1. Introduction
Since Cavell
et al. reported the first rare earth bis(iminophosphorano)methanediide complex, namely [Sm(BIPM
TMS)(NCy
2)(THF)] (BIPM
TMS = {C(PPh
2NSiMe
3)
2}
2−) [
1], a wide range of BIPM
R- derived rare earth complexes have been reported with the BIPM
R ligand being either mono-deprotonated ({BIPM
RH}
−) and coordinating as a methanide, or doubly deprotonated ({BIPM
R}
2−) and coordinating as either a methanediide or carbene [
2,
3,
4]. These complexes have shown extensive utility in a range of areas, including hydroamination/cyclisation, hydrosilylation and polymerisation reactions [
5,
6], and our work towards: (a) the preparation of rare earth heterobimetallics [
7]; (b) unusual reactivity towards small molecules [
8,
9,
10]; and (c) the stabilisation of the Ce(IV) oxidation state [
11]. In our studies we have employed primarily the BIPM
TMS and BIPM
Mes ({C(PPh
2NMes)
2}
2−; Mes = 2,4,6- trimethylphenyl) ligand frameworks [
7,
12,
13,
14,
15,
16], however, during our studies we have encountered several synthetic limitations in our attempts to prepare BIPM
R-derived rare earth complexes for the complete rare earth series. We have employed both alkane elimination and salt metathesis strategies towards the preparation of methanediide complexes of the general formula [Ln(BIPM
R)(X)(THF)
n] (R = TMS or Mes; X = I, CH
2SiMe
3 or CH
2Ph), with each approach having advantages and limitations.
The reaction of a range of rare earth tri-benzyl complexes [Ln(CH
2Ph)
3(THF)
3] (Ln = La, Ce, Pr, Nd, Sm, Gd, Dy, Er, Y) or [Y(CH
2SiMe
3)
3(THF)
3] with the protonated pro-ligand BIPM
TMSH
2 afforded different products depending on the size of the rare earth centre [
16,
17,
18]. In the case of the smaller rare earths, the methanediide alkyl complexes [Ln(BIPM
TMS)(R)(THF)] (R = CH
2Ph: Ln = Dy, Er, Y; R = CH
2SiMe
3: Ln = Y) were isolated [
16,
17,
18], however for the larger rare earths the only isolable products were the bis-BIPM complexes [Ln(BIPM
TMS)(BIPM
TMSH)] (Ln = La, Ce, Pr, Nd, Sm, Gd) [
16]. This variation in reactivity was ascribed to the lanthanide contraction [
19], with ligand scrambling occurring in the attempted preparations of the larger rare earth analogues despite the high steric demands of the BIPM
TMS ligand [
16]. By a similar methodology we prepared [Ln(BIPM
TMS)(I)(THF)
2] (Ln = Y, Er) via the reaction of [Ln(CH
2Ph)
2(I)(THF)
3] with BIPM
TMSH
2 [
15], but unfortunately this methodology could not be applied across the rare earth series due to the inaccessibility of [Ln(CH
2Ph)
2(I)(THF)
3] for rare earths larger than erbium [
12].
Salt metathesis strategies were first employed in the preparation of [Y(BIPM
TMSH)(I)
2(THF)], which was isolated in 64% yield via the reaction of [{K(BIPM
TMSH)}
2] with [Y(I)
3(THF)
3.5] in refluxing THF over 4 hours [
7]. Fine-tuning of the reaction conditions was required with excessive reaction times leading to decomposition. Perhaps surprisingly, applying a similar strategy to La with the reaction of [La(I)
3(THF)
4] and [{K(BIPM
TMSH)}
2] does not afford [La(BIPM
TMSH)(I)
2(THF)] as expected; no reaction is observed at ambient temperature and decomposition is observed at elevated temperatures [
12]. Gratifyingly, however, the reaction of [Ln(I)
3(THF)
4] (Ln = La, Ce) with [Rb(BIPM
TMSH)(THF)
n] affords [Ln(BIPM
TMSH)(I)
2(THF)] (Ln = La, Ce) in high yield with no forcing conditions required [
11,
13]. This variation exemplifies the importance of selecting the correct synthetic reagent for the reaction in hand. The reaction of [Ln(BIPM
TMSH)(I)
2(THF)] (Ln = Ce, Y) with [K(Bn)] affords the rare earth carbene complexes [Ln(BIPM
TMS)(I)(S)
n] (Ln = Y, S = THF,
n = 2; Ln = Ce, S = 1,2-dimethoxyethane,
n = 1) in good yield [
7,
11].
To fully investigate the importance of the steric demands of the
N-SiMe
3 group in BIPM
TMS, we employed a different BIPM ligand, namely the
N-aryl substituted BIPM
Mes system. In contrast to the difficulties experienced when employing salt metathesis strategies with the BIPM
TMS framework, we previously prepared [Ln(BIPM
MesH)(I)
2(THF)
2] (Ln = La, Ce, Pr, Nd, Sm) via the straight-forward reaction of [Ln(I)
3(THF)
n] with [{K(BIPM
MesH)}
2] [
12,
14]. Similarly to the BIPM
TMS system [La(BIPM
MesH)(I)
2(THF)
2] was converted to the rare earth carbene complex [La(BIPM
Mes)(I)(THF)
3] in 53% yield via reaction with [K(Bn)] [
12].
Whilst a range of complexes of the formula [{Ln(BIPM
TMSH)(Cl)(µ-Cl)}
2] have been reported previously by the reaction of [Ln(Cl)
3] and [{K(BIPM
TMSH)}
2] [
20], we have solely utilised iodide precursors. This is to reduce the likelihood of salt occlusion occurring as KI, RbI and CsI have a much lower propensity for salt occlusion in comparison to LiCl or KCl, and as salt occlusion can often negatively affect reactivity and promote unwanted side reactions this is a major advantage of utilising [Ln(I)
3(THF)
n] precursors. As ligand scrambling and Schlenk-type equilibra proved problematic in alkane elimination strategies, we focused on salt metathesis strategies in our attempts to complete the range of rare earth BIPM
RH (R = TMS, Mes) complexes of the general formula [Ln(BIPM
RH)(I)
2(THF)
n]. These complexes would provide an insight into the structure and bonding of rare earth complexes as well as having utility in the preparation of methanediide complexes of the general formula [Ln(BIPM
R)(I)(THF)
n]. With these complexes in hand we would be able to investigate what, if any, effect the varying metal size would have on their structure and reactivity.
3. Experimental Section
All manipulations were carried out using standard Schlenk techniques, or an MBraun UniLab glovebox, under an atmosphere of dry nitrogen. Solvents were dried by passage through activated alumina towers and degassed before use. All solvents were stored over potassium mirrors (with the exception of THF which was stored over activated 4 Å molecular sieves). Deuterated solvents were distilled from potassium, degassed by three freeze-pump-thaw cycles and stored under nitrogen. [Cs(BIPM
TMSH)] [
13], [Ln(I)
3(THF)
3.5] (Ln = Nd, Gd, Dy, Er, Yb) [
25], [{K(BIPM
MesH)}
2] [
6], [Ln(BIPM
MesH)(I)
2(THF)
2] (Ln = La, Ce, Pr) [
12,
14], [La(BIPM
TMSH)(I)
2(THF)] [
13], and [K(Bn)] [
30] were prepared according to published procedures. [Tb(I)
3(THF)
3.5] was prepared according to a modified literature procedure [
25]. [K(CHPh
2)] and [K(CPh
3)] were prepared by the reaction of KOBu
t/Bu
nLi with either diphenylmethane or triphenylmethane in hexanes, analogously to the preparation of [K(Bn)] [
30]. All other materials were purchased and dried before use.
1H, 13C and 31P NMR spectra were recorded on a Bruker 400 spectrometer operating at 400.2, 100.6 and 162.0 MHz, respectively; chemical shifts are quoted in ppm and are relative to tetramethylsilane (1H, 13C) or external 85% H3PO4 (31P). FTIR spectra were recorded on a Bruker Tensor 27 spectrometer. Elemental microanalyses were carried out by Mr. Stephen Boyer at the Microanalysis Service, London Metropolitan University, UK.
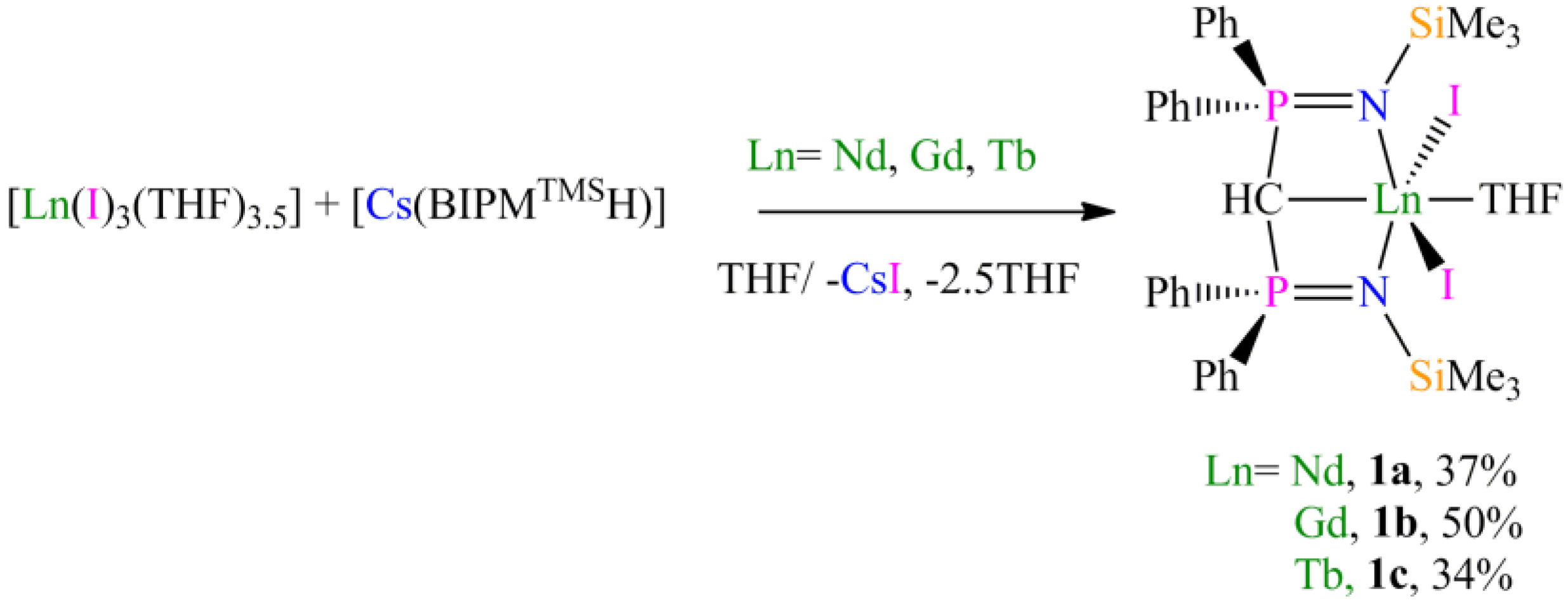
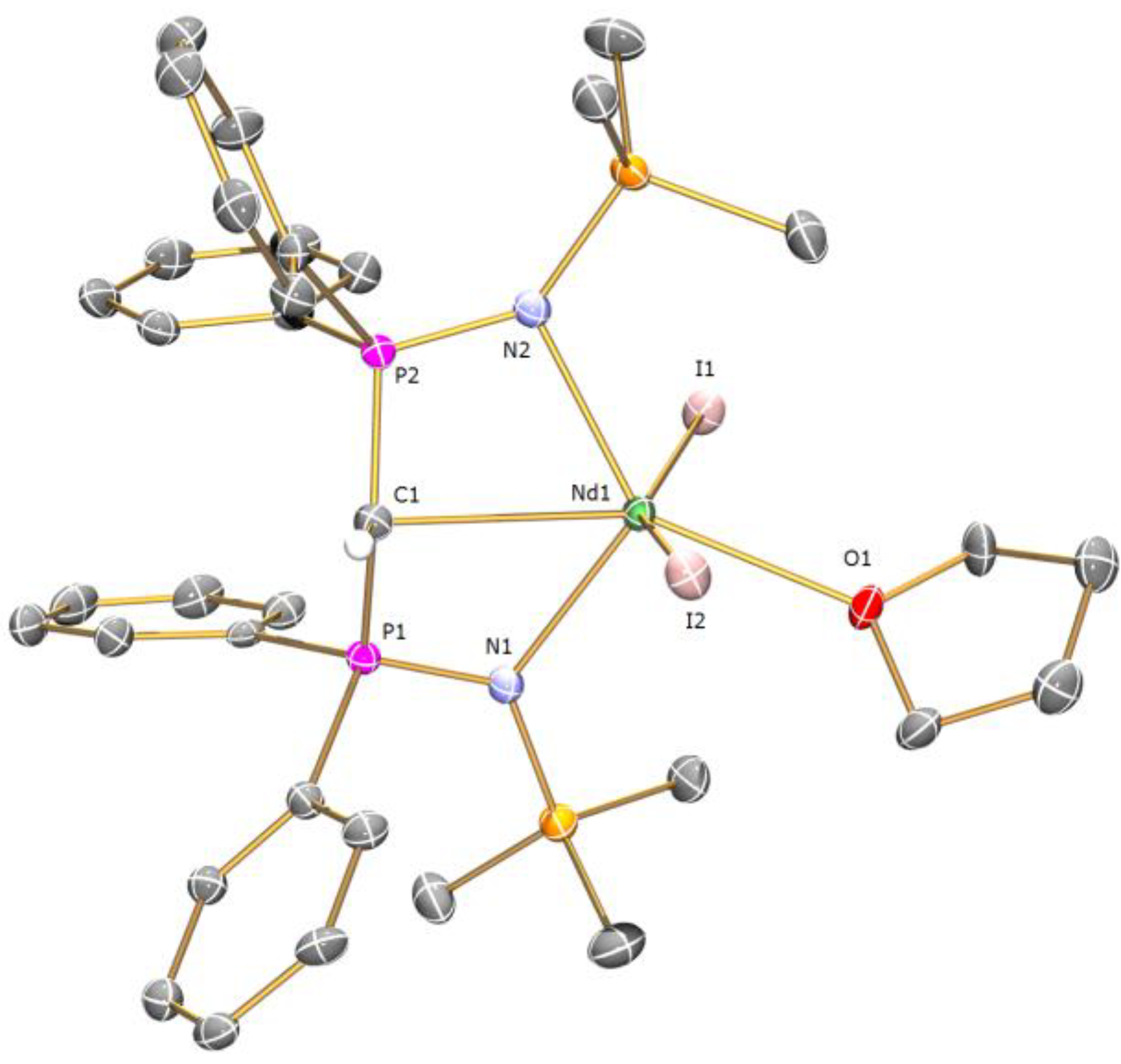
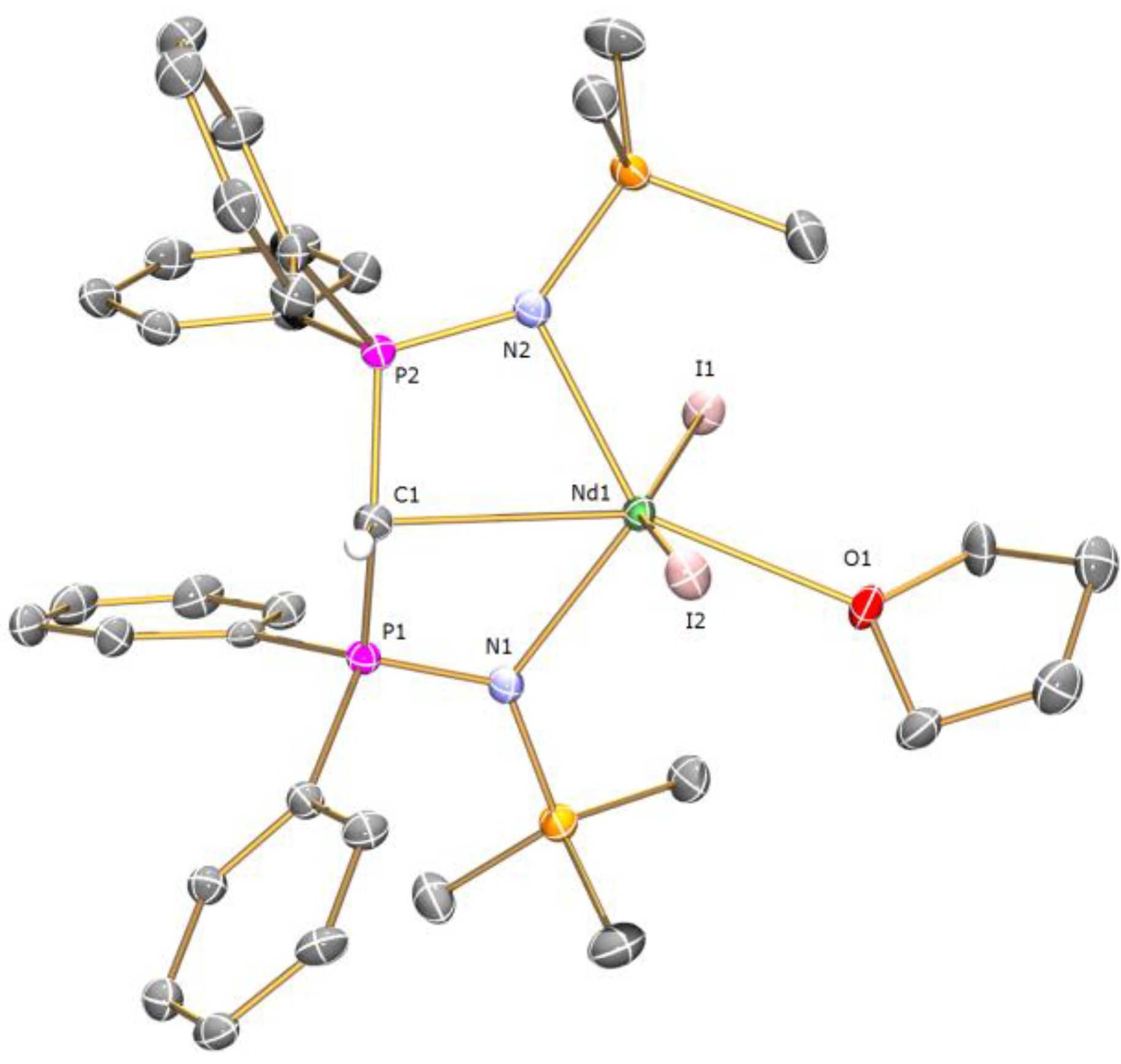
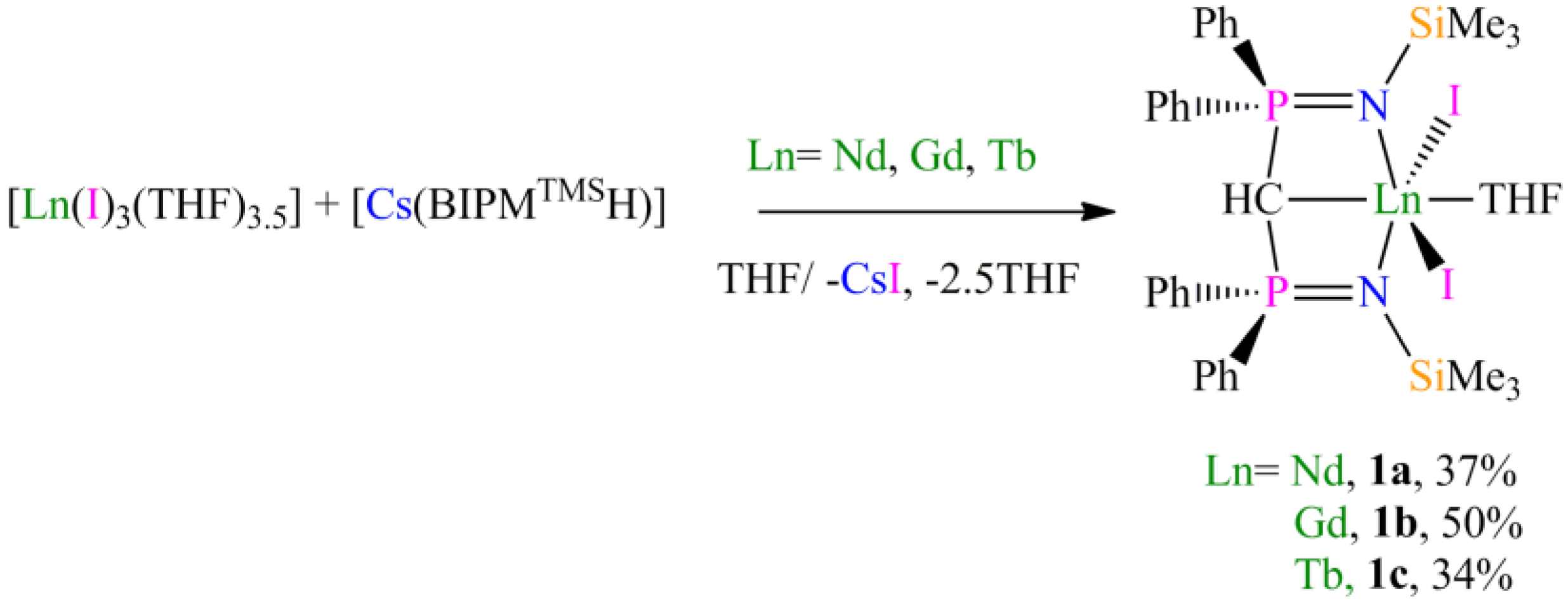
Preparation of [Nd(BIPMTMSH)(I)2(THF)] (1a): THF (40 mL) was added to a precooled (−78 °C) mixture of [Cs(BIPMTMSH)] (0.69 g, 1.00 mmol) and [Nd(I)3(THF)3.5] (0.78 g, 1.00 mmol) and the resulting mixture was allowed to warm to room temperature with stirring over 18 h. The mixture was filtered to remove CsI and all volatiles were removed in vacuo to afford a pale blue solid. Recrystallisation from toluene (5 mL) afforded 1a C7H8 as pale blue crystals. Yield 0.42 g, 37%. Anal. Calcd for C42H55I2N2NdOP2Si2: C, 45.05; H, 4.95; N, 2.50. Found: C, 44.89; H, 4.86; N, 2.58. FTIR ν/cm−1 (Nujol): 1377 (m), 1366 (w), 1214 (w), 1064 (m), 992 (w), 839 (m), 770 (w), 746 (w). µeff (Evans method, 298 K, C6D6): 4.14 µB.
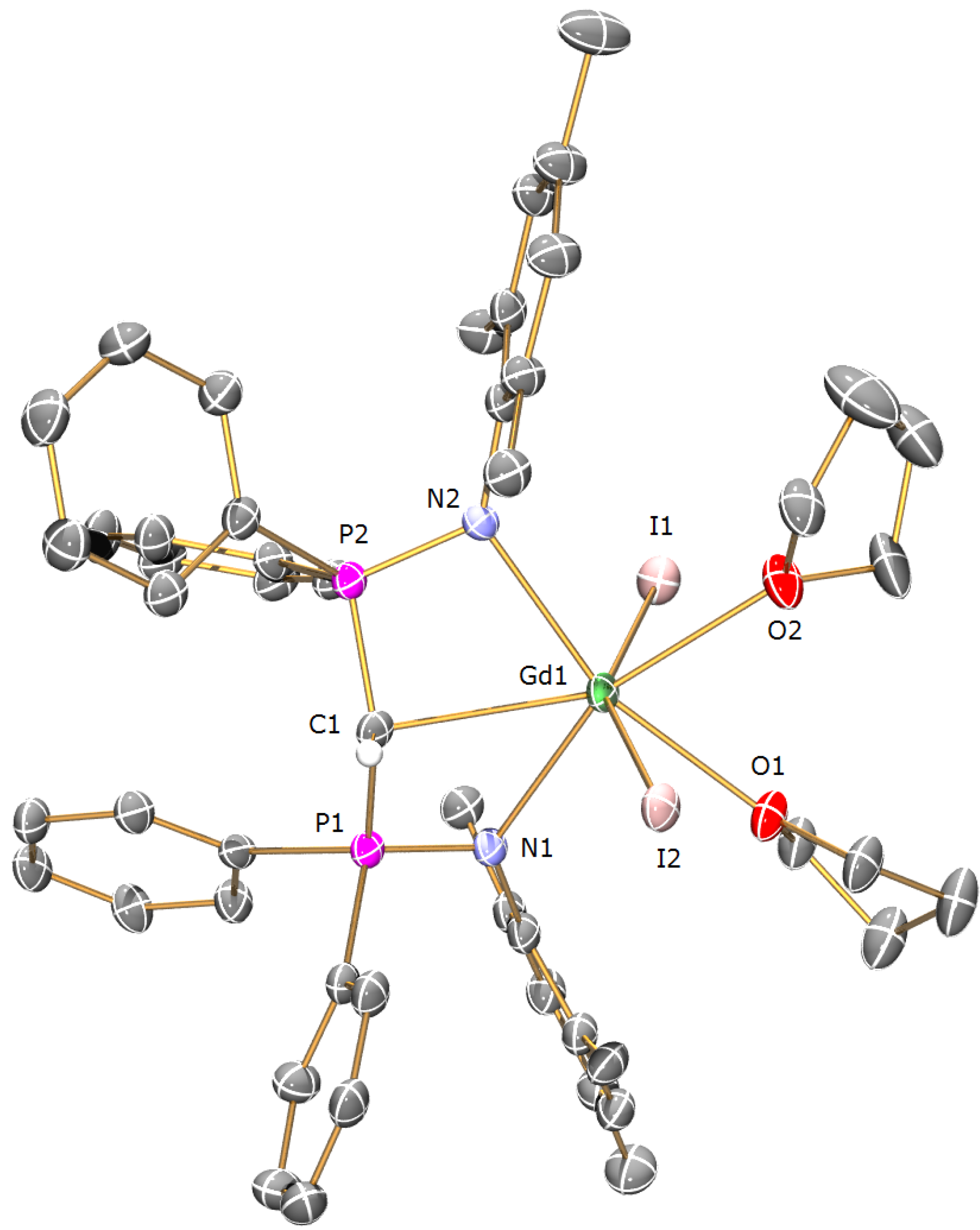
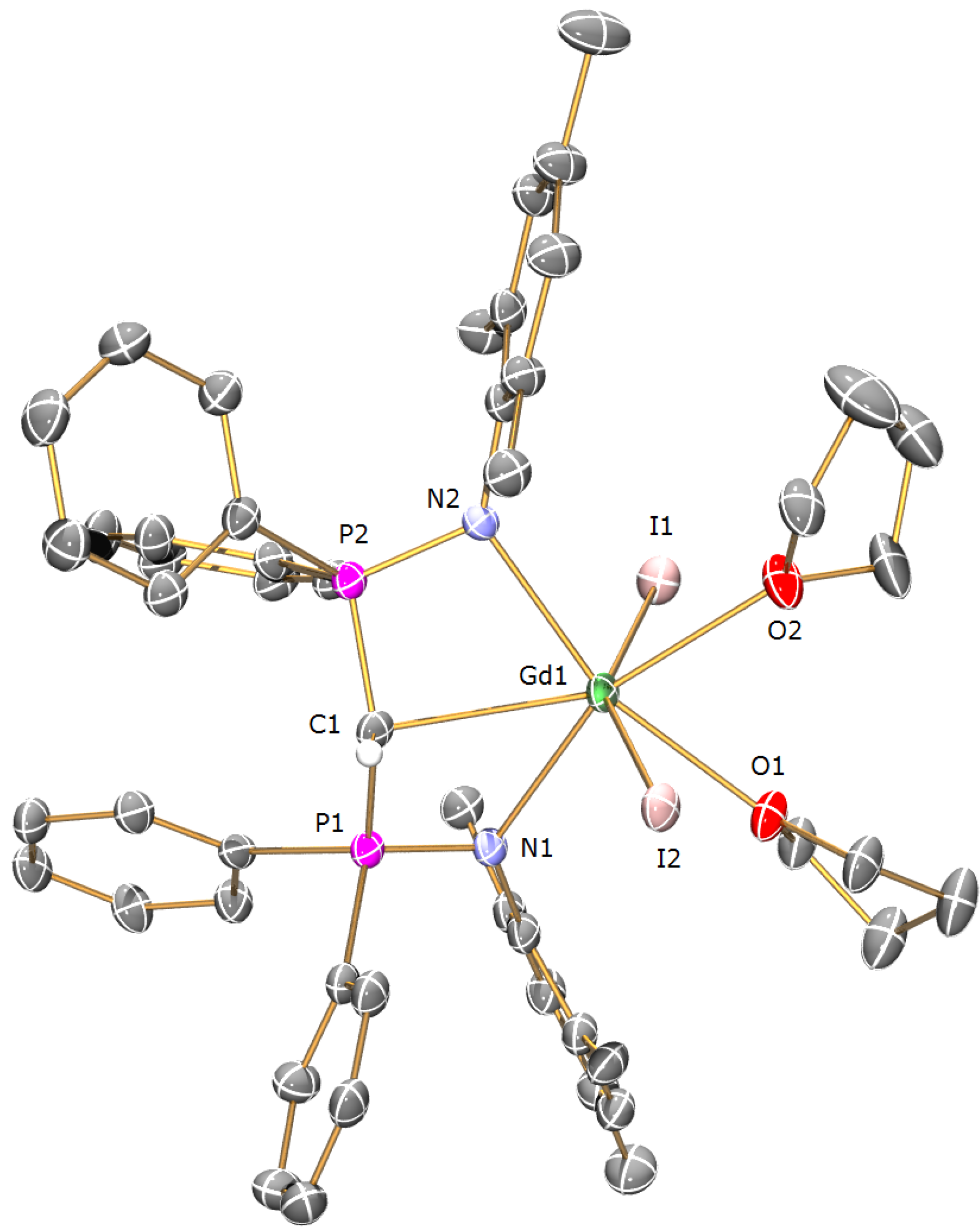
Preparation of [Gd(BIPMTMSH)(I)2(THF)] (1b): THF (40 mL) was added to a precooled (−78 °C) mixture of of [Cs(BIPMTMSH)] (0.69 g, 1.00 mmol) and [Gd(I)3(THF)3.5] (0.79 g, 1.00 mmol) and the resulting mixture was allowed to warm to room temperature with stirring over 18 h. The mixture was filtered to remove CsI and the solution reduced in volume to ca. 5 mL. Cooling of the solution to −30 °C afforded 1b OC4H8 as yellow crystals. Yield 0.56 g, 50%. Anal. Calcd for C39H55GdI2N2O2P2Si2: C, 42.09; H, 4.98; N, 2.52. Found: C, 42.07; H, 4.95; N, 2.61. FTIR ν/cm−1 (Nujol): 1437 (m), 1152 (m), 1114 (s), 1057 (s), 920 (w), 839 (m), 752 (w), 745 (w), 725 (w), 712 (w), 695 (w), 660 (w), 551 (m), 504 (w). µeff (Evans method, 298 K, C6D6): 8.47 µB.
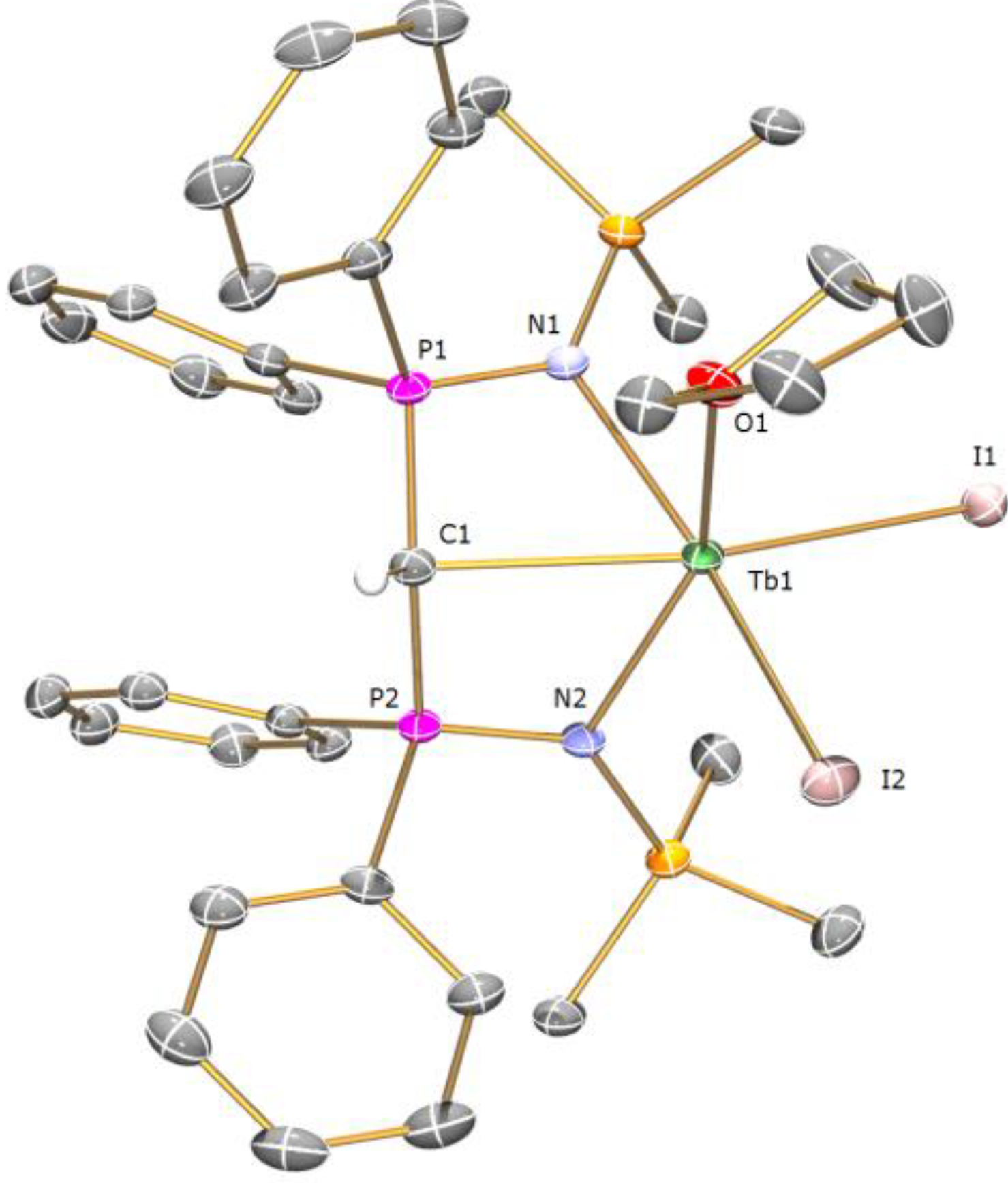
Preparation of [Tb(BIPMTMSH)(I)2(THF)] (1c): THF (40 mL) was added to a precooled (−78 °C) mixture of [Tb(I)3(THF)3.5] (0.79 g, 1.00 mmol) and [Cs(BIPMTMSH)] (0.69 g, 1.00 mmol) and the resulting mixture was allowed to warm to room temperature with stirring over 18 h. The mixture was filtered to remove CsI and the resulting solution reduced in volume to ca. 5 mL. Cooling of the solution to 4 °C afforded 1c OC4H8 as yellow crystals. Yield 0.38 g, 34%. Anal. Calcd for C39H55I2N2O2P2Si2Tb: C, 42.02; H, 4.97; N, 2.51. Found: C, 41.92; H, 4.88; N, 2.62. FTIR ν/cm−1 (Nujol): 1437 (m), 1154 (m), 1114 (m), 1056 (m), 938 (w), 920 (w), 840 (m), 771 (m), 753 (w), 725 (w), 712 (w), 695 (w), 551 (m). µeff (Evans method, 298 K, C6D6): 9.46 µB.
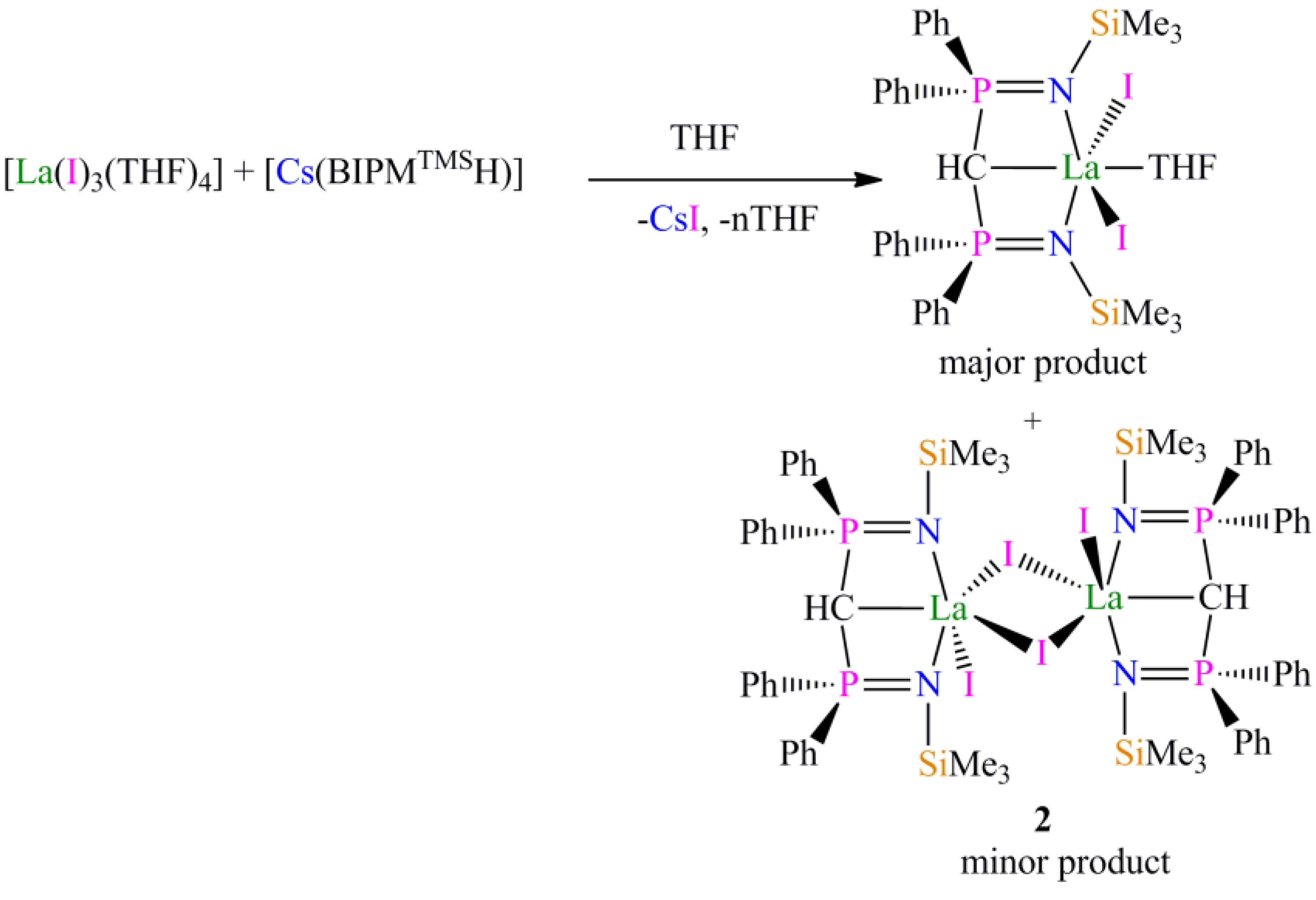
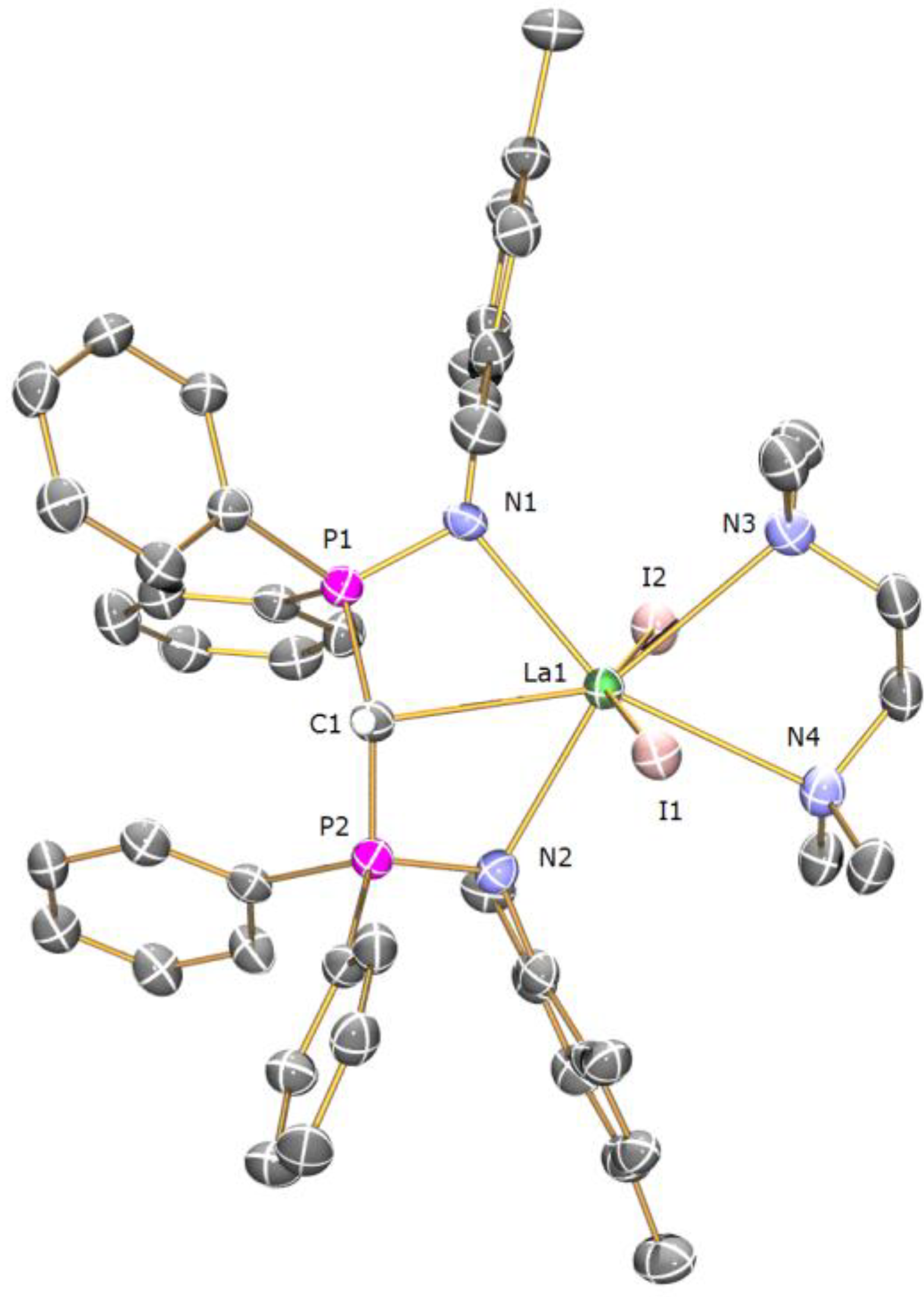
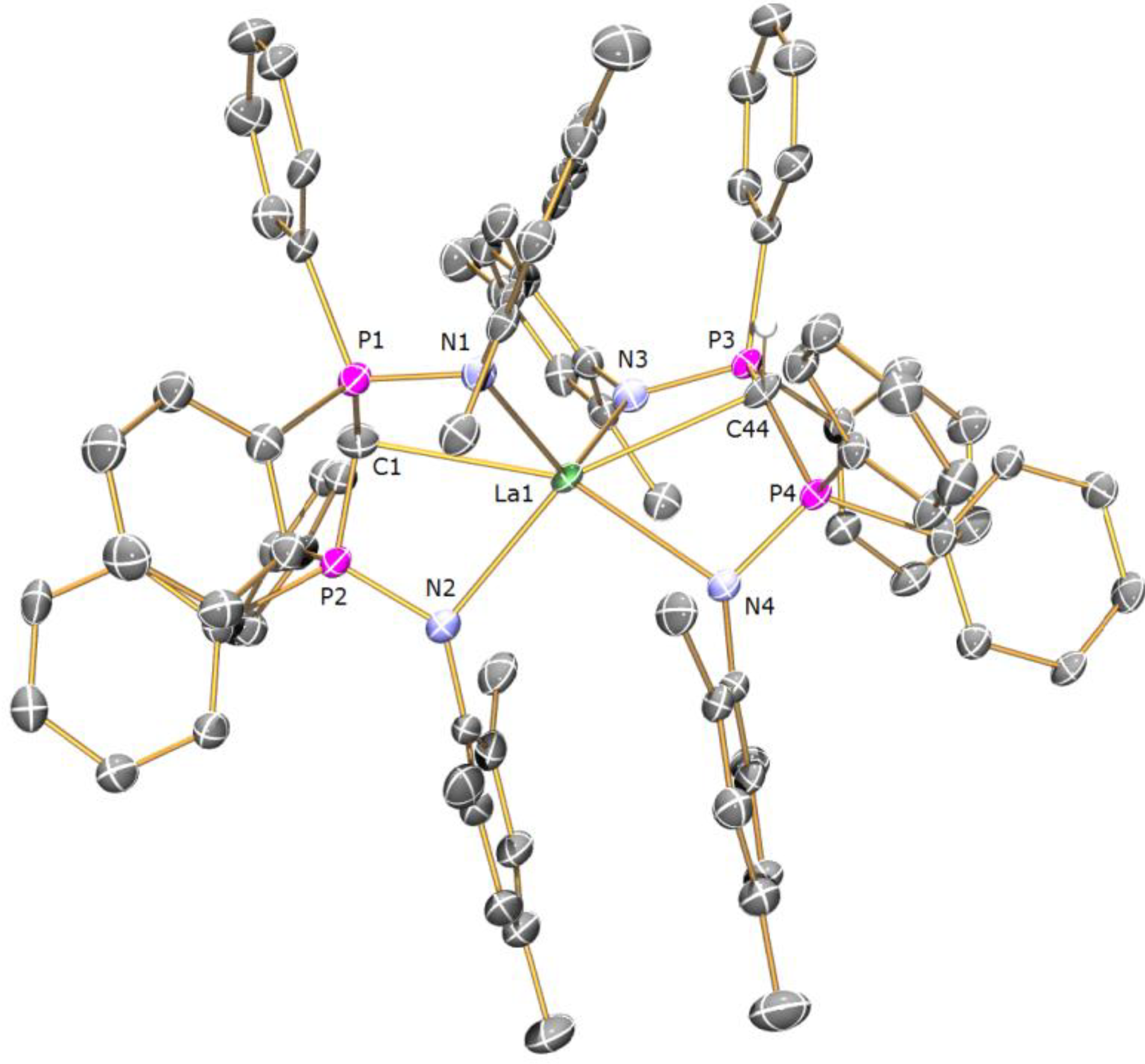
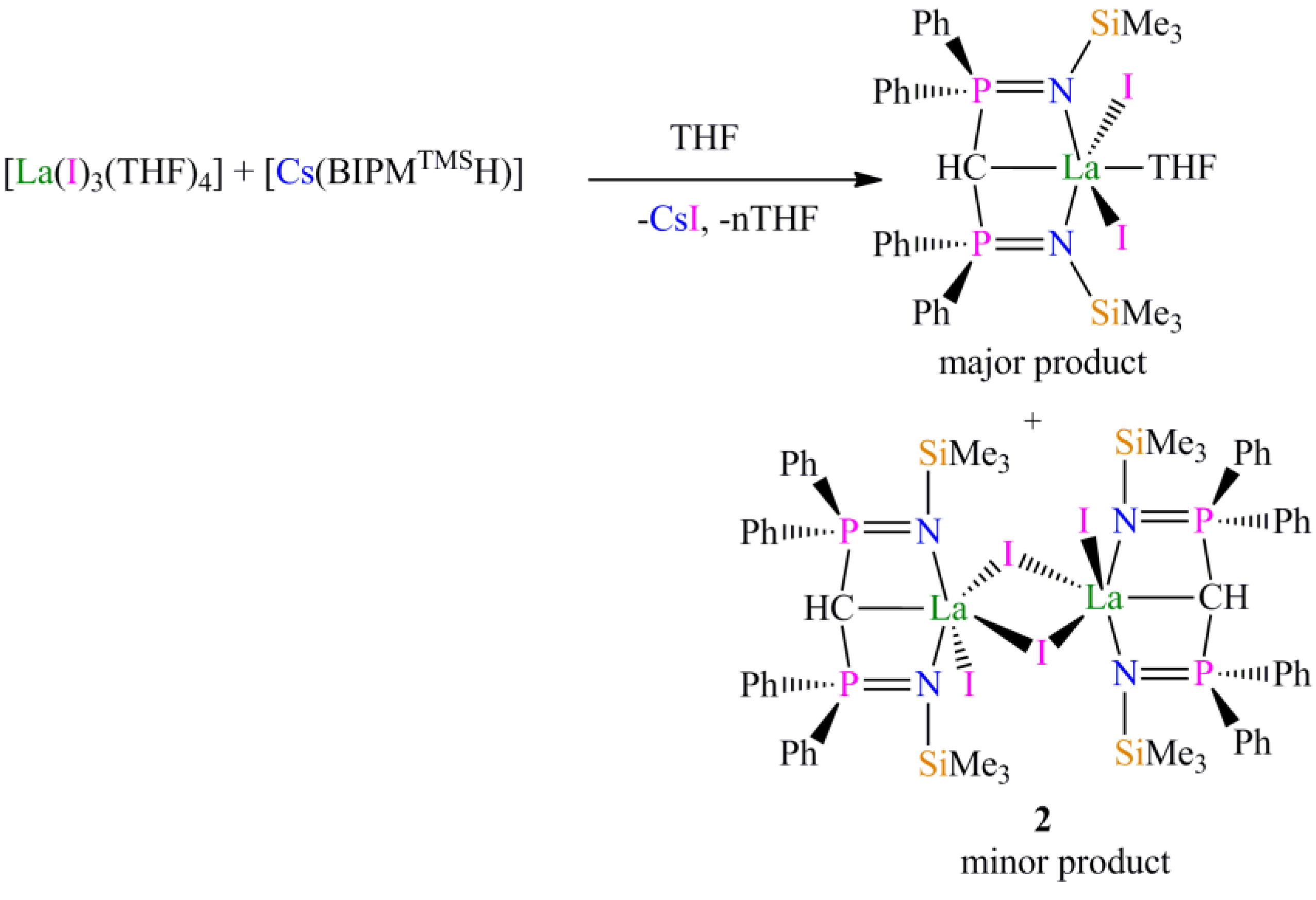
Preparation of [{La(BIPMTMSH)(I)(µ-I)}2] (2): During our studies we prepared many samples of [La(BIPM
TMSH)(I)
2(THF)], and on one occasion, following routine workup and recrystallisation, isolated a crop of crystals of
2 2C
7H
8 suitable for single crystal X-ray diffraction studies. Anal. Calcd for C
76H
94I
4La
2N
4P
4Si
4: C, 43.78; H, 4.54; N, 2.69. Found: C, 43.77; H, 4.49; N, 2.53. FTIR ν/cm
−1 (Nujol): 3054 (w), 1438 (m), 1415 (w), 1261 (m), 1214 (w), 1181 (w), 1116 (m), 1070 (m), 991 (m), 839 (m), 770 (m), 745 (m), 695 (m) 407 (m). The low solubility of
2 in arene solvents precluded NMR spectroscopy being performed in
d6-benzene or
d8-toluene, while dissolution in donor solvents such as
d8-THF afforded the solvated complex [La(BIPM
TMSH)(I)
2(
d8-THF)] [
13].
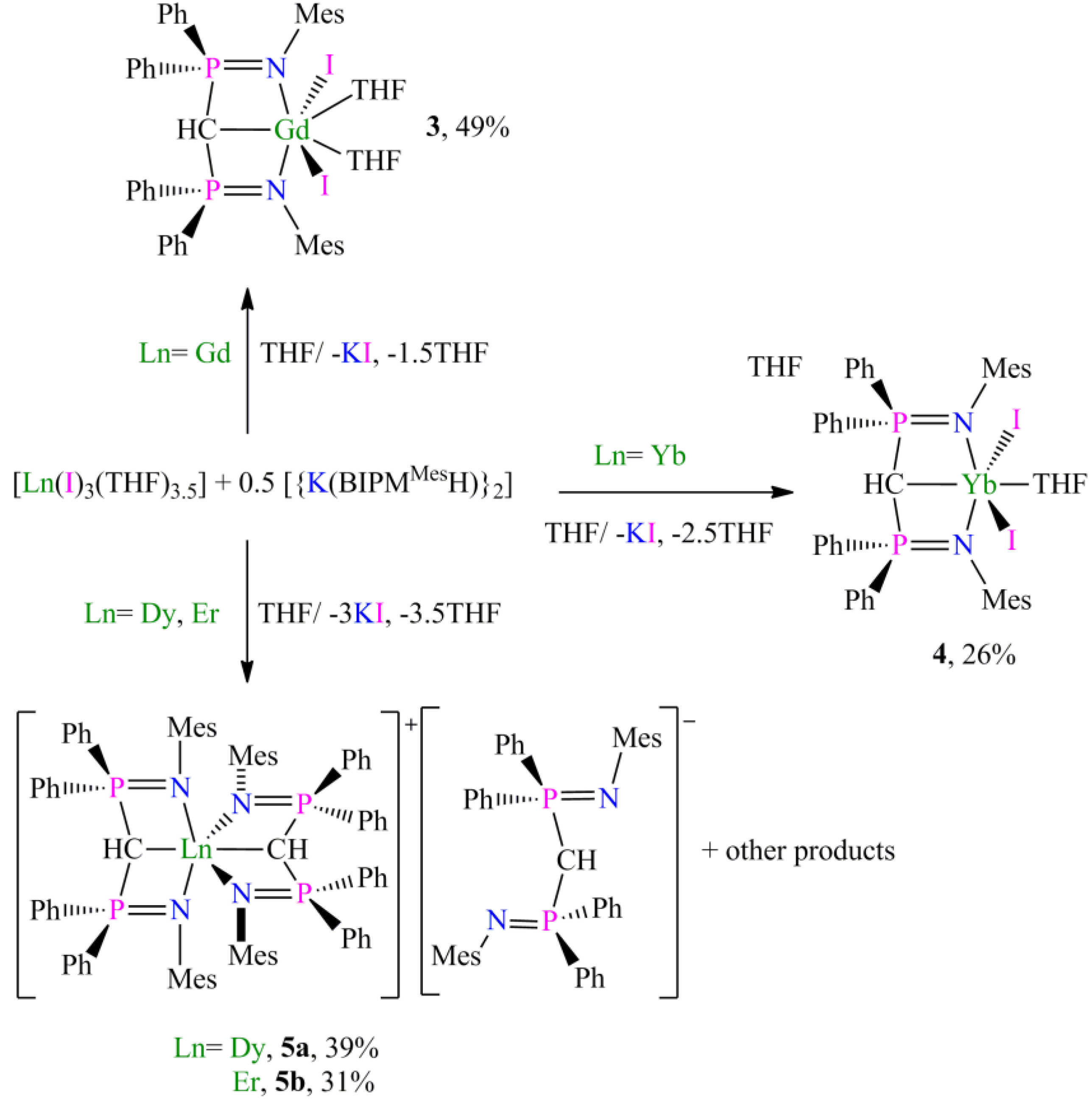
Preparation of [Gd(BIPMMesH)(I)2(THF)2] (3):THF (40 mL) was added to a precooled (−78 °C) mixture of [{K(BIPMMesH)}2] (0.69 g, 0.50 mmol) and [Gd(I)3(THF)3.5] (0.79 g, 1.00 mmol) and the resulting mixture was raised to room temperature with stirring over 18 h. The mixture was filtered to remove KI and all volatiles were removed in vacuo to afford a yellow solid. Recrystallisation from hot toluene (3 ml) afforded 3a 3C7H8 as yellow crystals on cooling to 4 °C. Yield 0.72 g, 49%. Anal. Calcd for C72H83GdI2N2O2P2: C, 58.37; H, 5.65; N, 1.89. Found: C, 58.17; H, 5.57; N, 1.74. FTIR ν/cm−1 (Nujol): 1435 (m), 1213 (m), 1155 (m), 940 (w), 854 (m), 741 (m), 695 (m), 663 (w), 590 (w), 560 (w), 535 (m). µeff (Evans method, 298 K, C6D6): 8.00 µB.
Preparation of [Yb(BIPMMesH)(I)2(THF)] (4):THF (40 mL) was added to a precooled (−78 °C) mixture of [Yb(I)3(THF)3.5] (1.21 g, 1.50 mmol) and [{K(BIPMMesH)}2] (1.05 g, 0.75 mmol), and the resulting orange mixture was raised to room temperature with stirring over 18 h. The mixture was filtered to remove KI and all volatiles removed in vacuo to afford an orange solid. Recrystallisation from hot toluene (15 ml) afforded 4 3C7H8 as orange crystals on cooling to −30 °C. Yield 0.55 g, 26%. Anal. Calcd for C47H51I2N2OP2Yb: C, 49.14; H, 4.48; N, 2.44. found: C, 49.34; H, 4.58; N, 2.10. FTIR ν/cm−1 (Nujol): 1588 (w), 1302 (w), 1261 (m), 1214 (m), 1156 (m), 1106 (s), 1014 (s), 859 (w), 792 (w), 771 (m), 733 (m), 588 (w), 572 (w), 536 (w). μeff (Evans method, 298 K, THF): 4.29 μB.
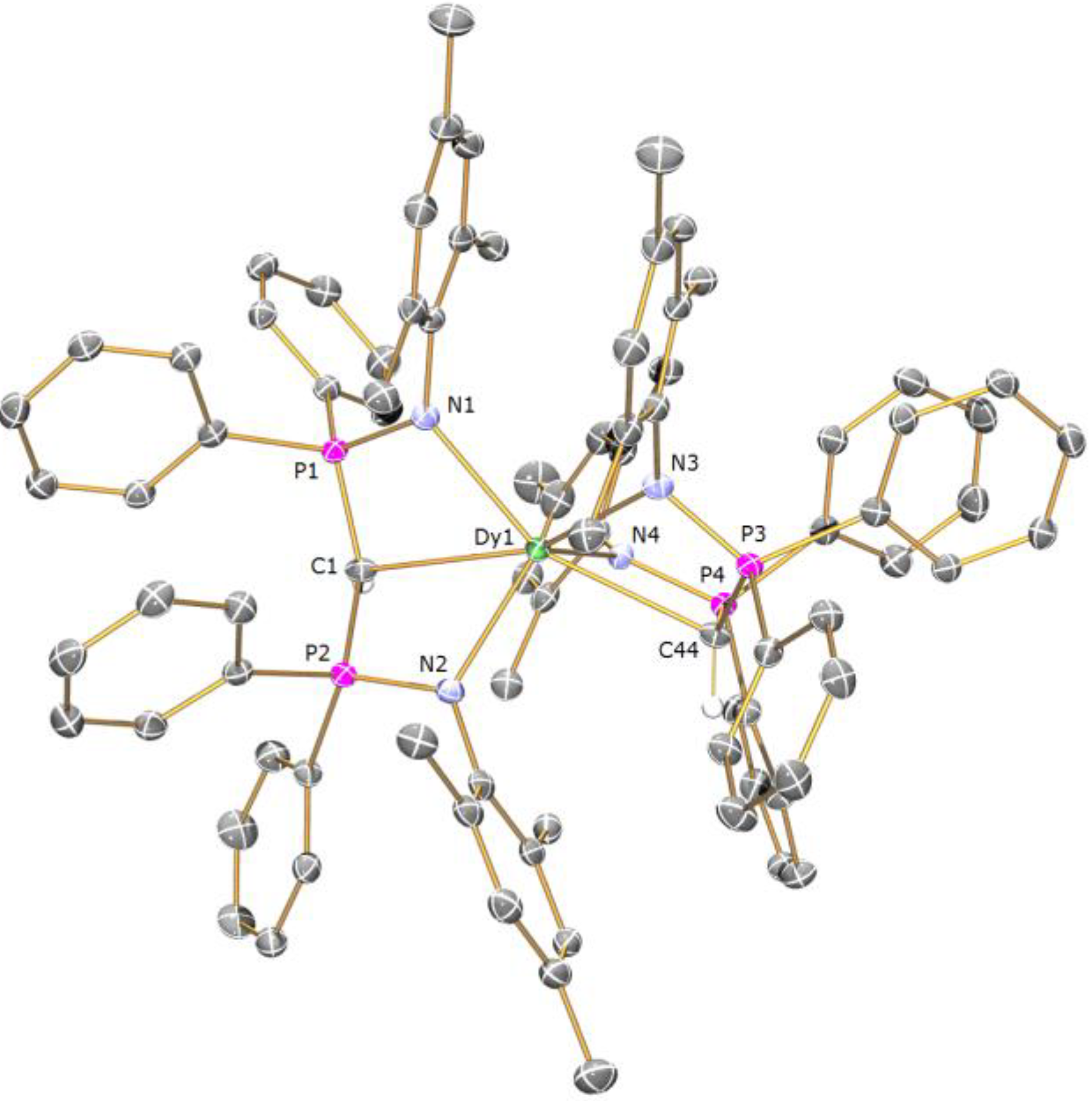
Preparation of [Dy(BIPMMesH)2][BIPMMesH] (5a): THF (20 mL) was added to a precooled (−78 °C) mixture of [{K(BIPM
MesH)}
2] (0.69 g, 0.50 mmol) and [Dy(I)
3(THF)
3.5] (0.80 g, 1.00 mmol) and the resulting mixture was allowed to raise to room temperature with stirring over 18 h. The mixture was filtered to remove KI and volatiles removed
in vacuo to yield a pale yellow solid. Recrystallisation from a hot toluene solution (3 ml) afforded
5a 4C
7H
8 as yellow crystals on cooling to ambient temperature. Yield 0.32 g, 39%. Anal. Calcd for C
157H
161DyN
6P
6: C, 76.03; H, 6.54; N, 3.39. Found: C, 69.03; H, 6.72; N, 3.58. Despite repeated attempts more satisfactory elemental analyses could not be obtained, with the low carbon value ascribed to carbide formation [
31] causing incomplete combustion during analysis. FTIR ν/cm
−1 (Nujol): 1604 (w), 1587 (w), 1573 (w), 1435 (m), 1332 (w), 1208 (m), 1154 (w), 972 (w), 940 (w), 853 (w) 739 (m), 694 (m). µ
eff (Evans method, 298 K, C
6D
6): 9.81 µ
B.
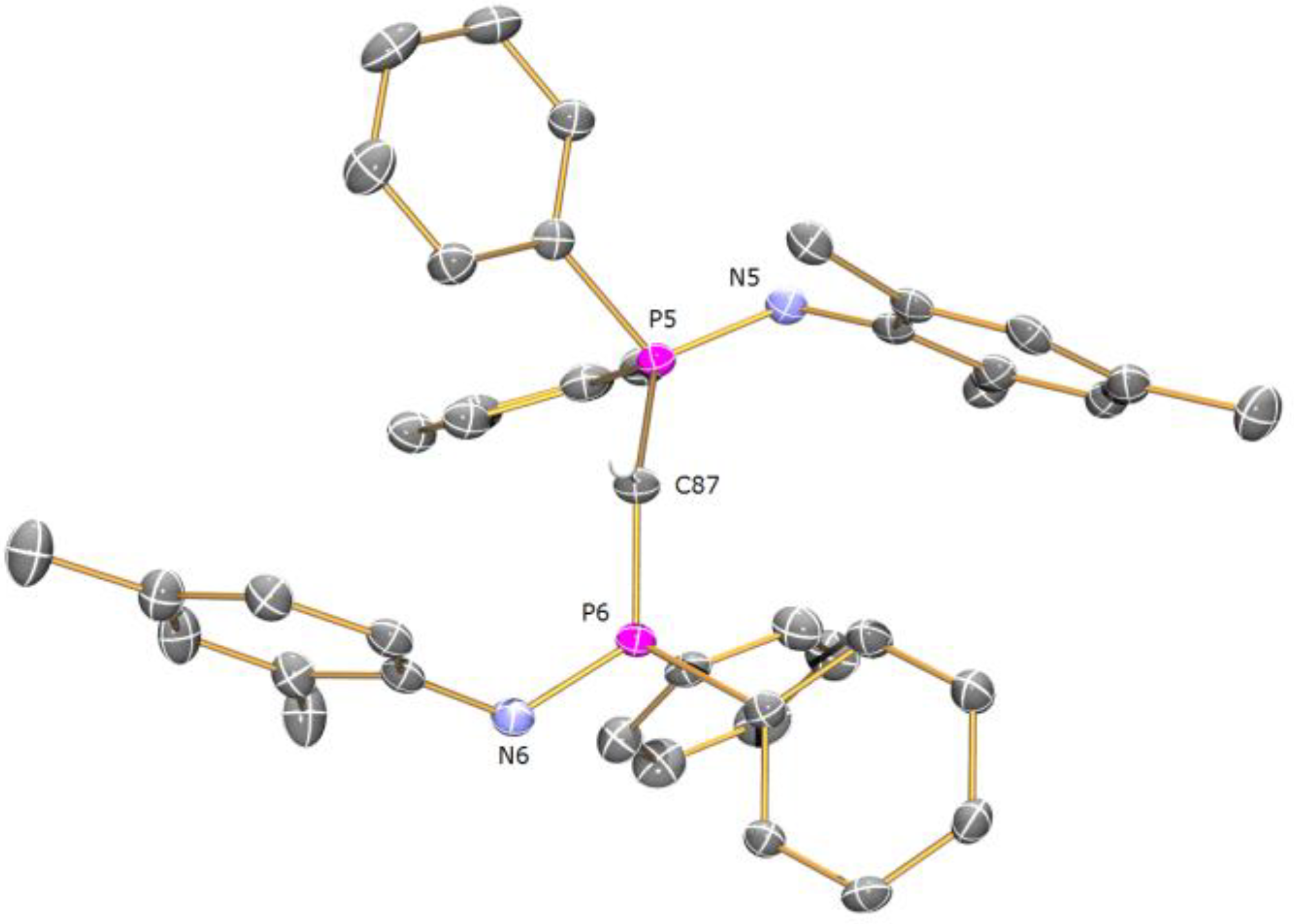
Preparation of [Er(BIPMMesH)2][BIPMMesH] (5b): THF (20 mL) was added to a precooled (−78 °C) mixture of [Er(I)3(THF)3.5] (1.20 g, 1.50 mmol) and [{K(BIPMMesH)}2] (1.05 g, 0.75 mmol), and the resultant beige mixture allowed to raise to room temperature with stirring over 72 h. The resulting suspension was filtered and volatiles were removed in vacuo to afford a pink solid. The solid was washed with hexanes and recrystallised from hot toluene (15 mL) to afford 5b 5C7H8 as pale yellow crystals on cooling to −30 °C. Yield 0.40 g, 31%. Anal. Calcd for C164H169ErN6P6: C, 76.43; H, 6.61; N, 3.26. Found: C, 76.32: H, 6.48: N, 3.37. FTIR ν/cm−1 (Nujol): 1261 (s), 1205 (w), 1098 (s), 1020 (s), 971 (w), 853 (w), 800 (s), 694 (m), 525 (m). μeff (Evans method, 298 K, THF): 10.25 μB.
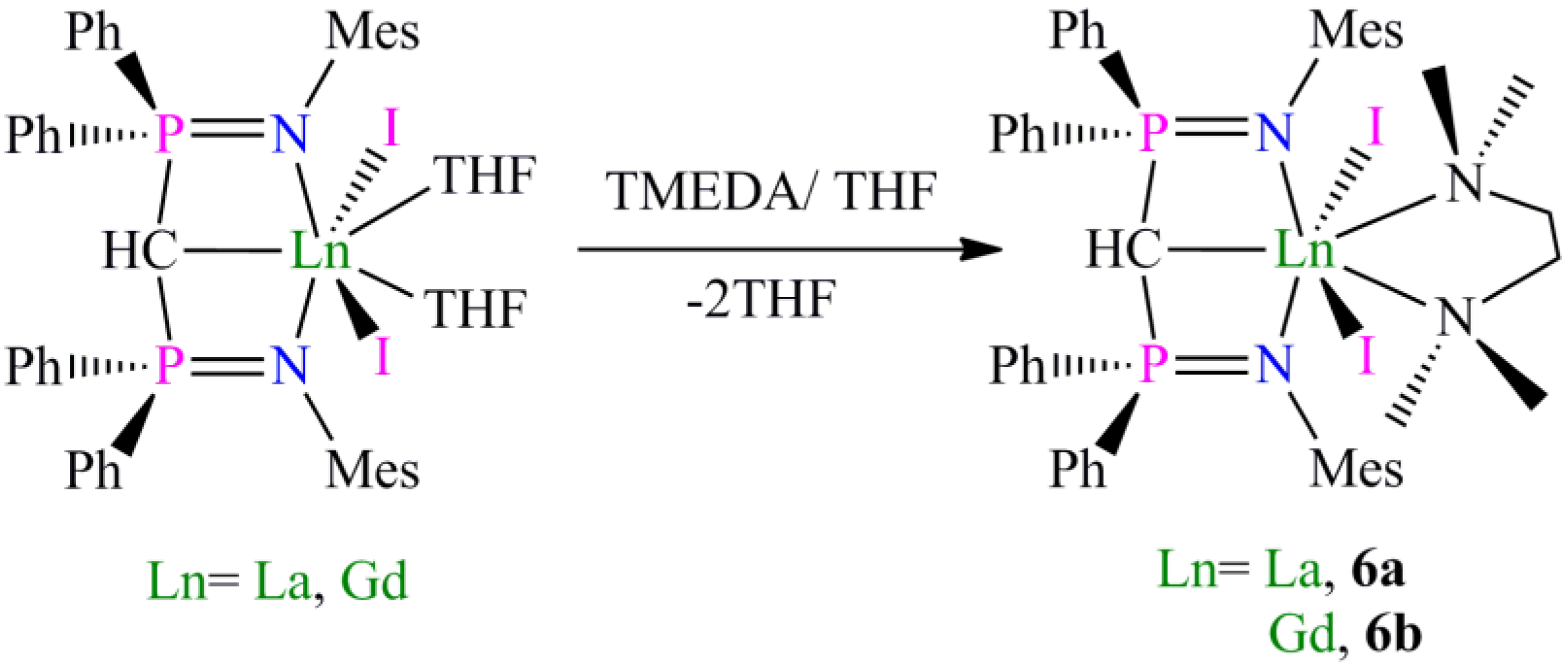
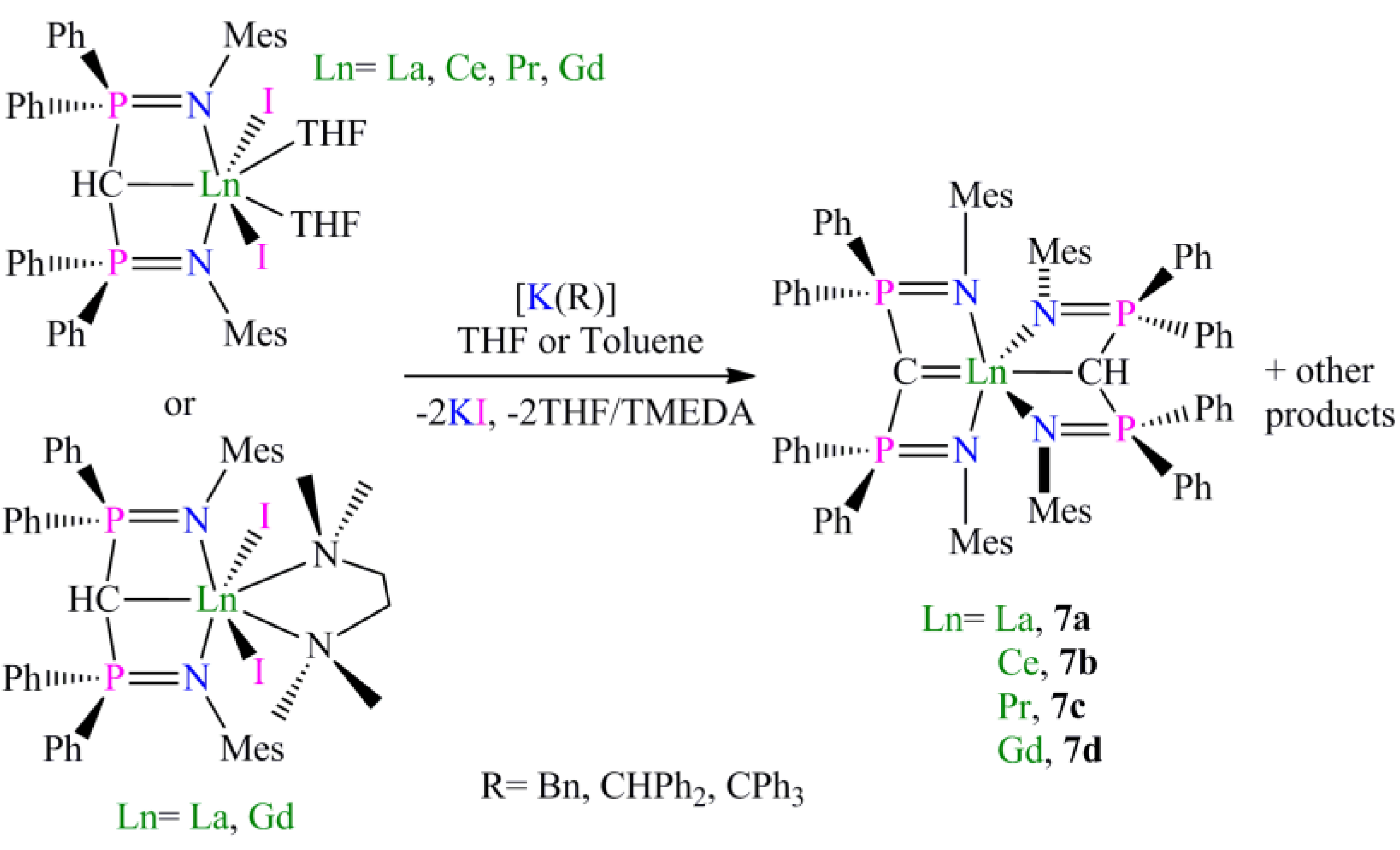
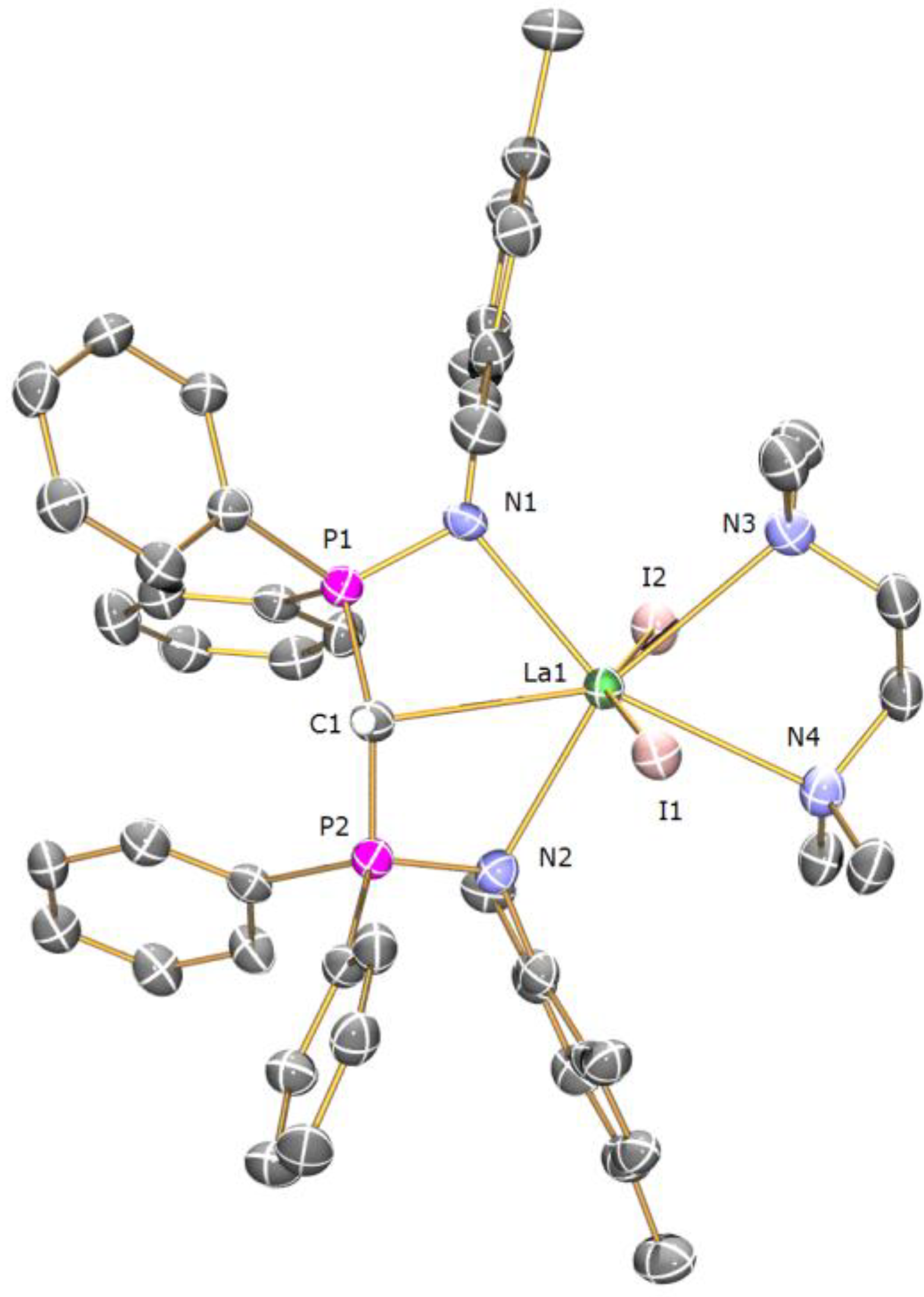
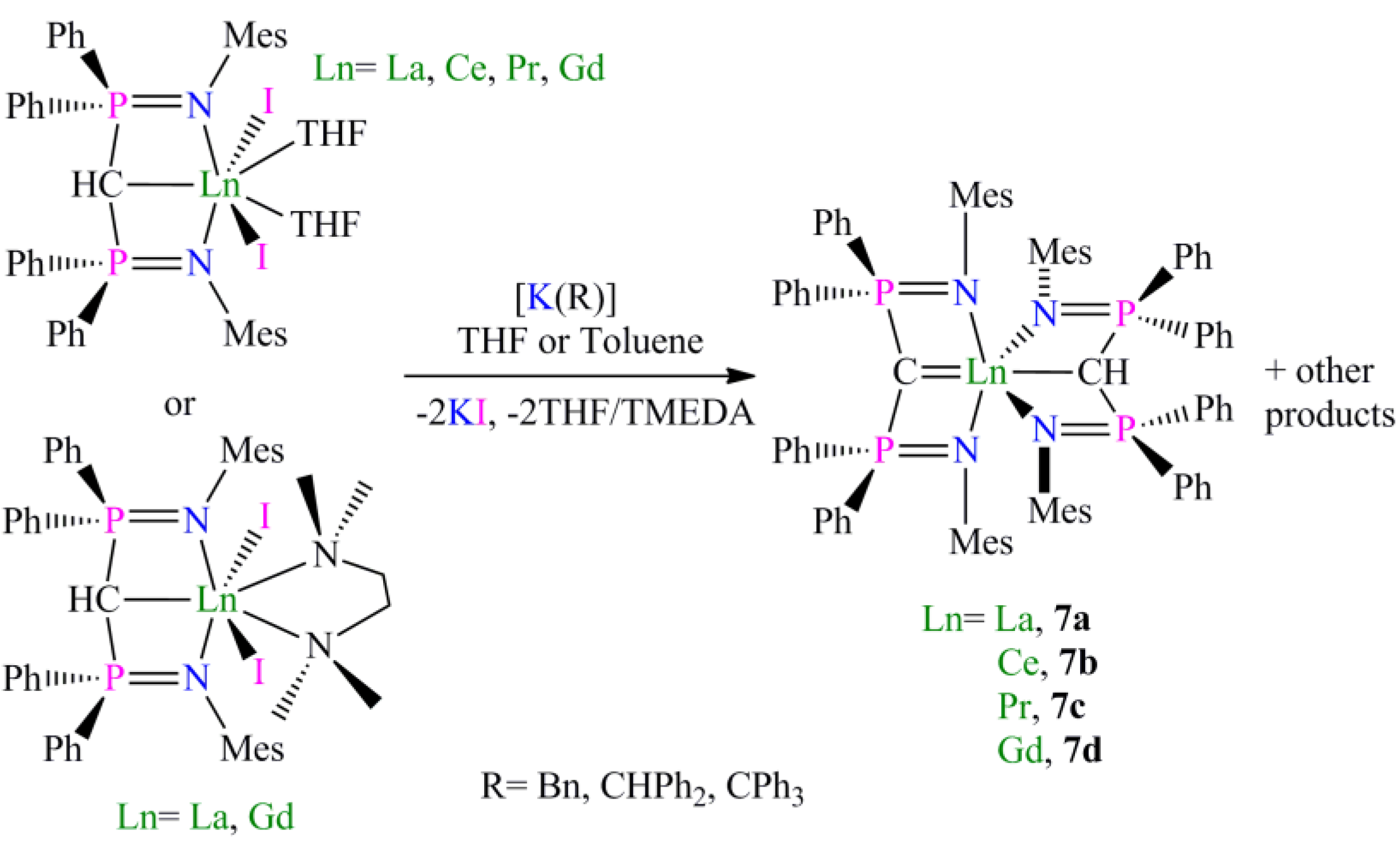
Preparation of [La(BIPMMesH)(I)2(TMEDA)] (6a) and [Gd(BIPMMesH)(I)2(TMEDA)] (6b): Complexes 6a and 6b were prepared by the dissolution of [Ln(BIPMMesH)(I)2(THF)] (Ln = La, Gd) in a THF/TMEDA solution followed by stirring for 18 h. In each case all volatiles were removed in vacuo to afford 6a (Ln = La) or 6b (Ln = Gd) as off-white and yellow solids, respectively, in quantitative yield. Recrystallisation of a small portion of each sample from toluene afforded crops of crystals of 6a 3C7H8 and 6b 2C7H8, respectively, suitable for single crystal X-ray diffraction studies. Data for 6a 3C7H8: Anal. Calcd for C70H83I2LaN4P2: C, 58.59; H, 5.83; N, 3.90. Found: C, 58.47; H, 5.93; N, 4.02. 1H NMR (298 K, C6D6): 1.65–2.40 (34 H, m, br, Mes-CH3, NCH2, NCH3), 3.91 (1 H, t, 2JPH = 8.8 Hz, HCP2), 6.55–7.15 (24 H, m, br, Ar-H). 13C{1H} NMR (298 K, C6D6): 8.44 (CH, t, JPC =138 Hz, HCP2), 20.51 (Mes-CH3), 20.65 (Mes-CH3), 21.18 (Mes-CH3), 21.44 (Mes-CH3), 49.84 (NCH3), 57.87 (NCH2), 125.44 (Ar-CH), 125.82 (Ar-C), 129.08 (Ar-CH), 129.81 (Ar-CH), 130.61 (Ar-CH), 131.11 (Ar-CH), 131.31 (Ar-CH), 132.17 (Ar-C), 132.58 (Ar-CH), 135.53 (Ar-C), 137.64 (Ar-C), 142.50 (Ar-C). 31P{1H} NMR (298 K, C6D6): 15.70 (s). FTIR ν/cm−1 (Nujol): 3053 (w), 1436 (m), 1215 (m), 1158 (m), 401 (m). As full spectroscopic data supported the formulation of 6a, full spectroscopic data was not obtained for paramagnetic 6b which was subsequently prepared and utilised in situ. During our investigations we attempted many preparations of [Ln(BIPMMes)(I)(THF)n] (Ln = La, Ce, Pr, Gd) utilising a range of methanide precursors including [Ln(BIPMMes)(I)2(THF)2] (Ln = La, Ce, Pr, Gd) and [Ln(BIPMMes)(I)2(TMEDA)] (Ln = La, 6a; Gd, 6b) and bases including [K(Bn)], [K{CHPh2}] and [K{C(Ph)3}]. In each case [Ln(BIPMMes)(I)(THF)n] was not isolated cleanly with mixtures of products observed in each case. The sole isolable products from these reactions were [Ln(BIPMMesH)(BIPMMes)] (Ln = La, 7a; Ce, 7b; Pr, 7c; Gd, 7d), with representative preparations of 7a and 7b given below.
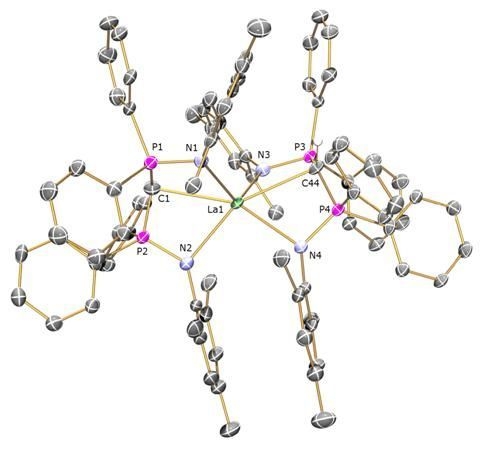
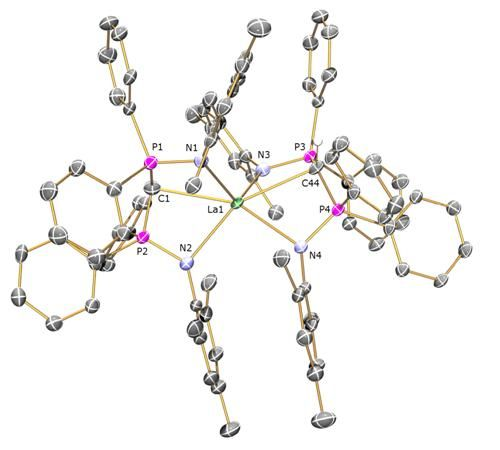
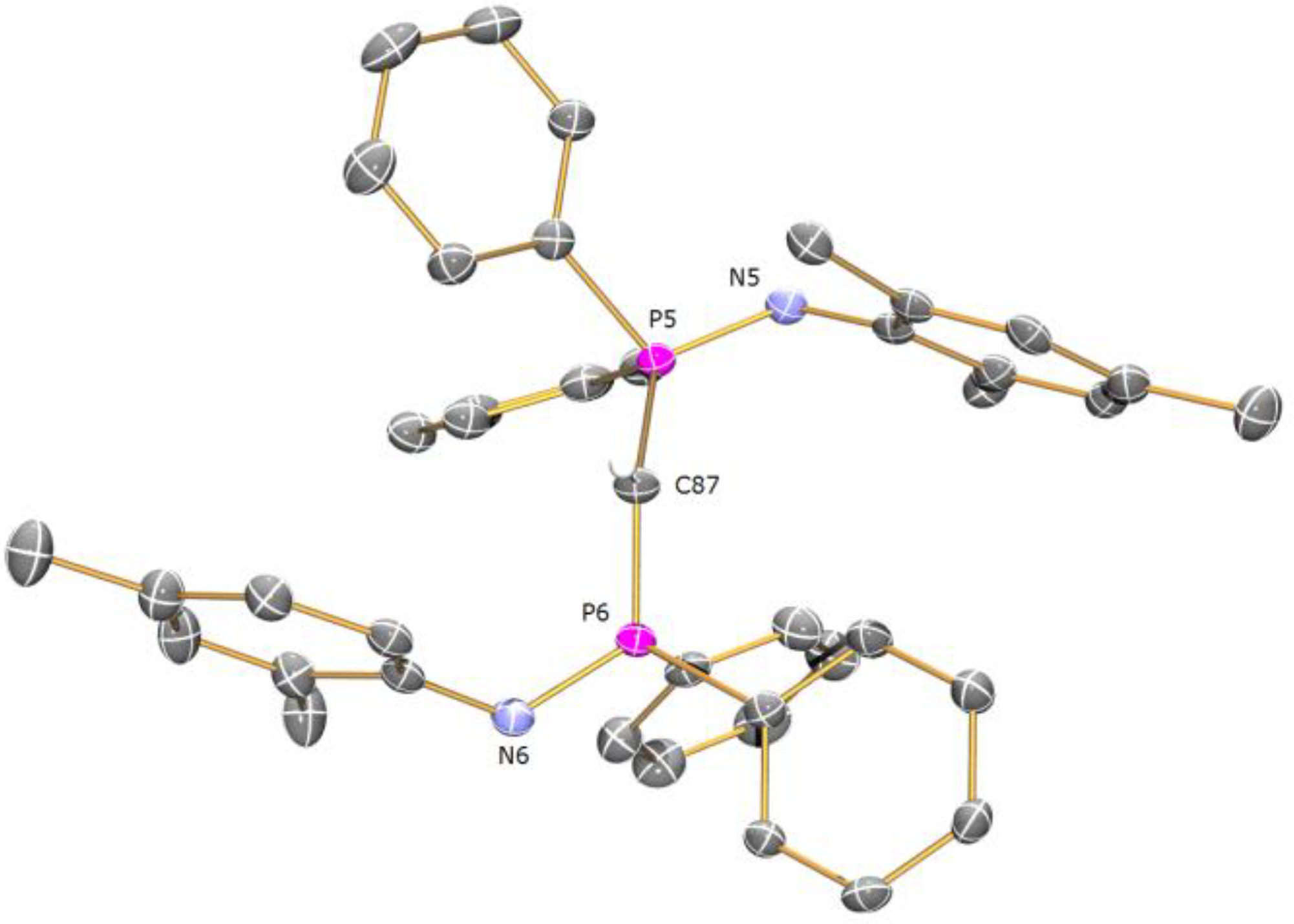
Preparation of [La(BIPMMesH)(BIPMMes)] (7a): THF (15 mL) was added to a pre-cooled (−78 °C) mixture of [La(BIPMMesH)(I)2(THF)] 3toluene (0.71 g, 0.49 mmol) and [K(CHPh2)] (0.10 g, 0.49 mmol). The resulting orange suspension was stirred at this temperature for 10 m and then raised to room temperature with stirring over 18 h. The resulting deep brown suspension was filtered to remove KI and all volatiles removed in vacuo to afford a brown solid. Recrystallisation from toluene (4 mL) afforded colourless crystals of 7a 3C7H8. Yield 70 mg, 8%. Anal. Calcd for C107H109LaN4P4: C, 74.99; H, 6.41; N, 3.27. Found: C, 74.77; H, 6.78; N, 3.17. 1H NMR (d6-benzene, 298 K): δ 2.10 (12 H, s, br, o-Mes-CH3), 2.26 (1 H, s, HCP2), 2.37 (12 H, s, br, o-Mes-CH3), 2.46 (6 H, s, p-Mes-CH3), 2.57 (6 H, s, p-Mes-CH3), 6.6-7.1 (28 H, m, br, Ar-CH), 7.37 (8 H, s, br, Ar-CH), 7.6–7.9 (12 H, m, br, Ar-CH). 13C{1H} NMR (d6-benzene, 298 K): δ 4.59 (t, JPC = 127 Hz, HCP2), 20.67 (Mes-CH3), 20.83 (Mes-CH3), 21.20 (Mes-CH3), 22.80 (Mes-CH3), 45.06 (s, CP2), 125.45 (Ar-C), 126.09 (Ar-C), 126.75 (Ar-CH), 128.09 (m, Ar-CH), 129.03 (Ar-CH), 129.64 (Ar-CH), 130.34 (Ar-CH), 130.57 (Ar-CH), 131.12 (Ar-CH), 132.17 (Ar-CH), 134.54 (Ar-C), 135.18 (br, Ar-C), 137.65 (Ar-C), 143.07 (Ar-C). 31P{1H} NMR (d6-benzene, 298 K): 2.88 (CP2), 10.51 (HCP2). FTIR ν/cm-1 (Nujol): 1589 (w), 1261 (s), 1098 (s), 1020 (s), 854 (w), 800 (s), 663 (m).
Preparation of [Ce(BIPMMesH)(BIPMMes)] (7b): THF (25 mL) was added to a pre-cooled (−78 °C) mixture of [Ce(BIPM
MesH)(I)
2(THF)] (3.00 g, 2.54 mmol) and [K(Bn)] (0.33 g, 2.54 mmol) and the resulting mixture allowed to warm to room temperature with stirring over 20 h. The resulting mixture was filtered and all volatiles removed
in vacuo to afford a brown solid. The solid was washed with hexanes and the resulting brown powder was recrystallised from toluene (5 mL) to afford
7b 3C
7H
8 as yellow crystals, as identified by unit cell check [
32]. Yield 0.10 g, 3%. On standing, the hexanes extract yielded
7b 2C
6H
14 as yellow crystals suitable for single crystal X-ray diffraction studies. Yield 0.14 g, 4%. Anal. Calcd for C
86H
85CeN
4P
4: C, 71.80; H, 5.96; N, 3.89. Found: C, 71.67; H, 5.88; N, 3.79. FTIR ν/cm-1 (Nujol): 1261 (m), 1212 (m), 1153 (m), 1099 (s), 1019 (s), 800 (s), 721 (w), 694 (w), 519 (w). μ
eff (Evans method, 298 K, THF): 1.88 μ
B.
X-ray Crystallography
Crystal data for compounds
1-7d are given in Table S2. Bond lengths and angles are listed in Table S1. Crystals were examined variously on a Bruker APEX CCD area detector diffractometer using graphite-monochromated MoKα radiation (λ = 0.71073 Å), or on an Oxford Diffraction SuperNova Atlas CCD diffractometer using mirror-monochromated CuKα radiation (λ = 1.5418 Å). Intensities were integrated from data recorded on 0.3 (APEX) or 1° (SuperNova) frames by
ω rotation. Cell parameters were refined from the observed positions of all strong reflections in each data set. Semi-empirical absorption correction based on symmetry-equivalent and repeat reflections (APEX) or Gaussian grid face-indexed absorption correction with a beam profile correction (Supernova), were applied. The structures were solved variously by direct and heavy atom methods and were refined by full-matrix least-squares on all unique
F2 values, with anisotropic displacement parameters for all non-hydrogen atoms, and with constrained riding hydrogen geometries;
Uiso(H) was set at 1.2 (1.5 for methyl groups) times
Ueq of the parent atom. The largest features in final difference syntheses were close to heavy atoms and were of no chemical significance. The data-set obtained for
4 is of low quality, and while the connectivity is clear, no assessment could be made of the geometric parameters, and despite exhaustive attempts, a better data-set could not be obtained. Highly disordered solvent molecules of crystallisation in
4 and
7a–
d could not be modelled and were treated with the Platon SQUEEZE procedure [
33]. Programs were Bruker AXS SMART [
34] and CrysAlisPro (control) [
35], Bruker AXS SAINT [
34] and CrysAlisPro (integration) [
35], and SHELXTL [
36] and OLEX2 [
37] were employed for structure solution and refinement and for molecular graphics. Crystal data have been deposited with the Cambridge Structural Database CCDC numbers 970500-970513.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}