
Cancer Vaccines in Ovarian Cancer: How Can We Improve?


Abstract
:
1. Rationale for Immunotherapy in Ovarian Cancer
2. Therapeutic Vaccines in Ovarian Cancer
2.1. Cell-Based Vaccines
2.2. Peptide/Protein-Based Vaccines
2.3. Genetic Vaccines
2.4. Epigenetic Vaccines
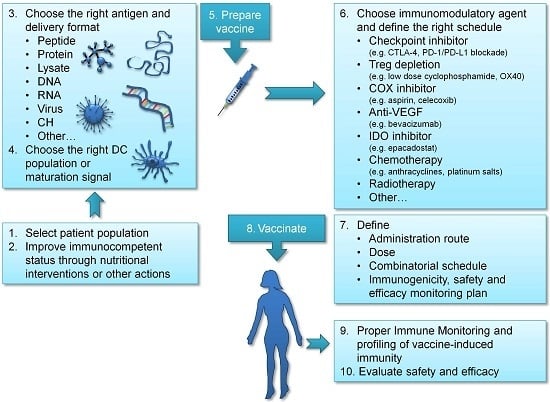
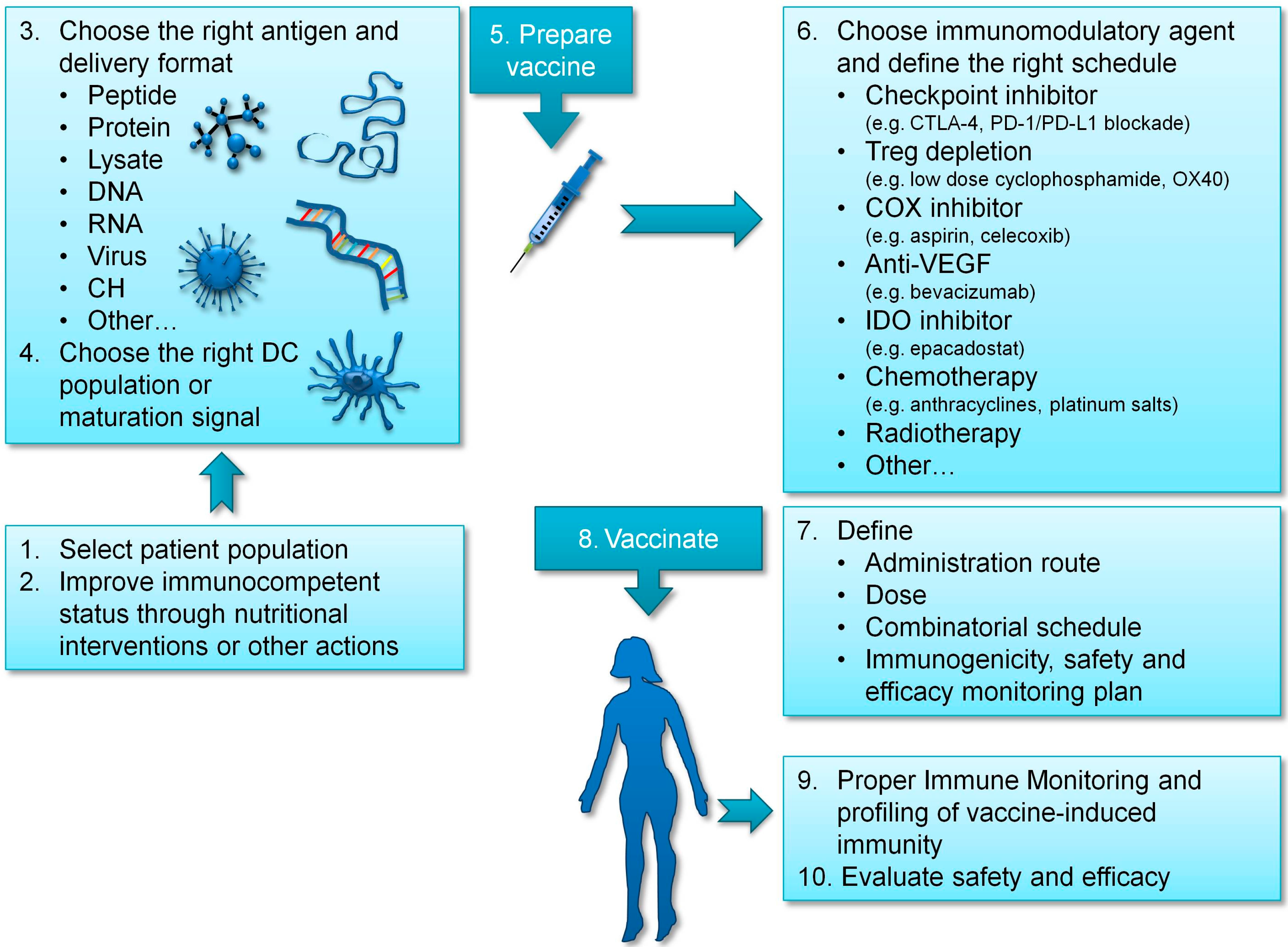
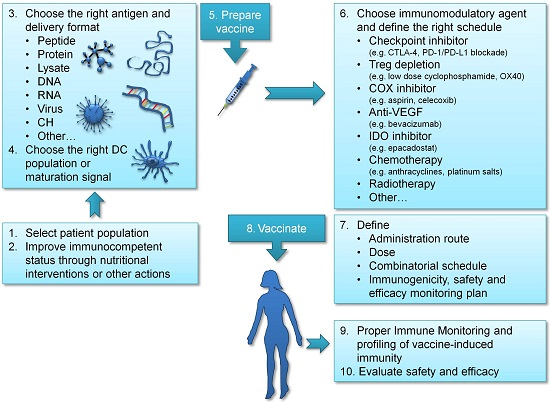
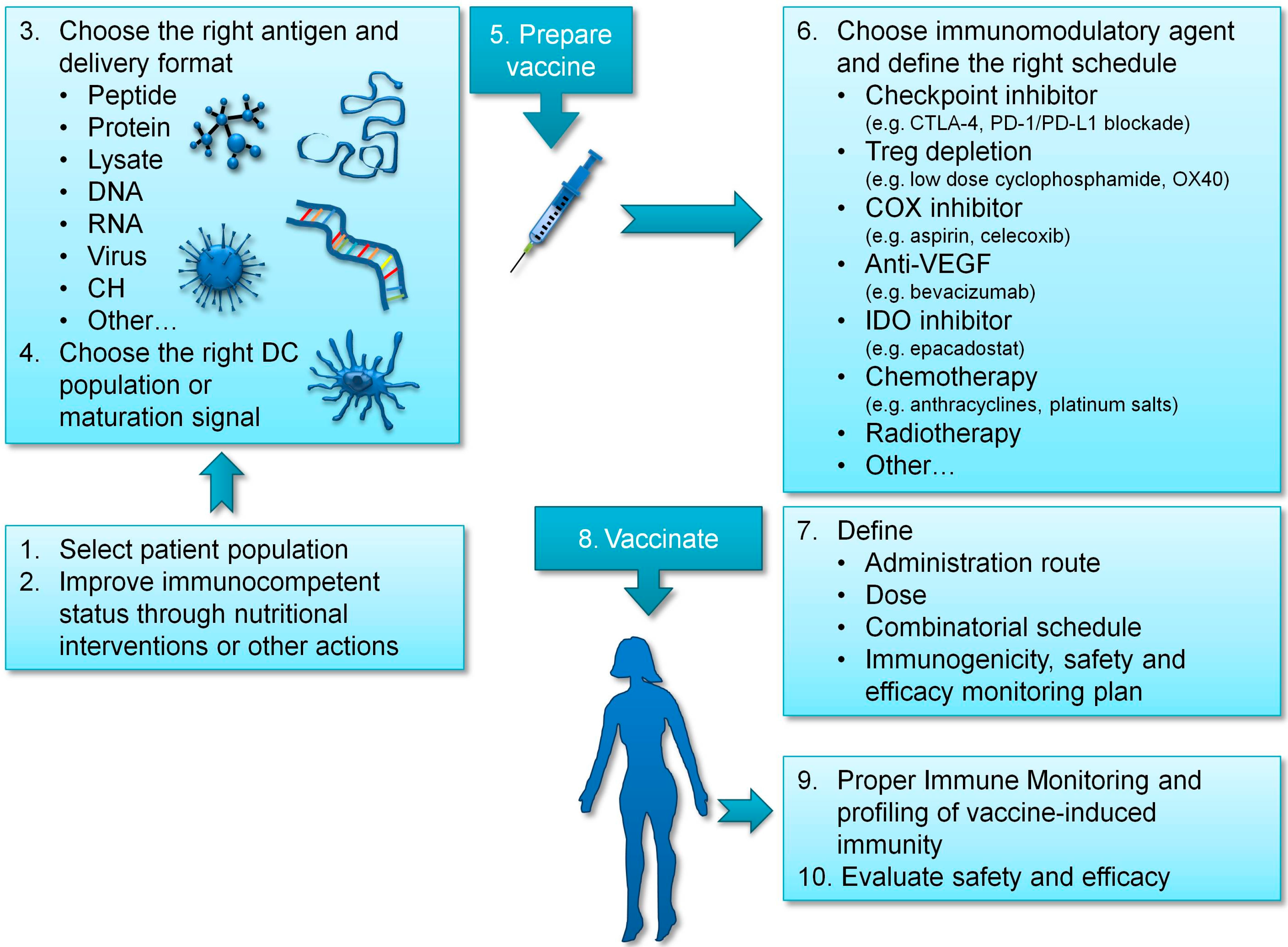
3. Improving Vaccination Strategies in Ovarian Cancer
3.1. Choosing the Right Antigen
3.2. Providing DC Maturation Signals to Enhance T Cell Activation
3.3. Targeting the Right DC
3.4. Improving the Immunocompetent Status of Vaccinated Patients
4. Immunomodulatory and Combinatorial Strategies in Ovarian Cancer
5. Concluding Remarks
Conflicts of Interest
Abbreviations
| BC | breast cancer |
| CEA | carcinoembryonic antigen |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic T-lymphocyte-associated antigen 4 |
| DC | dendritic cells |
| EOC | epithelial ovarian cancer |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| i.p. | intraperitoneal |
| IDO1 | indoleamine 2,3-dioxygenase |
| IFN | interferon |
| KLH | keyhole limpet hemocyanin |
| LC | lung cancer |
| MDSC | myeloid derived suppressor cells |
| MEL | melanoma |
| MES | mesothelioma |
| MHC | major histocompatibility complex |
| moAb | monoclonal antibody |
| mOC | metastatic ovarian cancer |
| NED | no evidence of disease |
| NSCLC | non-small cell lung cancer |
| OC | ovarian cancer |
| OCDC | DC loaded with oxidized tumor lysate |
| PCRC | pancreatic cancer |
| PC | prostate cancer |
| PD-(L)1 | programmed death-(ligand)1 |
| PGE | prostaglandine E |
| PPV | personalized peptide vaccine |
| ROC | recurrent ovarian cancer |
| SAR | sarcoma |
| shRNA | short hairpin RNA |
| TAA | tumor-associated antigen |
| TGF | transforming growth factor |
| TIL | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| Tregs | regulatory T cells |
| VEGF(R) | vascular endothelial growth factor (receptor) |
References
- Ovarian Cancer: Statistics. Available online: http://www.cancer.net/cancer-types/ovarian-cancer/statistics (accessed on 22 January 2016).
- IARC. GLOBOCAN 2012. Incidence, Mortality and Prevalence Worldwide (Ovary). Available online: http://globocan.iarc.fr/old/burden.asp?selection_pop=224900&Text-p=World&selection_cancer=22182&Text-c=Ovary&pYear=8&type=0&window=1&submit=%C2%A0Execute (accessed 18 February 2016).
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral t cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, K.; Chu, C.S.; Woo, E.Y.; Rivers, P.M.; Toll, A.J.; Hudson, B.; Maus, M.V.; Riley, J.L.; Choi, Y.; Coukos, G.; et al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin. Cancer Res. 2003, 9, 1517–1527. [Google Scholar] [PubMed]
- Goodell, V.; Salazar, L.G.; Urban, N.; Drescher, C.W.; Gray, H.; Swensen, R.E.; McIntosh, M.W.; Disis, M.L. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J. Clin. Oncol. 2006, 24, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Bellone, S.; Roman, J.J.; Smith, C.V.; Pecorelli, S.; Radominska-Pandya, A.; Cannon, M.J.; Parham, G.P. Phenotypic and functional analysis of tumor-infiltrating lymphocytes compared with tumor-associated lymphocytes from ascitic fluid and peripheral blood lymphocytes in patients with advanced ovarian cancer. Gynecol. Obstet. Investig. 2001, 51, 254–261. [Google Scholar] [CrossRef]
- Gnjatic, S.; Ritter, E.; Buchler, M.W.; Giese, N.A.; Brors, B.; Frei, C.; Murray, A.; Halama, N.; Zornig, I.; Chen, Y.T.; et al. Seromic profiling of ovarian and pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 5088–5093. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory t cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C. The expression of the regulatory T cell-specific forkhead box transcription factor foxp3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Inaba, T.; Ino, K.; Kajiyama, H.; Yamamoto, E.; Shibata, K.; Nawa, A.; Nagasaka, T.; Akimoto, H.; Takikawa, O.; Kikkawa, F. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol. Oncol. 2009, 115, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Nikaido, T.; Ochiai, K.; Takakura, S.; Saito, M.; Aoki, Y.; Ishii, N.; Yanaihara, N.; Yamada, K.; Takikawa, O.; et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin. Cancer Res. 2005, 11, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Mechanism of action of immunotherapy. Semin. Oncol. 2014, 41 (Suppl. 5), S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Hoption Cann, S.A.; van Netten, J.P.; van Netten, C. Dr william coley and tumour regression: A place in history or in the future. Postgrad. Med. J. 2003, 79, 672–680. [Google Scholar] [PubMed]
- D’Amelio, E.; Salemi, S.; D’Amelio, R. Anti-infectious human vaccination in historical perspective. Int. Rev. Immunol. 2015, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Lowy, D.R. Virus infection and human cancer: An overview. Recent Res. Cancer 2014, 193, 1–10. [Google Scholar]
- Szarewski, A.; Poppe, W.A.; Skinner, S.R.; Wheeler, C.M.; Paavonen, J.; Naud, P.; Salmeron, J.; Chow, S.N.; Apter, D.; Kitchener, H.; et al. Efficacy of the human papillomavirus (hpv)-16/18 as04-adjuvanted vaccine in women aged 15-25 years with and without serological evidence of previous exposure to hpv-16/18. Int. J. Cancer 2012, 131, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines: Past, present and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [PubMed]
- Seya, T.; Shime, H.; Takeda, Y.; Tatematsu, M.; Takashima, K.; Matsumoto, M. Adjuvant for vaccine immunotherapy of cancer - focusing on toll-like receptor 2 and 3 agonists for safely enhancing antitumor immunity. Cancer Sci. 2015, 106, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Coukos, G.; Kandalaft, L.E. Whole tumor antigen vaccines: Where are we? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Guidance Development Review Committee; Working Group for Clinical Studies of Cancer Immunotherapy; Working Group for Effector Cell Therapy; Working Group for CMC/Non-clinical Studies; Working Group for Cancer Vaccines and Adjuvants; Working Group for Anti-immune Checkpoint Therapy and Comprehensive Cancer Immunotherapy; Biostatistics Subcommittee. 2015 guidance on cancer immunotherapy development in early-phase clinical studies. Cancer Sci. 2015, 106, 1761–1771. [Google Scholar]
- Bapsy, P.P.; Sharan, B.; Kumar, C.; Das, R.P.; Rangarajan, B.; Jain, M.; Suresh Attili, V.S.; Subramanian, S.; Aggarwal, S.; Srivastava, M.; et al. Open-label, multi-center, non-randomized, single-arm study to evaluate the safety and efficacy of dendritic cell immunotherapy in patients with refractory solid malignancies, on supportive care. Cytotherapy 2014, 16, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Nisenbaum, H.L.; Levine, B.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J.; Chiang, C.L.; Tanyi, J.; Kim, S.; Bosch, M.; Montone, K.; Mick, R.; Levine, B.L.; Torigian, D.A.; et al. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated t cells in recurrent ovarian cancer. Oncoimmunology 2013, 2, e22664. [Google Scholar] [CrossRef] [PubMed]
- Hernando, J.; Park, T.-W.; Kübler, K.; Offergeld, R.; Schlebusch, H.; Bauknecht, T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: Clinical and immunological evaluation of a phase i trial. Cancer Immunol. Immunother. 2002, 51, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Pietersz, G.A.; Apostolopoulos, V.; Vaughan, H.; Karanikas, V.; et al. Mannan-MUC1–pulsed dendritic cell immunotherapy: A phase I trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Peethambaram, P.P.; Melisko, M.E.; Rinn, K.J.; Alberts, S.R.; Provost, N.M.; Jones, L.A.; Sims, R.B.; Lin, L.R.C.; Frohlich, M.W.; Park, J.W. A phase I trial of immunotherapy with lapuleucel-T (APC8024) in patients with refractory metastatic tumors that express her-2/neu. Clin. Cancer Res. 2009, 15, 5937–5944. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Wirths, S.; Stuhler, G.; Reichardt, V.L.; Kanz, L.; Brugger, W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood 2000, 96, 3102–3108. [Google Scholar] [PubMed]
- Chu, C.; Boyer, J.; Schullery, D.; Gimotty, P.; Gamerman, V.; Bender, J.; Levine, B.; Coukos, G.; Rubin, S.; Morgan, M.; et al. Phase I/II randomized trial of dendritic cell vaccination with or without cyclophosphamide for consolidation therapy of advanced ovarian cancer in first or second remission. Cancer Immunol. Immunother. 2012, 61, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Chiba, A.; Izawa, H.; Yanagida, E.; Okamoto, M.; Shimodaira, S.; Yonemitsu, Y.; Shibamoto, Y.; Suzuki, N.; Nagaya, M. The feasibility and clinical effects of dendritic cell-based immunotherapy targeting synthesized peptides for recurrent ovarian cancer. J. Ovarian Res. 2014, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAifurin/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, N.; Mochizuki, K.; Harada, M.; Sukehiro, A.; Kawano, K.; Yamada, A.; Ushijima, K.; Sugiyama, T.; Nishida, T.; Yamana, H.; et al. Vaccination with predesignated or evidence-based peptides for patients with recurrent gynecologic cancers. J. Immunother. 2004, 27, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Secord, A.A.; Blackwell, K.; Hobeika, A.C.; Sinnathamby, G.; Osada, T.; Hafner, J.; Philip, M.; Clay, T.M.; Lyerly, H.K.; et al. MHC class I–presented tumor antigens identified in ovarian cancer by immunoproteomic analysis are targets for T-cell responses against breast and ovarian cancer. Clin. Cancer Res. 2011, 17, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Chianese-Bullock, K.A.; Irvin, W.P., Jr.; Petroni, G.R.; Murphy, C.; Smolkin, M.; Olson, W.C.; Coleman, E.; Boerner, S.A.; Nail, C.J.; Neese, P.Y.; et al. A multipeptide vaccine is safe and elicits T-cell responses in participants with advanced stage ovarian cancer. J. Immunother. 2008, 31, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Schiffman, K.; Cheever, M.A.; Disis, M.L. Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, p369–377, results in short-lived peptide-specific immunity. Clin. Cancer Res. 2002, 8, 1014–1018. [Google Scholar] [PubMed]
- Disis, M.L.; Gooley, T.A.; Rinn, K.; Davis, D.; Piepkorn, M.; Cheever, M.A.; Knutson, K.L.; Schiffman, K. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide–based vaccines. J. Clin. Oncol. 2002, 20, 2624–2632. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Rinn, K.; Knutson, K.L.; Davis, D.; Caron, D.; dela Rosa, C.; Schiffman, K. Flt3 ligand as a vaccine adjuvant in association with HER-2/neu peptide-based vaccines in patients with HER-2/neu–overexpressing cancers. Blood 2002, 99, 2845–2850. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Goodell, V.; Schiffman, K.; Knutson, K.L. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J. Clin. Immunol. 2004, 24, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Schiffman, K.; Guthrie, K.; Salazar, L.G.; Knutson, K.L.; Goodell, V.; dela Rosa, C.; Cheever, M.A. Effect of dose on immune response in patients vaccinated with an HER-2/neu intracellular domain protein—based vaccine. J. Clin. Oncol. 2004, 22, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Qian, F.; Matsuzaki, J.; Mhawech-Fauceglia, P.; Andrews, C.; Hoffman, E.W.; Pan, L.; Ritter, G.; Villella, J.; Thomas, B.; et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12837–12842. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.M.; Gnjatic, S.; Sabbatini, P.; Aghajanian, C.; Hensley, M.L.; Spriggs, D.R.; Iasonos, A.; Lee, H.; Dupont, B.; Pezzulli, S.; et al. Safety and immunogenicity study of NY-ESO-1b peptide and montanide ISA-51 vaccination of patients with epithelial ovarian cancer in high-risk first remission. Clin. Cancer Res. 2008, 14, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; Tsuji, T.; Ferran, L.; Ritter, E.; Sedrak, C.; Tuballes, K.; Jungbluth, A.A.; Ritter, G.; Aghajanian, C.; Bell-McGuinn, K.; et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 2012, 18, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Ashtar, E.; Czystowska, M.; Szajnik, M.E.; Wieckowski, E.; Bernstein, S.; Herrin, V.E.; Shams, M.A.; Steinberg, S.M.; Merino, M.; et al. A gynecologic oncology group phase ii trial of two p53 peptide vaccine approaches: Subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol. Immunother. 2012, 61, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Lambeck, A.J.A.; Gooden, M.J.M.; Hoogeboom, B.-N.; Wolf, R.; Hamming, I.E.; Hepkema, B.G.; Willemse, P.H.B.; Molmans, B.H.W.; Hollema, H.; et al. Immunization with a p53 synthetic long peptide vaccine induces p53-specific immune responses in ovarian cancer patients, a phase II trial. Int. J. Cancer 2009, 125, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Vermeij, R.; Hoogeboom, B.-N.; Schulze, U.R.; Wolf, R.; Hamming, I.E.; van der Zee, A.G.; Melief, K.J.; van der Burg, S.H.; Daemen, T.; et al. Long-term clinical and immunological effects of p53-slp® vaccine in patients with ovarian cancer. Int. J. Cancer 2012, 130, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, R.; Leffers, N.; Hoogeboom, B.-N.; Hamming, I.L.E.; Wolf, R.; Reyners, A.K.L.; Molmans, B.H.W.; Hollema, H.; Bart, J.; Drijfhout, J.W.; et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int. J. Cancer 2012, 131, E670–E680. [Google Scholar] [CrossRef] [PubMed]
- Kawano, K.; Tsuda, N.; Matsueda, S.; Sasada, T.; Watanabe, N.; Ushijima, K.; Yamaguchi, T.; Yokomine, M.; Itoh, K.; Yamada, A.; et al. Feasibility study of personalized peptide vaccination for recurrent ovarian cancer patients. Immunopharmacol. Immunotoxicol. 2014, 36, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.S.; Vadhan-Raj, S.; Butts, C.; Savary, C.; Melichar, B.; Verschraegen, C.; Kavanagh, J.J.; Hicks, M.E.; Levy, L.B.; Folloder, J.K.; et al. Pilot study of Flt3 ligand comparing intraperitoneal with subcutaneous routes on hematologic and immunologic responses in patients with peritoneal carcinomatosis and mesotheliomas. Clin. Cancer Res. 2003, 9, 5228–5237. [Google Scholar] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.-Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. A pilot study to evaluate the safety and clinical outcomes of vaccination with recombinant CEA-MUC-1-TRICOM (PANVAC) poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Jäger, E.; Karbach, J.; Gnjatic, S.; Neumann, A.; Bender, A.; Valmori, D.; Ayyoub, M.; Ritter, E.; Ritter, G.; Jäger, D.; et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc. Natl. Acad. Sci. USA 2006, 103, 14453–14458. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Matsuzaki, J.; Karbach, J.; Neumann, A.; Mhawech-Fauceglia, P.; Miller, A.; Beck, A.; Morrison, C.D.; Ritter, G.; Godoy, H.; et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc. Natl. Acad. Sci. USA 2012, 109, 5797–5802. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, L.A.; Oparin, D.V.; Gooley, T.; Sandmaier, B.M. The role of cancer vaccines following autologous stem cell rescue in breast and ovarian cancer patients: Experience with the STn-KLH vaccine (theratope®). Clin. Breast Cancer 2003, 3 (Suppl. 4), S144–S151. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.J.; Kudryashov, V.; Ragupathi, G.; Danishefsky, S.J.; Livingston, P.O.; Bornmann, W.; Spassova, M.; Zatorski, A.; Spriggs, D.; Aghajanian, C.; et al. Immunization of ovarian cancer patients with a synthetic lewis(y)-protein conjugate vaccine: A phase I trial. Int. J. Cancer 2000, 87, 79–85. [Google Scholar] [CrossRef]
- EMBL-EBI. IPD-IMGT/HLA. 2016 [The IPD-IMGT/HLA Database allows you to retrieve information upon a specific HLA allele as named in the WHO Nomenclature Committee Reports]. Available online: http://www.ebi.ac.uk/ipd/imgt/hla/allele.html (accessed on 25 April 2016).
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Draube, A.; Klein-González, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed]
- Leonhartsberger, N.; Ramoner, R.; Falkensammer, C.; Rahm, A.; Gander, H.; Holtl, L.; Thurnher, M. Quality of life during dendritic cell vaccination against metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2012, 61, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautès-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [PubMed]
- Neller, M.A.; Lopez, J.A.; Schmidt, C.W. Antigens for cancer immunotherapy. Semin. Immunol. 2008, 20, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Fratesi, P.; Reese, D.M.; Strang, G.; Laus, R.; Peshwa, M.V.; Valone, F.H. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; van den Eeden, S.J.; Franken, K.L.; Melief, C.J.; van der Burg, S.H.; Offringa, R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Prac. 2012, 2012, 743193. [Google Scholar] [CrossRef] [PubMed]
- Tuefferd, M.; Couturier, J.; Penault-Llorca, F.; Vincent-Salomon, A.; Broët, P.; Guastalla, J.-P.; Allouache, D.; Combe, M.; Weber, B.; Pujade-Lauraine, E.; et al. HER2 status in ovarian carcinomas: A multicenter gineco study of 320 patients. PLoS ONE 2007, 2, e1138. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Ciliberto, G. Genetic cancer vaccines: Current status and perspectives. Exp. Opin. Biol. Ther. 2012, 12, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Kannagi, R.; Yin, J.; Miyazaki, K.; Izawa, M. Current relevance of incomplete synthesis and neo-synthesis for cancer-associated alteration of carbohydrate determinants—hakomori’s concepts revisited. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.I.; Toyota, M.; Kawashima, R.; Hagiwara, T.; Suzuki, H.; Imai, K.; Shinomura, Y.; Tokino, T.; Kannagi, R.; Dohi, T. DNA hypermethylation contributes to incomplete synthesis of carbohydrate determinants in gastrointestinal cancer. Gastroenterology 2008, 135, 142–151.e3. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Deng, G. Aberrant expression of carbohydrate antigens in cancer: The role of genetic and epigenetic regulation. Gastroenterology 2008, 135, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Dall’Olio, F.; Malagolini, N.; Trinchera, M.; Chiricolo, M. Mechanisms of cancer-associated glycosylation changes. Front. Biosci. 2012, 17, 670–699. [Google Scholar] [CrossRef]
- Avci, F.Y.; Li, X.; Tsuji, M.; Kasper, D.L. Carbohydrates and t cells: A sweet twosome. Semin. Immunol. 2013, 25, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S. Tumor-associated carbohydrate antigens defining tumor malignancy: Basis for development of anti-cancer vaccines. Adv. Exp. Med. Biol. 2001, 491, 369–402. [Google Scholar] [PubMed]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 1235681. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.F.; Kudryashov, V.; Saigo, P.E.; Finstad, C.L.; Lloyd, K.O. Selection of carbohydrate antigens in human epithelial ovarian cancers as targets for immunotherapy: Serous and mucinous tumors exhibit distinctive patterns of expression. Int. J. Cancer 1999, 81, 193–198. [Google Scholar] [CrossRef]
- Kinney, A.Y.; Sahin, A.; Vernon, S.W.; Frankowski, R.F.; Annegers, J.F.; Hortobagyi, G.N.; Buzdar, A.U.; Frye, D.K.; Dhingra, K. The prognostic significance of sialyl-tn antigen in women treated with breast carcinoma treated with adjuvant chemotherapy. Cancer 1997, 80, 2240–2249. [Google Scholar] [CrossRef]
- Kobayashi, H.; Terao, T.; Kawashima, Y. Serum sialyl Tn as an independent predictor of poor prognosis in patients with epithelial ovarian cancer. J. Clin. Oncol. 1992, 10, 95–101. [Google Scholar] [PubMed]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-Tn (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cordon-Cardo, C.; Zhang, H.S.; Reuter, V.E.; Adluri, S.; Hamilton, W.B.; Lloyd, K.O.; Livingston, P.O. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int. J. Cancer 1997, 73, 42–49. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.L.; Haynes, L.; Parker, C.; Iversen, P. Interdisciplinary critique of sipuleucel-T as immunotherapy in castration-resistant prostate cancer. J. Natl. Cancer Inst. 2012, 104, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Ophir, E.; Bobisse, S.; Coukos, G.; Harari, A.; Kandalaft, L.E. Personalized approaches to active immunotherapy in cancer. Biochim. Biophys. Acta 2016, 1865, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wolfel, C.; Huber, C.; Wolfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific t cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, P.; Merrick, A.E.; West, E.; O‘Donnell, D.; Selby, P.; Vile, R.; Melcher, A.A. Optimization of dendritic cell loading with tumor cell lysates for cancer immunotherapy. J. Immunother. 2008, 31, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.Y.; Yang, W.K.; Lee, H.C.; Hsu, D.M.; Lin, H.L.; Lin, S.Z.; Chen, C.C.; Harn, H.J.; Liu, C.L.; Lee, W.Y.; et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase ii clinical trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Reyes, D.; Salazar, L.; Espinoza, E.; Pereda, C.; Castellon, E.; Valdevenito, R.; Huidobro, C.; Ines Becker, M.; Lladser, A.; Lopez, M.N.; et al. Tumour cell lysate-loaded dendritic cell vaccine induces biochemical and memory immune response in castration-resistant prostate cancer patients. Br. J. Cancer 2013, 109, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Von Euw, E.M.; Barrio, M.M.; Furman, D.; Levy, E.M.; Bianchini, M.; Peguillet, I.; Lantz, O.; Vellice, A.; Kohan, A.; Chacón, M.; et al. A phase I clinical study of vaccination of melanoma patients with dendritic cells loaded with allogeneic apoptotic/necrotic melanoma cells. Analysis of toxicity and immune response to the vaccine and of IL-10 -1082 promoter genotype as predictor of disease progression. J. Transl. Med. 2008, 6, 6. [Google Scholar] [PubMed]
- Zappasodi, R.; Pupa, S.M.; Ghedini, G.C.; Bongarzone, I.; Magni, M.; Cabras, A.D.; Colombo, M.P.; Carlo-Stella, C.; Gianni, A.M.; Di Nicola, M. Improved clinical outcome in indolent B-cell lymphoma patients vaccinated with autologous tumor cells experiencing immunogenic death. Cancer Res. 2010, 70, 9062–9072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-N.; Choi, I.-K.; Huang, J.-H.; Yoo, J.-Y.; Choi, K.-J.; Yun, C.-O. Optimizing DC vaccination by combination with oncolytic adenovirus coexpressing IL-12 and GM-CSF. Mol. Ther. 2011, 19, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Knocke, S.; Mundt, B.; Gürlevik, E.; Strüver, N.; Kloos, A.; Boozari, B.; Schache, P.; Manns, M.P.; Malek, N.P.; et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J. Clin. Invest. 2011, 121, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Kashiwagi, S.; Reeves, P.; Nezivar, J.; Yang, Y.; Arrifin, N.H.; Nguyen, M.; Jean-Mary, G.; Tong, X.; Uppal, P.; et al. A novel mycobacterial Hsp70-containing fusion protein targeting mesothelin augments antitumor immunity and prolongs survival in murine models of ovarian cancer and mesothelioma. J. Hematol. Oncol. 2014, 7, 15–15. [Google Scholar] [CrossRef] [PubMed]
- Darrasse-Jeze, G.; Deroubaix, S.; Mouquet, H.; Victora, G.D.; Eisenreich, T.; Yao, K.H.; Masilamani, R.F.; Dustin, M.L.; Rudensky, A.; Liu, K.; et al. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J. Exp. Med. 2009, 206, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Turley, S.; Mellman, I.; Inaba, K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000, 191, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Lefebvre, F.; Filali-Mouhim, A.; Cameron, M.J.; Goulet, J.P.; Haddad, E.K.; Breton, G.; Trumpfheller, C.; Pollak, S.; Shimeliovich, I.; et al. Synthetic double-stranded rna induces innate immune responses similar to a live viral vaccine in humans. J. Exp. Med. 2011, 208, 2357–2366. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Wang, B.; Shivji, G.M.; Toto, P.; Amerio, P.; Tomai, M.A.; Miller, R.L.; Sauder, D.N. Imiquimod, a topical immune response modifier, induces migration of langerhans cells. J. Investig. Dermatol. 2000, 114, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; O’Neill, D.W.; Nonaka, D.; Hardin, E.; Chiriboga, L.; Siu, K.; Cruz, C.M.; Angiulli, A.; Angiulli, F.; Ritter, E.; et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using toll-like receptor 7 agonist imiquimod as vaccine adjuvant. J. Immunol. 2008, 181, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Dietsch, G.N.; Matthews, M.A.; Yang, Y.; Ghanekar, S.; Inokuma, M.; Suni, M.; Maino, V.C.; Henderson, K.E.; Howbert, J.J.; et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012, 18, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Von Hoff, D.D.; Weiss, G.J.; Dietsch, G.N.; Manjarrez, K.L.; Randall, T.D.; Hershberg, R.M. A phase I dose-finding study of the novel toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin. Cancer Res. 2014, 20, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Sem. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Oh, S.; Gharagozlou, S.; Vedi, R.N.; Ericson, K.; Low, W.C.; Chen, W.; Ohlfest, J.R. In vivo vaccination with tumor cell lysate plus cpg oligodeoxynucleotides eradicates murine glioblastoma. J. Immunother. 2007, 30, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sfondrini, L.; Rossini, A.; Besusso, D.; Merlo, A.; Tagliabue, E.; Menard, S.; Balsari, A. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 2006, 176, 6624–6630. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, M.M.; DeVeer, M.J.; Edling, A.; Oates, R.K.; Simons, B.; Lindner, D.; Williams, B.R. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes enhanced antitumor activity. Cancer Res. 2004, 64, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology 2011, 133, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdörfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to cpg oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Rothenfusser, S.; Hornung, V.; Jahrsdorfer, B.; Blackwell, S.; Ballas, Z.K.; Endres, S.; Krieg, A.M.; Hartmann, G. Identification of CpG oligonucleotide sequences with high induction of IFN-α/β in plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 2154–2163. [Google Scholar] [CrossRef]
- Gibson, S.J.; Lindh, J.M.; Riter, T.R.; Gleason, R.M.; Rogers, L.M.; Fuller, A.E.; Oesterich, J.L.; Gorden, K.B.; Qiu, X.; McKane, S.W.; et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell. Immunol. 2002, 218, 74–86. [Google Scholar] [CrossRef]
- King, J.W.; Taylor, E.M.; Crow, S.D.; White, M.C.; Todd, J.R.; Poe, M.B.; Conrad, S.A.; Gelder, F.B. Comparison of the immunogenicity of hepatitis b vaccine administered intradermally and intramuscularly. Rev. Infect. Dis. 1990, 12, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Tureci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding rna elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, N.; Torensma, R.; Tel, J. Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am. J. Pathol. 2012, 181, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Nakai, N.; Hartmann, G.; Kishimoto, S.; Katoh, N. Dendritic cell vaccination in human melanoma: Relationships between clinical effects and vaccine parameters. Pigment Cell Melanoma Res. 2010, 23, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific t-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Schreibelt, G.; Skold, A.E.; Figdor, C.G.; De Vries, I.J. Paradigm shift in dendritic cell-based immunotherapy: From in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front. Immunol. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguille, S.; Lion, E.; Van den Bergh, J.; Van Acker, H.H.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F.; Berneman, Z.N. Interleukin-15 dendritic cells as vaccine candidates for cancer immunotherapy. Hum. Vaccines Immunother. 2013, 9, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rossi, M.; Ratzinger, G.; de Cos, M.A.; Chung, D.J.; Panageas, K.S.; Wolchok, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-loaded langerhans cells, despite increased IL15 secretion and T-cell activation in vitro, elicit antitumor T-cell responses comparable to peptide-loaded monocyte-derived dendritic cells in vivo. Clin. Cancer Res. 2011, 17, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Lion, E.; Smits, E.L.J.M.; Berneman, Z.N.; Van Tendeloo, V.F.I. NK cells: Key to success of DC-based cancer vaccines? Oncologist 2012, 17, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.-J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ dendritic cells derived in vitro from CD34+ progenitors closely resemble blood dendritic cells, including their adjuvant responsiveness, contrary to monocyte-derived dendritic cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef] [PubMed]
- Kooi, S.; Zhang, H.Z.; Patenia, R.; Edwards, C.L.; Platsoucas, C.D.; Freedman, R.S. HLA class I expression on human ovarian carcinoma cells correlates with T-cell infiltration in vivo and T-cell expansion in vitro in low concentrations of recombinant interleukin-2. Cell. Immunol. 1996, 174, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Maine, C.J.; Aziz, N.H.; Chatterjee, J.; Hayford, C.; Brewig, N.; Whilding, L.; George, A.J.; Ghaem-Maghami, S. Programmed death ligand-1 over-expression correlates with malignancy and contributes to immune regulation in ovarian cancer. Cancer Immunol. Immunother. 2014, 63, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Bouzin, C.; Brouet, A.; De Vriese, J.; Dewever, J.; Feron, O. Effects of vascular endothelial growth factor on the lymphocyte-endothelium interactions: Identification of caveolin-1 and nitric oxide as control points of endothelial cell anergy. J. Immunol. 2007, 178, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Dolcetti, L.; Serafini, P.; Zanovello, P.; Bronte, V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol. Rev. 2008, 222, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 induction by IFNγ and TNFα self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Derhovanessian, E.; Solana, R.; Larbi, A.; Pawelec, G. Immunity, ageing and cancer. Immun. Ageing 2008, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Boyer, J.D.; Jawad, A.; McDonald, K.; Rogers, W.T.; Prak, E.T.L.; Sullivan, K.E. Immunologic consequences of chemotherapy for ovarian cancer: Impaired responses to the influenza vaccine. Vaccine 2013, 31. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A.; Brondstetter, T.I.; English, C.A.; Lee, H.E.; Virts, E.L.; Thoman, M.L. Il-7 gene therapy in aging restores early thymopoiesis without reversing involution. J. Immunol. 2004, 173, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.G.; Ahmed, T.; Bunnell, S.C.; Wu, D.; Chung, H.; Huber, B.T.; Meydani, S.N. Age-associated decline in effective immune synapse formation of CD4+ T cells is reversed by vitamin e supplementation. J. Immunol. 2007, 178, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.; van der Wielen, R.P.; Brink, E.J.; Hendriks, H.F.; Dorovska-Taran, V.N.; Mohede, I.C. Effects of cis-9, trans-11 and trans-10, cis-12 conjugated linoleic acid (CLA) isomers on immune function in healthy men. Eur. J. Clin. Nutr. 2003, 57, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Dupuis, G.; Fortin, C.; Douziech, N.; Larbi, A. T cell response in aging: Influence of cellular cholesterol modulation. Adv. Exp. Med. Biol. 2006, 584, 157–169. [Google Scholar] [PubMed]
- Kensler, T.W.; Spira, A.; Garber, J.E.; Szabo, E.; Lee, J.J.; Dong, Z.; Dannenberg, A.J.; Hait, W.N.; Blackburn, E.; Davidson, N.E.; et al. Transforming cancer prevention through precision medicine and immune-oncology. Cancer Prev. Res. 2016, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lollini, P.-L.; Cavallo, F.; Nanni, P.; Quaglino, E. The promise of preventive cancer vaccines. Vaccines 2015, 3, 467–489. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Lyerly, H.K. Checkpoint blockade in combination with cancer vaccines. Vaccine 2015, 33, 7377–7385. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Co-expression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Tamada, K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci. 2015, 106, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Butler, M.; Oble, D.A.; Seiden, M.V.; Haluska, F.G.; Kruse, A.; MacRae, S.; Nelson, M.; Canning, C.; Lowy, I.; et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective re-induction therapy with an anti-PD-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [PubMed]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Chhabra, A.; Chachoua, A.; Adams, S.; Donach, M.; Fenton-Kerimian, M.; Friedman, K.; Ponzo, F.; Babb, J.S.; Goldberg, J.; et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: A proof-of-principle trial. Lancet Oncol. 2015, 16, 795–803. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. Tgfbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, M.; Kohrt, H.; Levy, R.; Jones, J.; Camphausen, K.; Dicker, A.; Demaria, S.; Formenti, S. Current clinical trials testing combinations of immunotherapy and radiation. Semin. Radiat. Oncol. 2015, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; de Vries, I.J.; Aarntzen, E.A.; de Boer, A.; Scharenborg, N.M.; van de Rakt, M.; van Spronsen, D.J.; Preijers, F.W.; Figdor, C.G.; Adema, G.J.; et al. A pilot study on the immunogenicity of dendritic cell vaccination during adjuvant oxaliplatin/capecitabine chemotherapy in colon cancer patients. Br. J. Cancer 2010, 103, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium fasl establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Dranitsaris, G.; Cohen, R.B.; Acton, G.; Keltner, L.; Price, M.; Amir, E.; Podack, E.R.; Schreiber, T.H. Statistical considerations in clinical trial design of immunotherapeutic cancer agents. J. Immunother. 2015, 38, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Perez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Vaccine Class | Vaccine Name | Description | Clinical Development: Phase | No. of Pts (OvCa Pts) | Clinical Outcome |
|---|---|---|---|---|---|
| DCs | APCEDEN | DCs loaded with whole-tumor lysate | Phase II; (Bapsy, 2014 [24]) | 38 pts (9 OvCa pts) | No CR observed; ORR was 28.9% (11/38) and irRC was 42.1% (16/38) |
| OCDC | DCs loaded with autologous oxidized tumor lysate | Pilot; (Chiang, 2013 [25]) | 5 OvCa pts | 2/5 pts (40%) demonstrated PFS2 > PFS1 | |
| DCVax-L | DCs loaded with autologous oxidized tumor lysate, combined with bevacizumab and metronomic Cy | Pilot; (Kandalaft, 2013 [26]) | 6 OvCa pts | 4/6 pts (66%) achieved clinical benefit (including 2 PR and 2 SD) | |
| DC-wtl | DCs loaded with crude whole tumor lysate | Phase I; (Hernando, 2002 [27]) | 8 pts (6 OvCa pts) | Data suggested a positive correlation with disease stabilization | |
| DC-MFP | DCs loaded with mannan-MUC1 fusion protein (MFP) | Phase I; (Loveland, 2006 [28]) | 9 pts (2 OvCa pts) | 2/9 pts (22%) in progression at entry were stable after therapy, for at least 3 years | |
| Lapuleucel-T, Neuvenge, APC 8024 | DCs loaded with BA7072, a fusion protein HER-2/neu linked to GM-CSF | Phase I; HER-2+ tumors; (Peethambaram, 2009 [29]) | 18 pts (4 OvCa pts) | 2/18 pts (11%) had SD lasting > 48 weeks | |
| HER-2/neu; MUC1 peptides | DCs loaded with synthetic peptides derived from HER-2/neu or MUC1 peptides | Phase I; HER-2+ or MUC1+ tumors; (Brossart, 2000 [30]) | 10 pts (3 OvCa pts), HLA-A*02+ | No data | |
| hTERT; HER-2/neu; PADRE peptides | DCs loaded with synthetic peptides derived from hTERT; HER-2/neu; PADRE | Phase I/II; (Chu, 2012 [31]) | 14 OvCa pts, HLA-A*02+ | 3 years-OS was 90%; 3 years-PFS was 80% (with Cy) | |
| WT-1; MUC1; CA125 | DCs loaded with synthetic peptides derived from WT-1; MUC1; CA125 | Phase II; (Kobayashi, 2014 [32]) | 56 OvCa pts | DCR and ORR were 29% and 3.6%, respectively | |
| Whole tumor cells | Fang vaccine, Vigil™ Ovarian, Gemogenovatucel-T | Autologous tumor cells eletroporated with FANG vector, a plasmid encoding GM-CSF and a bi-shRNA targeting furin convertase, thereby downregulating TGF-b1 and b2 | Phase I; (Senzer, 2012 [33]) | 27 pts (5 OvCa pts) | 23/26 pts (88%) showed SD at month 2 or later |
| Listeria monocy togenes | CRS-207 | Lm strain engineered to express human mesothelin | Phase I; (Le, 2012 [34]) | 17 pts (2 OvCa pts) | 37% of subjects lived ≥ 15 mo (months) |
| Peptides/proteins | Mixture of peptides (comparison) | Predesigned peptides vs. PPV (personalized peptide vaccine); admixed with Montanide ISA-51 | Pilot; (Tsuda, 2004 [35]) | 14 pts (5 OvCa pts), HLA-A*02+ or HLA-A*24+ | No clinical response with predesigned; 3/5 cervical cancer pts (60%) showed objective tumor regression |
| Mixture OvCa-associated peptides | OvCa-associated peptides admixed with Montanide ISA-51 and GM-CSF | Pilot; (Morse, 2011 [36]) | 15 pts (8 OvCa pts); HLA-A*02+ | With median follow-up of 492 days, 4 OvCa pts had relapsed and 3 died (expected relapse rate 18–22 mo in 75% of pts) | |
| Mixture of different peptides | OvCa-associated peptides plus a helper peptide from tetanus toxoid protein, admixed with Montanide ISA-51 and GM-CSF | Phase I; (Chianese-Bullock, 2008 [37]) | 9 OvCa pts, HLA-A*01+, -A*02+ or A*03+ | One participant remained disease-free at 19 months after active treatment | |
| HER-2/neu | Epitope p369–377, admixed with GM-CSF | Phase I; HER-2/neu++ Tu (Knutson, 2002 [38]) | 6 pts (2 OvCa pts), HLA-A*02+ | No data | |
| - | Multiple peptides derived from either the extracellular domain (ECD) or the ICD, admixed with GM-CSF | Phase I; HER-2/neu++ Tu (Disis1, 2002 [39]) | 38 pts (5 OvCa pts), HLA-A*02+ | No data | |
| - | Peptides from the ICD, admixed with GM-CSF | Phase I; HER-2/neu++ Tu (Disis, 2002 [40]) | 10 pts (1 OvCa pts) | No data | |
| - | Multiple peptides derived from either the ECD, the ICD, or both, admixed with GM-CSF | Phase I; HER-2/neu++ Tu (Disis, 2004 [41]) | 38 pts (5 OvCa pts) | No data | |
| HER-2/neu-ICD | ICD protein, aas 676–1255, His-tagged | Phase I; HER-2/neu++ Tu (Disis, 2004 [42]) | 29 pts (1 OvCa pt) | No data | |
| NY-ESO-1 | Epitope p157–170, admixed with Montanide ISA-51 | Phase I; (Odunsi, 2007 [43]) | 18 OvCa pts, HLA-DPB1*0401+ or *0402+ | Median PFS of 19.0 mo (vs. 16–18 weeks in pts receiving 2nd line chemo) | |
| - | Epitope p157–165, admixed with Montanide ISA-51 | Phase I; NY-ESO-1+ or LAGE-1+ Tu; (Diefenbach, 2008 [44]) | 9 OvCa pts, HLA-A*02:01+ | Median PFS of 13 mo. 3/9 pts (33%) remained in CR at 25, 38, and 52 mo | |
| NY-ESO-1 OLP | NY-ESO-1 overlapping long peptides, +/− Montanide and Poly-ICLC | Phase I; (Sabbatini, 2012 [45]) | 28 OvCa pts (HLA indep) | Pts NY-ESO-1+ receiving OLP + Montanide + Poly-ICLC showed delayed time to recurrence | |
| NY-ESO-1 protein | NY-ESO-1 protein + Montanide + CM-CSF +/− decitabine | Phase I; (Odunsi, 2014 [46]) | 12 OvCa pts | 5/10 (50%) pts had SD (median duration 6.3 mo), and 1/10 (10%) had PR (duration 5.8 mo) | |
| P53 | Wt p53: 264–272 peptide admixed with GM-CSF and Montanide ISA-51, either SC (Arm A) or loaded into DCs (Arm B) | Phase II; p53++ Tu; (Rahma, 2012 [47]) | 21 OvCa pts, HLA-A*02:01+ | No significant difference between arms in median OS (40.8 mo vs. 29.6 mo, p = 0.26), nor in PFS (4.2 mo vs. 8.7 mo, p = 0.94) | |
| P53-SLP | Ten synthetic peptides 25–30 aa long overlapping peptides (aas 70–248 in wt-p53) admixed in Montanide ISA-51 | Phase II; (Leffers, 2009 [48]) | 18 OvCa pts (HLA indep) | 2/18 (11%) of pts with SD, not clearly attributable to vaccination | |
| - | - | Phase II; (Leffers, 2012 [49]) | 20 OvCa pts (HLA indep) | No difference in survival between p53-SLP treated pts and historical controls (median 44.0 mo vs. 47.4 mo, p = 0.601) | |
| - | Same, but two days before vaccination, 300 mg/m2 Cy i.v. was given | Phase II; (Vermeij, 2012 [50]) | 10 OvCa pts (HLA indep) | No data | |
| PPV | Personalized peptide vaccine: mixture of 4 peptides (from a panel of 31) previously tested for immunity in each pt, admixed in Montanide ISA51VG | Phase II; (Kawano, 2014 [51]) | 42 OvCa pts (HLA-dep) | Median survival time (MST) was 39.2 mo in platinum-sensitive pts, vs. 16.2 mo in platinum-resistant | |
| Flt3-L | Truncated glycoprotein Flt3-L (Fms-like tyr kinase-3-ligand, which increases DCs and monocytes), either i.p. or s.c. | Pilot; (Freedman, 2003 [52]) | 15 pts (9 OvCa pts) | No objective responses were observed | |
| Genetic vaccines | PANVAC-C + PANVAC-V | Poxviral vaccine: CEA-MUC1-TRICOM (B7.1, ICAM-1, LFA-3) engineered into vaccinia (PANVAC-V) as prime and fowlpox (PANVAC-C) as booster vaccination | Pilot; CEA+ or MUC1+ Tu; (Gulley, 2008 [53]) | 25 pts (3 OvCa pts) | 1 OvCa pt (1/25: 4%) had durable (18 mo) clinical response |
| rV-NY-ESO-1 + rF-NY-ESO-1 | NY-ESO-1 engineered into vaccinia (rV) as prime and fowlpox (rF) as booster vaccination | Phase I; NY-ESO-1+ Tu; (Jager, 2006 [54]) | 36 pts (1 OvCa pt) | 7/9 pts with stage II/IV MEL survived 17–63+ mo | |
| - | - | Phase II; NY-ESO-1+ Tu; (Odunsi, 2012 [55]) | 47 pts (22 OvCa pts) | In OvCa pts, median TTP was 21 mo and median OS was 48 mo | |
| Epigenetic vaccines | Theratope® | Synthetic Syalyl-Tn-KLH (STn: carbohydrate associated with the MUC1 mucin), admixed with Detox-B, after autologous transplantation | Phase II/III; MUC1+ Tu; (Holmberg, 2003 [56]) | 70 pts (17 OvCa pts) | Decreased risk for relapse and death (p = 0.07 and p = 0.1 respectively), as compared to transplanted pts only |
| Lewis(y) | Synthetic Lewis(y) pentasaccharide coupled to KLH (Ley: carbohydrate epitopes overexpressed in OvCa), admixed with QS-21 | Phase I; (Sabbatini, 2000 [57]) | 25 OvCa pts | Median TTP was 6 mo (2–17 mo) |
| Type | Product Name | Description | Clinical Development: Phase Indication NCT |
|---|---|---|---|
| DC | DCVAC/OvCa | DCs activated with an ovarian tumor cell lysate | Phase II OC NCT02107378 |
| FRalphaDC vaccine | DCs loaded with five immunogenic peptide epitopes, derived from the tumor-associated antigen human folate receptor alpha (FR alpha or FOLR1), including FR30, FR56, FR76, FR113, and FR238 | Pilot OC NCT02111941 | |
| Ontak + DC vaccine | Ontak (denileukin diftitox): a cytotoxic recombinant protein consisting of interleukin-2 (IL-2) protein sequences fused to diphtheria toxin; the use of Ontak is followed by autologous DC vaccine to stimulate tumor killing immune cells | Phase II OC NCT00703105 | |
| Ovapuldencel-T | DCs loaded with autologous, lethally irradiated cancer cells and mixed with GM-CSF | Phase II OC NCT02033616 | |
| Dendritic cell/tumor fusion vaccine | DC/tumor fusion vaccine with GM-CSF and imiquimod (cytokine production stimulation) | Phase II OC NCT00799110 | |
| Whole tumor cells | Fang vaccine, Vigil™ ovarian, Gemogenovatucel-T | Autologous tumor cells eletroporated with FANG vector, a plasmid encoding GM-CSF and a bi-shRNA targeting furin convertase, thereby downregulating transforming growth factor (TGF)-β1 and β2 | Phase II/III OC NCT02346747 |
| Peptide/protein | OVax | Autologous ovarian cancer cell peptide antigens conjugated to the hapten 2,4-dinitrophenol (DNP) | Phase I/II OC NCT00660101 |
| HER-2 peptide vaccine | Combination of MVF-HER-2 (597–626) and MVF-HER-2 (266–296) emulsified with nor-MDP and ISA 720 | Phase I Solid TumorsNCT01376505 | |
| FBP E39/J65 | Two Folate Binding Protein Peptide Vaccines (E39 and J65) | Phase I/II BC/OC NCT02019524 | |
| WT2725 | Peptide derived from Wilms tumor gene 1 (WT1) protein | Phase I WT1++ Tumors NCT01621542 | |
| DSP-7888 Dosing emulsion | WT1 protein-derived peptide vaccine | Phase I Different Tumors NCT02498665 | |
| OC-L | Trial to test the addition of 2 investigational agents, Montanide and poly-ICLC (a TLR3 agonist) to a backbone of autologous oxidized tumor cell lysate vaccine (OC-L) administered with GMCSF | Phase I OC NCT02452775 | |
| Genetic | Ad-sig-hMUC-1/ecdCD40L | Ad-sig-hMUC-1/ecdCD40L adenoviral vector encodes a fusion protein in which the hMUC-1 epithelial antigen is attached to the CD40L (CD40 ligand), which binds to CD40 on DCs, stimulating internalization of hMUC-1 Ag | Phase I Epithelial Ca (LC/BC/OC/PC/CRC) NCT02140996 |
| AdV-tk + valacyclovir | AdV-tk: adenoviral vector expressing the herpes simplex virus thymidine kinase (HSV-tk) gene, which, when administered in conjunction with a synthetic acyclic guanosine analogue (valacyclovir), possesses potential antineoplastic activity. Release of TAAs by dying tumor cells may then stimulate an antitumor cytotoxic T lymphocyte (CTL) response | Phase I Epithelial Ca (LC/MES/BC/OC) NCT01997190 | |
| ID-LV305 | ID-LV305: An engineered lentiviral vector targeting DCs and containing nucleic acids encoding for the human tumor-associated cancer-testis antigen NY-ESO-1 | Phase I MEL/NSCLC/OC/SAR NCT02122861 | |
| p53MVA | p53MVA vaccine: modified vaccinia virus Ankara expressing tumor protein p53 | Phase I OC NCT02275039 | |
| Trovax® | Trovax®: modified vaccinia virus Ankara (MVA) vector, encoding the 5T4 antigen | Phase II OC NCT01556841 | |
| Epigenetic | OPT-822/OPT-821 | OPT-822/OPT-821: Two carbohydrate-based immunostimulants comprised of the Globo H hexasaccharide 1 (Globo H) epitope linked to KLH, which may stimulate a cytotoxic T-lymphocyte (CTL) response against Globo H-expressing tumor cells | Phase II OC NCT02132988 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin Lluesma, S.; Wolfer, A.; Harari, A.; Kandalaft, L.E. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines 2016, 4, 10. https://doi.org/10.3390/biomedicines4020010
Martin Lluesma S, Wolfer A, Harari A, Kandalaft LE. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines. 2016; 4(2):10. https://doi.org/10.3390/biomedicines4020010
Chicago/Turabian StyleMartin Lluesma, Silvia, Anita Wolfer, Alexandre Harari, and Lana E. Kandalaft. 2016. "Cancer Vaccines in Ovarian Cancer: How Can We Improve?" Biomedicines 4, no. 2: 10. https://doi.org/10.3390/biomedicines4020010