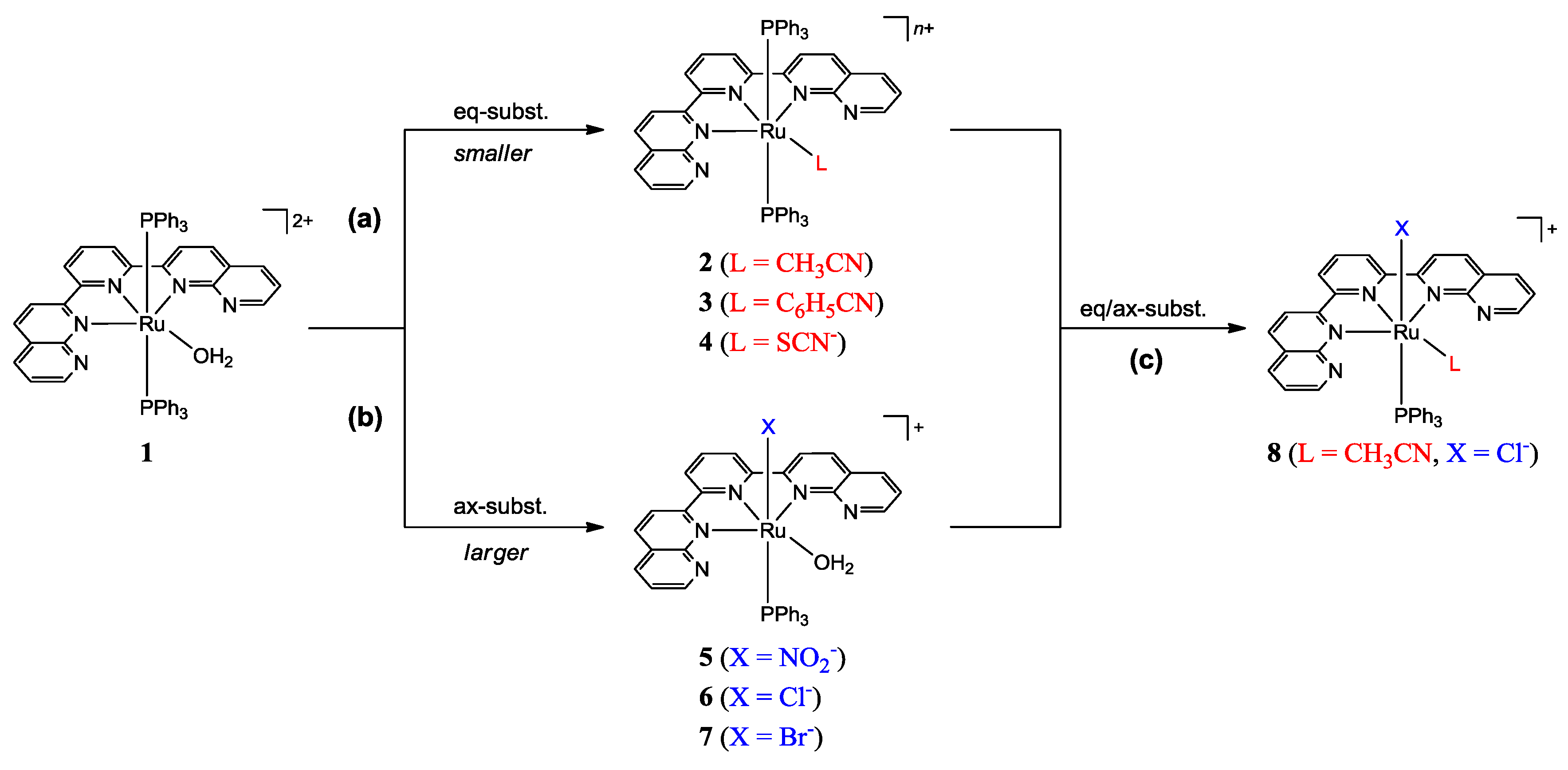
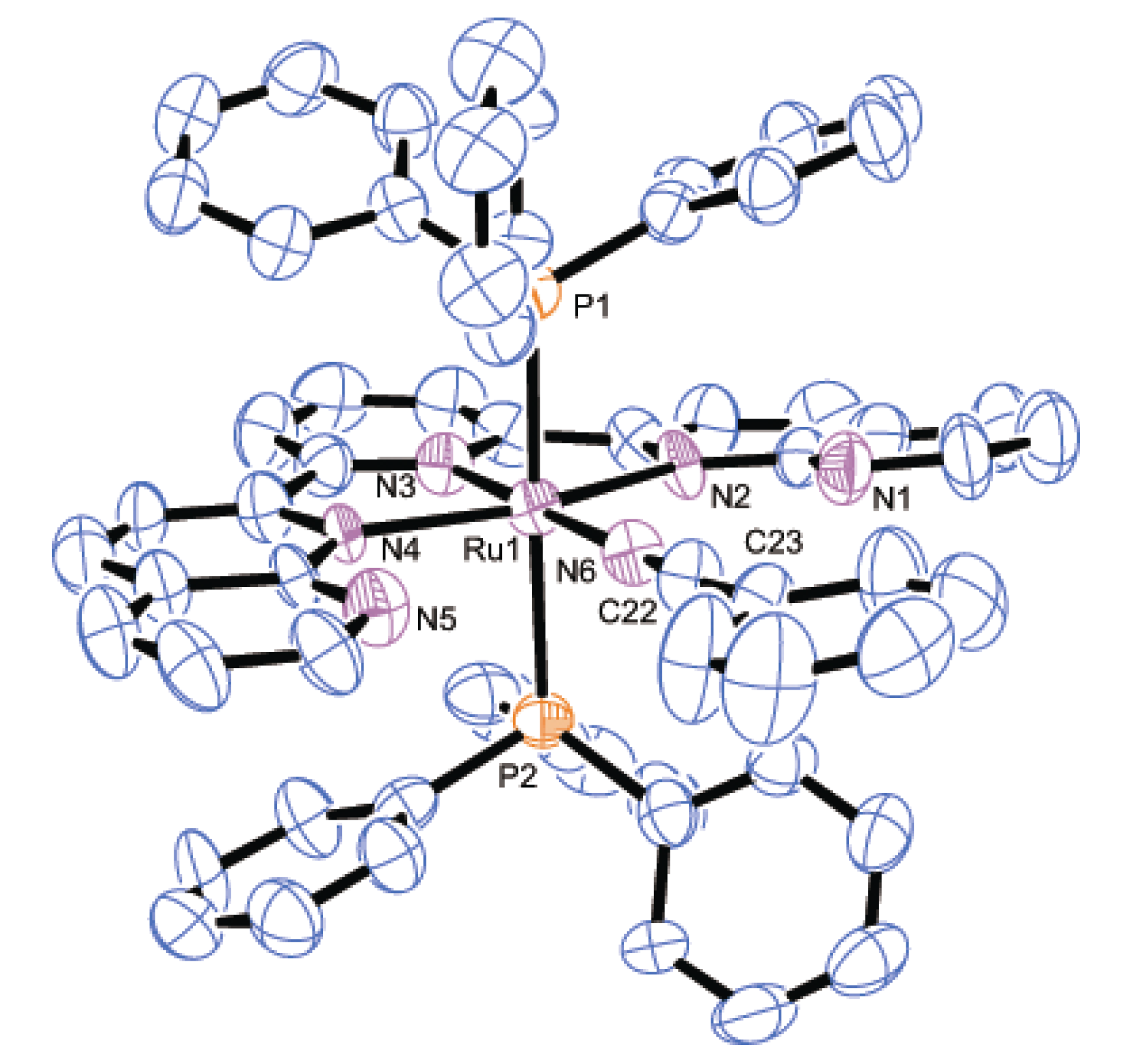
2.2. Spectral and Structural Features of the Complexes
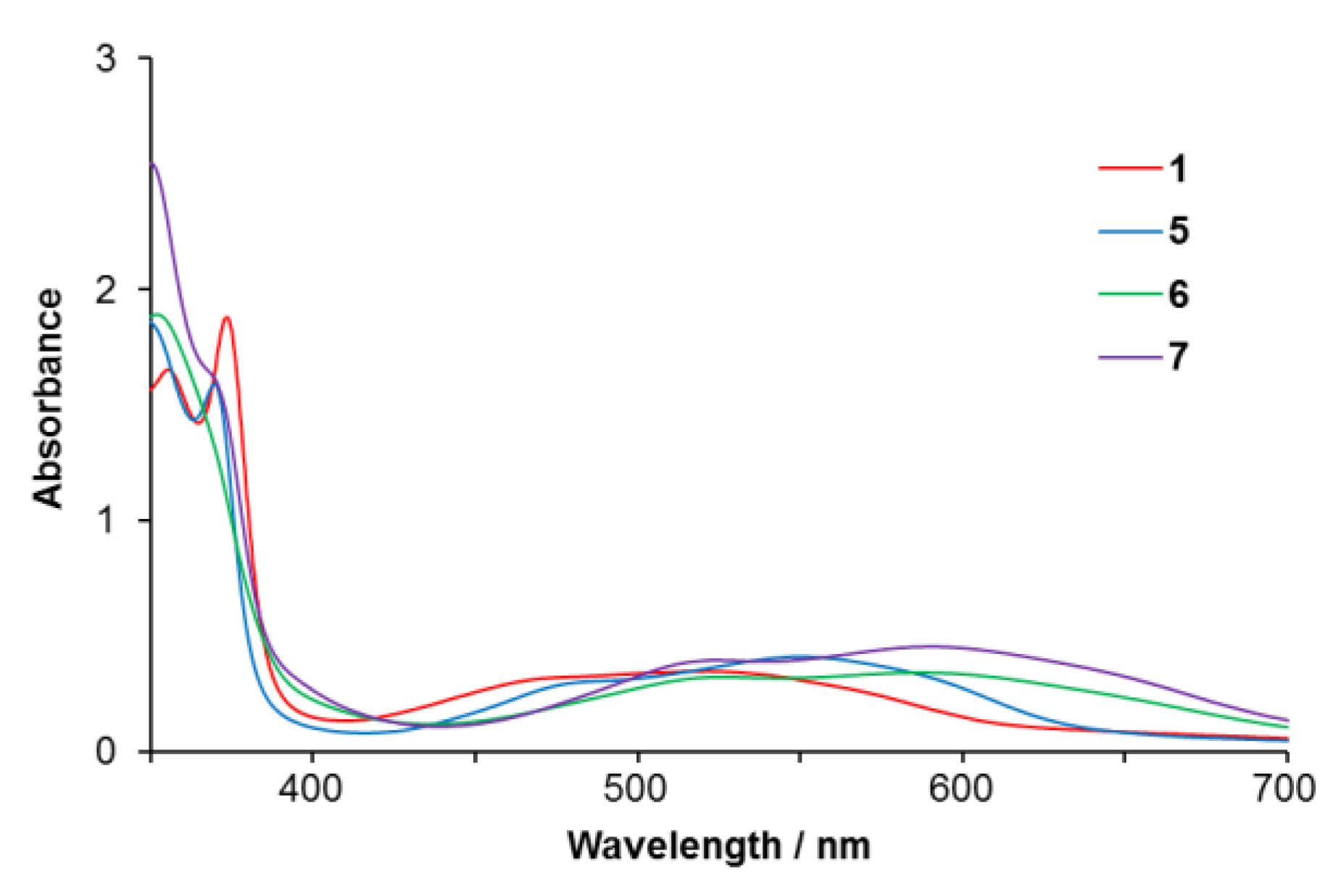
2.2.1. Electronic Absorption Spectra

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | λmax/nm (ε/M−1 cm−1) | Solvent | ||
|---|---|---|---|---|
| 1 | 523 (3500) | 373 (16700) | 355 (15800) | acetone |
| 327 (22400) | ||||
| 2 | 477 (3100) | 374 (20400) | 356 (16300) | methanol |
| 319 (24300) | 274 (42000) | |||
| 3 | 492 (4300) | 453 (4600) | 372 (18600) | methanol |
| 312 (28700) | ||||
| 4 | 547 (3400) | 373 (12800) | 321 (19100) | acetonitrile |
| 279 (28800) | ||||
| 5 | 551 (4100) | 370 (15900) | 343 (18800) | acetone |
| 327 (18800) | ||||
| 6 | 580 (3700) | 352 (19900) | 330 (27400) | acetone |
| 7 | 590 (4600) | 350 (25500) | 327 (21800) | acetone |
| 8 | 563 (3400) | 374 (15800) | 353 (18600) | methanol |
| 320 (25300) | 240 (37200) | |||
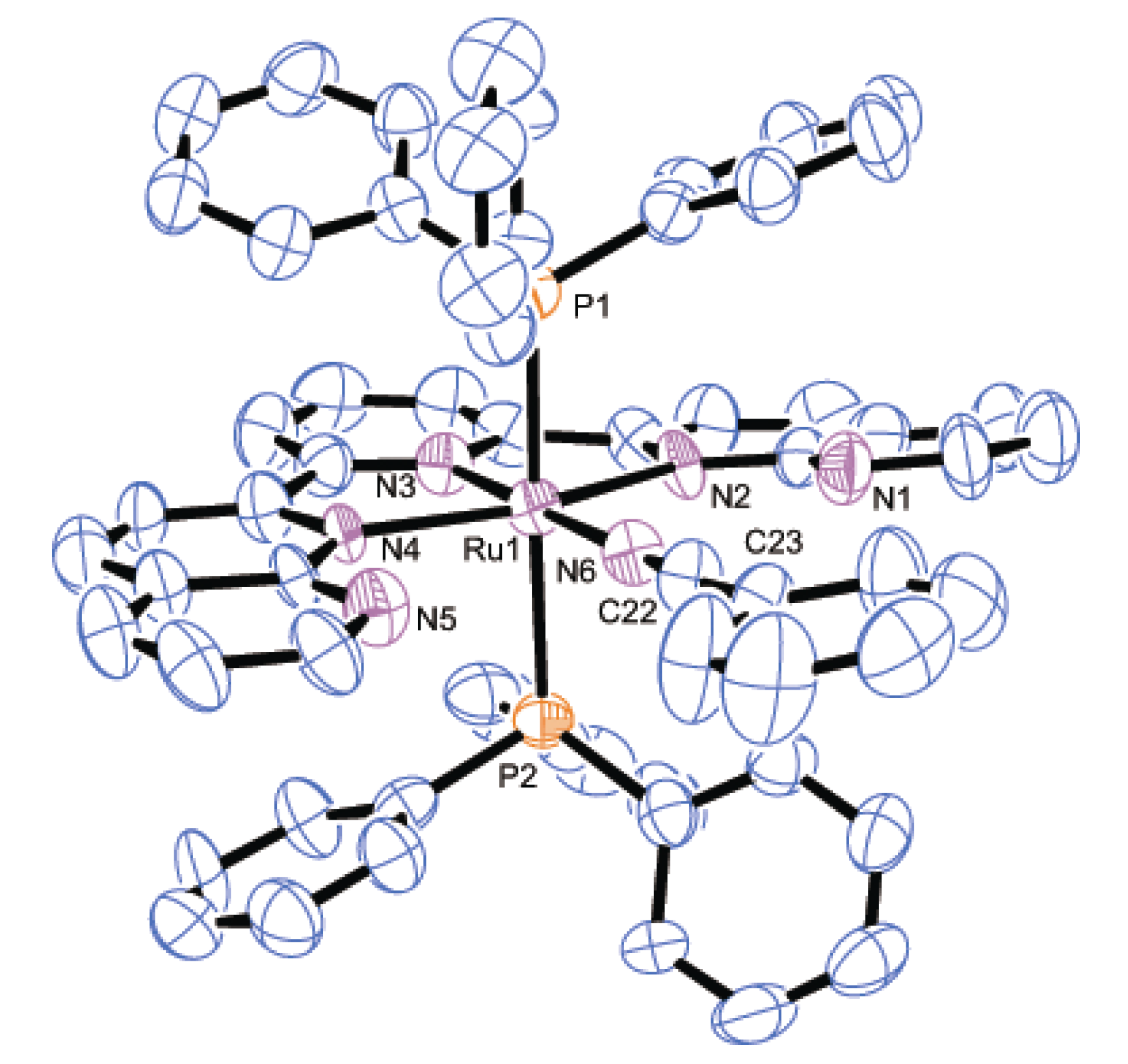
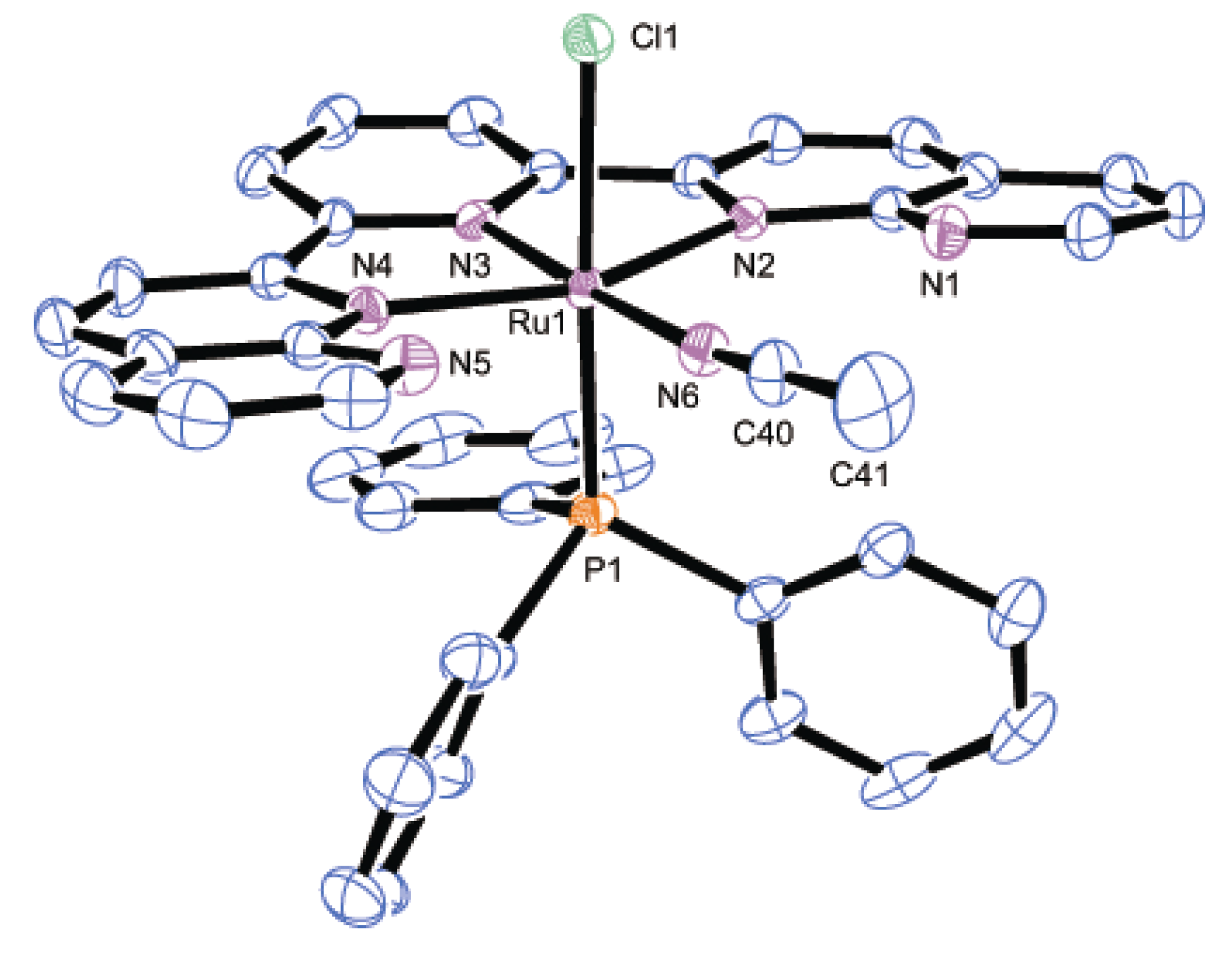
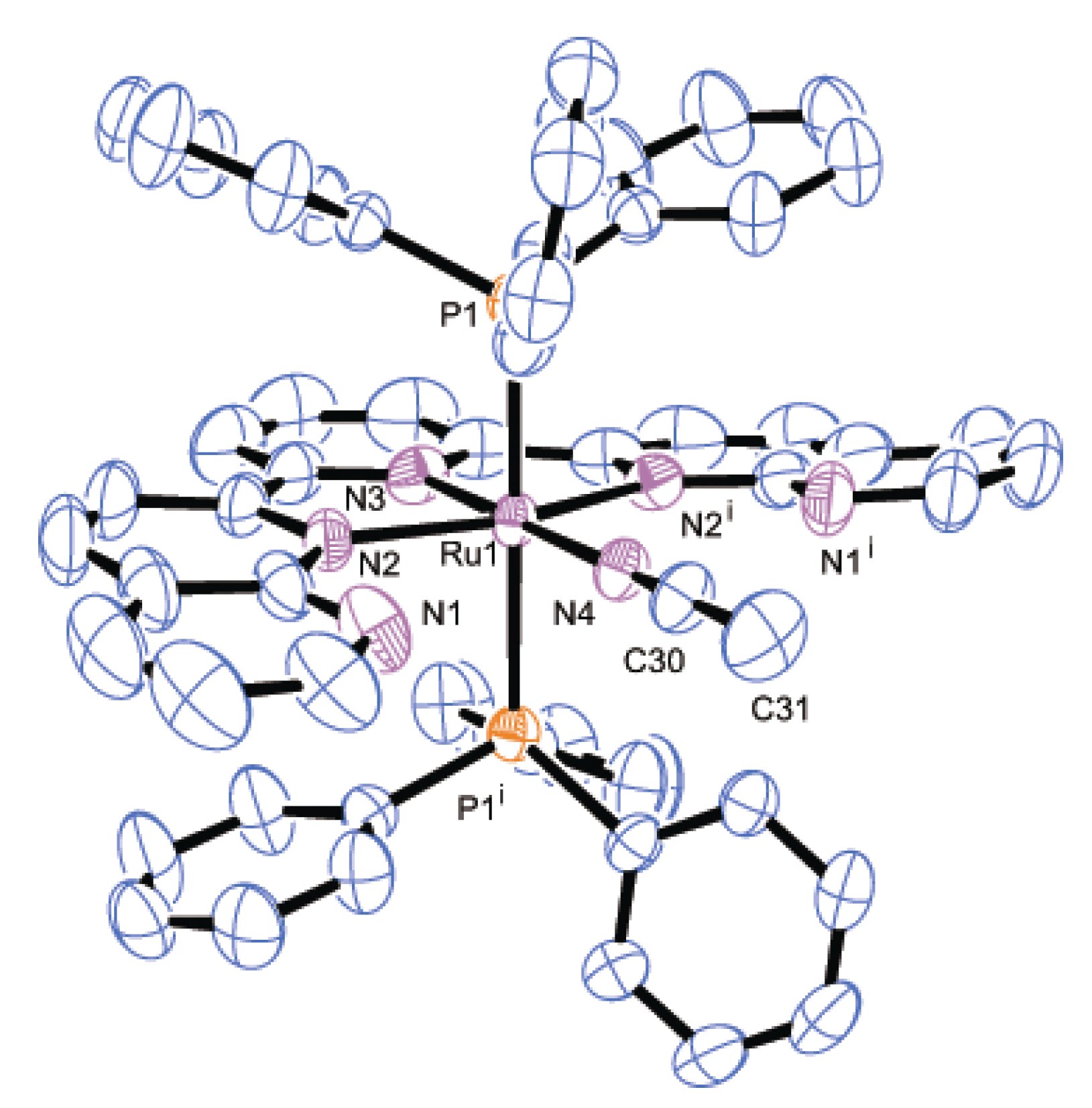
2.2.2. Molecular Structures
| Parameter | 2 | 3 | 8·2H2O | |||
|---|---|---|---|---|---|---|
| Bond distances | Ru1-P1 | 2.4306(7) | Ru1-P1 | 2.436(2) | Ru1-P1 | 2.3272(7) |
| Ru1-N2 | 2.118(2) | Ru1-P2 | 2.430(2) | Ru1-Cl1 | 2.4412(7) | |
| Ru1-N3 | 1.968(2) | Ru1-N2 | 2.167(6) | Ru1-N2 | 2.1206(15) | |
| Ru1-N4 | 2.051(2) | Ru1-N3 | 2.007(7) | Ru1-N3 | 1.9660(14) | |
| N4-C30 | 1.123(4) | Ru1-N4 | 2.163(6) | Ru1-N4 | 2.1442(15) | |
| C30-C31 | 1.464(5) | Ru1-N6 | 2.042(8) | Ru1-N6 | 2.0517(15) | |
| - | - | N6-C22 | 1.141(12) | N6-C40 | 1.130(3) | |
| - | - | C22-C23 | 1.438(14) | C40-C41 | 1.465(3) | |
| Bond angles | Ru1-N4-C30 | 180.0 | Ru1-N6-C22 | 179.3(6) | Ru1-N6-C40 | 174.2(3) |
| N4-C30-C31 | 180.0 | N6-C22-C23 | 176.3(9) | N6-C40-C41 | 178.2(3) | |
| Dihedral anglea | - | - | - | 13.0(4) | - | - |


3. Experimental Section
3.1. Material and Methods
3.2. Synthesis of the Complexes
3.2.1. Synthesis of [Ru(dnp)(PPh3)2(OH2)](PF6)2 (1)
3.2.2. Synthesis of [Ru(dnp)(PPh3)2(RCN)](PF6)2 (R = CH3 (2); R = C6H5 (3))
3.2.3. Synthesis of [Ru(dnp)(PPh3)2(SCN)](PF6) (4) and [Ru(dnp)(PPh3)(OH2)X](PF6) (X = NO2 (5), Cl (6), Br (7))
3.2.4. Synthesis of [Ru(dnp)(PPh3)(CH3CN)Cl]Cl (8)
3.3. X-ray Crystallographic Analyses
| Parameter | 2 | 3 | 8·2H2O |
|---|---|---|---|
| Chemical formula | C59H46N6O8Cl2P2Ru | C64H48N6F12P4Ru | C41H35N6O2Cl2PRu |
| Formula weight | 1200.97 | 1354.07 | 846.72 |
| Temperature (K) | 296(1) | 296(1) | 173(1) |
| Crystal system | monoclinic | monoclinic | Monoclinic |
| Space group | C2/c | Cc | C2/c |
| Unit cell parameters | |||
| a (Å) | 17.7754(3) | 17.1743(4) | 26.4297(5) |
| b (Å) | 13.2405(2) | 16.1763(4) | 16.2228(3) |
| c (Å) | 22.6972(4) | 21.0790(4) | 19.4965(4) |
| Β (°) | 96.4561(7) | 97.8336(7) | 114.9980(7) |
| V (Å3) | 5308.03(15) | 5801.5(2) | 7576.3(3) |
| Z | 4 | 4 | 8 |
| Calculated density (g cm−3) | 1.503 | 1.550 | 1.485 |
| μ (Mo Kα) (mm−1) | 0.520 | 0.468 | 0.642 |
| No. of measured reflections | 25463 | 46099 | 34745 |
| No. of observed reflections | 6075 | 12645 | 8632 |
| Refinement method | Full-matrix least-squares on F2 | ||
| Parameters | 356 | 785 | 474 |
| R1 (I > 2σ(I)) a | 0.0444 | 0.0580 | 0.0311 |
| w R2 (all data) b | 0.1292 | 0.2164 | 0.0803 |
| S | 1.050 | 1.126 | 1.038 |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chowdhury, A.D.; Das, A.; Irshad, K.; Mobin, S.M.; Lahiri, G.K. Isomeric complexes of [RuII(trpy)(L)Cl] (trpy = 2,2':6',2"-terpyridine and HL = quinaldic acid): Preference of isomeric structural form in catalytic chemoselective epoxidation process. Inorg. Chem. 2011, 50, 1775–1785. [Google Scholar] [CrossRef]
- Dakkach, M.; López, M.I.; Romero, I.; Rodríguez, M.; Atlamsani, A.; Parella, T.; Fontrodona, X.; Llobet, A. New Ru(II) complexes with anionic and neutral N-donor ligands as epoxidation catalysts: An evaluation of geometrical and electronic effects. Inorg. Chem. 2010, 49, 7072–7079. [Google Scholar] [CrossRef]
- Oyama, D.; Yamanaka, T.; Fukuda, A.; Takase, T. Modulation of intramolecular hydrogen bonding strength by axial ligands in ruthenium(II) complexes. Chem. Lett. 2013, 42, 1554–1555. [Google Scholar] [CrossRef]
- Campos-Fernández, C.S.; Thomson, L.M.; Galán-Mascarós, J.R.; Ouyang, X.; Dunbar, K.R. Homologous series of redox-active, dinuclear cations [M2(O2CCH3)2(pynp)2]2+ (M = Mo, Ru, Rh) with the bridging ligand 2-(2-pyridyl)-1,8-naphthyridine (pynp). Inorg. Chem. 2002, 41, 1523–1533. [Google Scholar]
- Tseng, H.-W.; Zong, R.; Muckerman, J.T.; Thummel, R. Mononuclear ruthenium(II) complexes that catalyze water oxidation. Inorg. Chem. 2008, 47, 11763–11773. [Google Scholar] [CrossRef]
- Al-Far, A.M.; Slaughter, L.M. cis-cis-trans-Bis(acetonitrile-κN)dichloridobis(triphenylphosphine-κP)ruthenium(II) acetonitrile disolvate. Acta Crystallogr. 2008, E64, m184. [Google Scholar]
- Kaveevivitchai, N.; Zong, R.; Tseng, H.-W.; Chitta, R.; Thummel, R.P. Further observations on water oxidation catalyzed by mononuclear Ru(II) complexes. Inorg. Chem. 2012, 51, 2930–2939. [Google Scholar] [CrossRef]
- Suen, H.F.; Wilson, S.W.; Pomerantz, M.; Walsh, J.L. Photosubstitution reactions of terpyridine complexes of ruthenium(II). Inorg. Chem. 1989, 28, 786–791. [Google Scholar] [CrossRef]
- Zong, R.; Naud, F.; Segal, C.; Burke, J.; Wu, F.; Thummel, R. Design and study of bi[1,8]naphthyridine ligands as potential photooxidation mediators in Ru(II) polypyridyl aquo complexes. Inorg. Chem. 2004, 43, 6195–6202. [Google Scholar] [CrossRef]
- Oyama, D.; Yamanaka, Y.; Watanabe, Y.; Takase, T.; Fukushima University, Fukushima, Japan. Unpublished work. 2013.
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; John Wiley & Sons: New York, NY, USA, 1986; pp. 221–227. [Google Scholar]
- Ooyama, D.; Nagao, N.; Nagao, H.; Miura, Y.; Hasegawa, A.; Ando, K.-I.; Howell, F.S.; Mukaida, M.; Tanaka, K. Redox- and thermally-induced nitro-nitrito linkage isomerizations of ruthenium(II) complexes having nitrosyl as a spectator ligand. Inorg. Chem. 1995, 34, 6024–6033. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) polypyridine complexes: Photophysics, photochemistry, electrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Thummel, R.P.; Decloitre, Y. Polyaza cavity-shaped molecules. VIII. Ruthenium(II) complexes with 3,3'-annelated derivatives of 2-(2'-pyridyl)quinoline and 2-(2'-pyridyl)-1,8-naphthyridine. Inorg. Chim. Acta 1987, 128, 245–249. [Google Scholar] [CrossRef]
- Ooyama, D.; Saito, M. Synthesis, characterization and reactivity of polypyridyl ruthenium(II) carbonyl complexes with phosphine derivatives: ruthenium-carbon bond labilization based on steric and electronic effects. Inorg. Chim. Acta 2006, 359, 800–806. [Google Scholar] [CrossRef]
- Chen, J.; Szalda, D.J.; Fujita, E.; Creutz, C. Iron(II) and ruthenium(II) complexes containing P, N, and H ligands: structure, spectroscopy, electrochemistry, and reactivity. Inorg. Chem. 2010, 49, 9380–9391. [Google Scholar] [CrossRef]
- Ooyama, D.; Sato, M. A synthetic precursor for hetero-binuclear metal complexes, [Ru(bpy)(dppy)2(CO)2](PF6)2 (bpy = 2,2'-bipyridine, dppy = 2-(diphenylphosphino)pyridine). Appl. Organomet. Chem. 2004, 18, 380–381. [Google Scholar] [CrossRef]
- Ooyama, D.; Sato, M. Crystal structure of (2,2'-bipyridine-N,N′)(diphenyl-2-phosphinopyridine-P)chloro(dicarbonyl)ruthenium(II) hexafluorophosphate. Anal. Sci. 2003, 19, x39–x40. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Methyl protons of both complexes could not be assigned due to their overlapping with solvent signals.
- Rigaku. CrystalStructure, Version 3.8; Rigaku and Rigaku Americas: Woodlands, TX, USA, 2007.
- Sheldrick, G.M. SHELX, SHELXL-97; University of Göttingen: Göttingen, Germany, 1997.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar]
- Altomare, A.; Burla, M.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Molitern, A.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar]
- Higashi, T. ABSCOR; Rigaku Corporation: Tokyo, Japan, 1995. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oyama, D.; Fukuda, A.; Yamanaka, T.; Takase, T. Facile and Selective Synthetic Approach for Ruthenium Complexes Utilizing a Molecular Sieve Effect in the Supporting Ligand. Inorganics 2013, 1, 32-45. https://doi.org/10.3390/inorganics1010032
Oyama D, Fukuda A, Yamanaka T, Takase T. Facile and Selective Synthetic Approach for Ruthenium Complexes Utilizing a Molecular Sieve Effect in the Supporting Ligand. Inorganics. 2013; 1(1):32-45. https://doi.org/10.3390/inorganics1010032
Chicago/Turabian StyleOyama, Dai, Ayumi Fukuda, Takashi Yamanaka, and Tsugiko Takase. 2013. "Facile and Selective Synthetic Approach for Ruthenium Complexes Utilizing a Molecular Sieve Effect in the Supporting Ligand" Inorganics 1, no. 1: 32-45. https://doi.org/10.3390/inorganics1010032