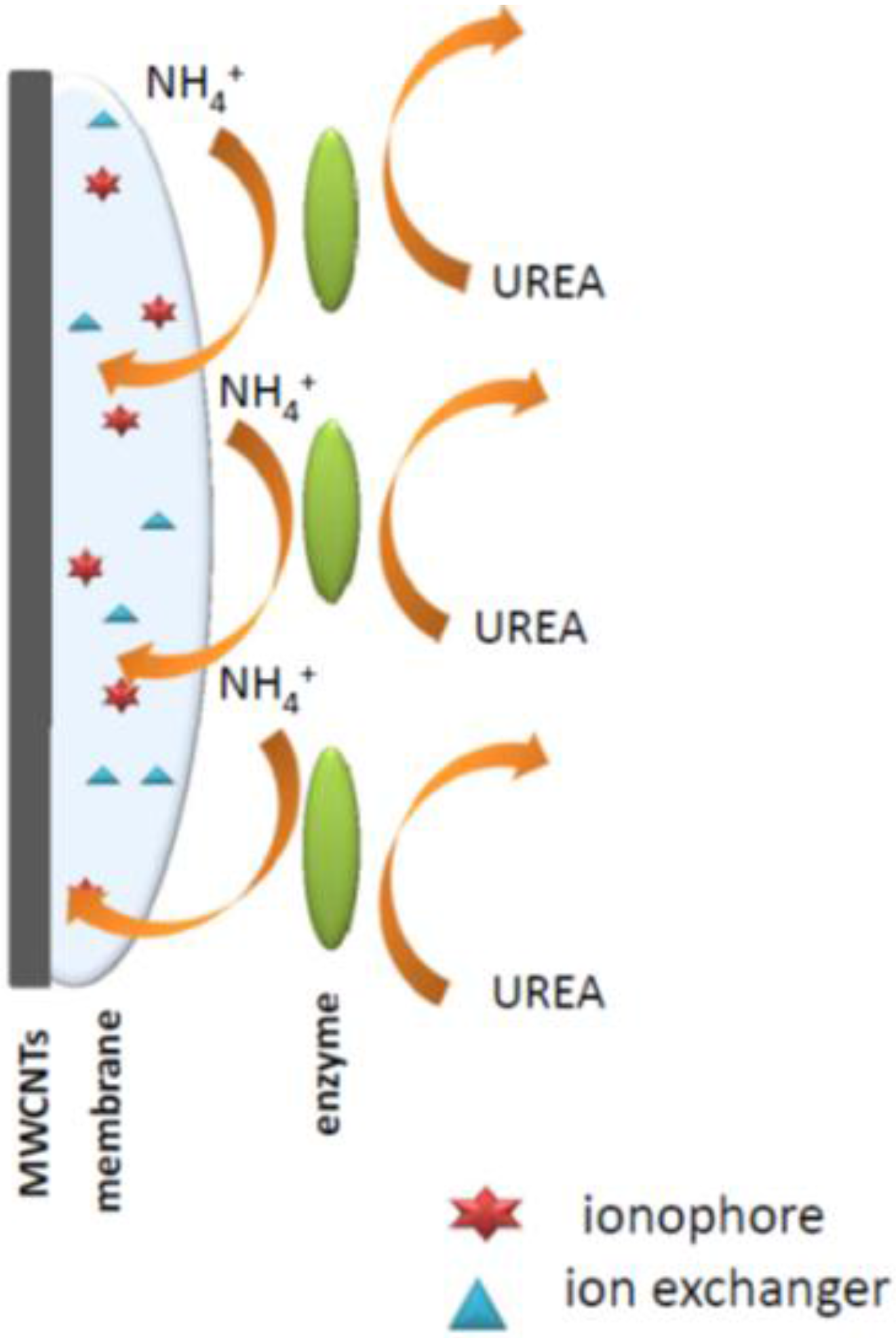
1. Introduction
Determination of urea is important in terms of clinical research, as well as other environmental and industrial applications. One of the attractive, and often considered, methods for the determination of urea is to use potentiometric biosensors based on ion-selective electrodes [
1,
2,
3]. This approach offers potentially fast and relatively simple, in terms of the apparatus applied, quantification of urea. The other benefit of potentiometric approach is that ion-selective electrodes are well-established tools of clinical and biological analysis.
In the presence of the enzyme, hydrolysis of urea is observed resulting, ultimately, in pH change.
In principle, two sensing modes can be used to follow the enzymatic-driven decomposition of urea, either following an H+ ion concentration decrease or the increase of the NH4+ ion content.
Both ammonium ion- and hydrogen ion-plastic, solvent polymeric (
i.e., plasticized poly(vinyl chloride)-based), ion-selective membrane electrodes are well known sensors. (e.g., [
4,
5,
6]). Their high sensitivity, low detection limit, and fast response time provide an opportunity to use them for preparation of urea biosensors. Moreover, both H
+ and NH
4+ ion-selective electrodes can and have been prepared in the internal-solution free arrangement, which is generally highly promising for miniaturization and disposable sensors. Among other potential materials tested, in recent years for all-solid state transducers for potentiometric electrodes, carbon nanotubes (CNTs) have attracted significant research attention [
7,
8,
9,
10,
11]. Carbon nanotubes can be applied on the glassy carbon surface and then covered by an ion-selective membrane [
7,
8,
9]. Alternatively, carbon nanotubes can be also useful to prepare (nominally) disposable sensors, where CNTs are used both as a transducer and as an electrical lead [
12,
13,
14]. The latter concept is especially attractive to develop further in order to obtain disposable potentiometric biosensors. The general idea of such devices requires that the bio-molecule should be immobilized at the outer interface of the ion-selective membrane. In this way the biological reaction (e.g., enzymatic hydrolysis of urea) would generate a product close to the outer interface of the ion-selective membrane, allowing fast detection and assuring low detection limits.
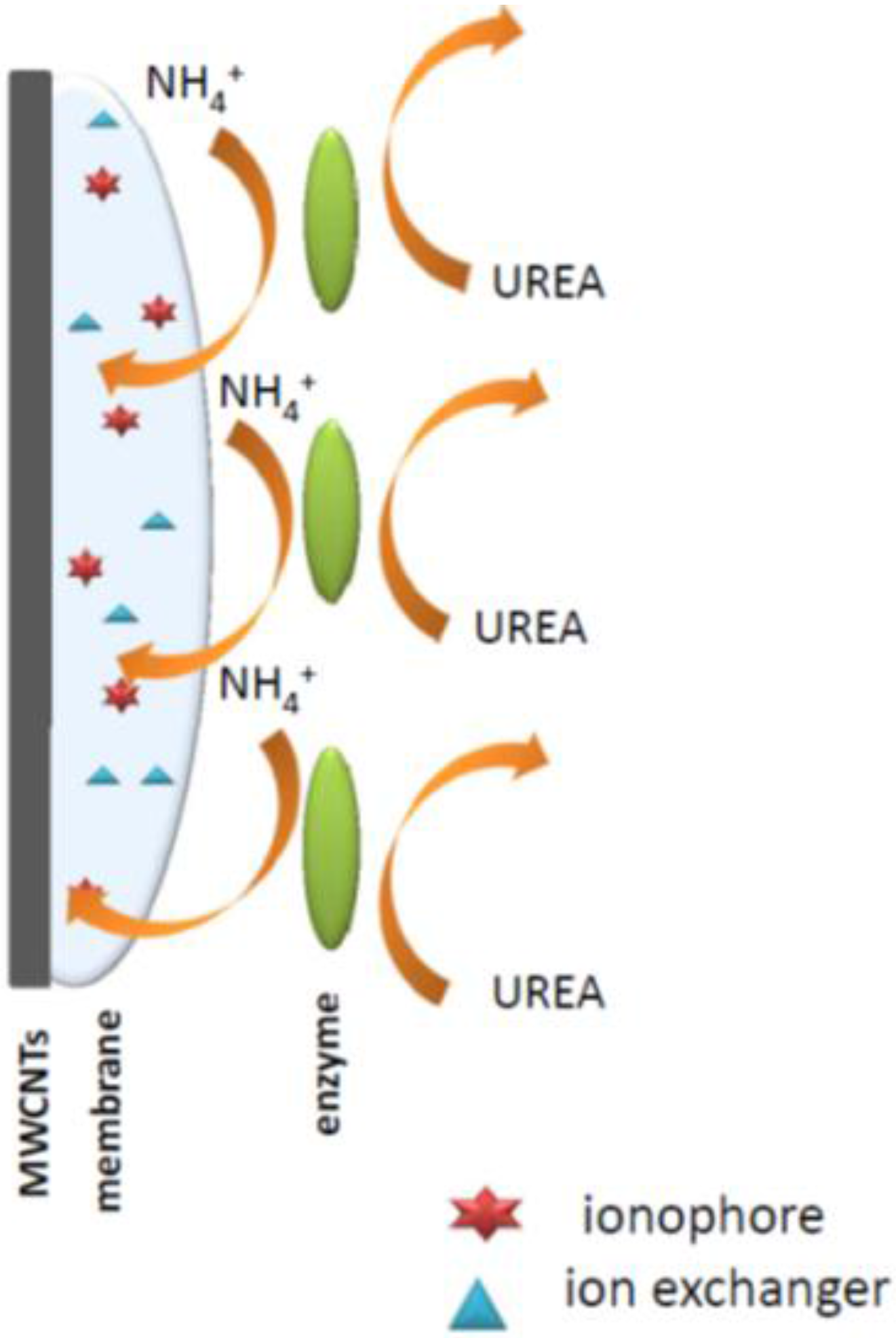
Thus, an important aspect of potentiometric enzymatic biosensor construction is the choice of the appropriate method of the enzyme immobilization on the ion-selective membrane surface. The major issues here are preservation of the activity of the enzyme as well as the preferred analytical parameters. The literature describes the physical (adhesion, inclusion) and chemical (cross-linking [
15], covalent linking [
16]) methods of enzymes immobilization. It is possible to use cellulose or poly(vinyl chloride) as a medium to urease immobilization on the surface of glass pH-electrodes [
17]. The covalent linking of the enzyme to a poly(vinyl alcohol) matrix [
18] or covalent attachment of enzyme molecules directly on the membrane using carboxylated poly(vinyl chloride) [
19] have been reported as effective methods of enzyme immobilization. This material has been also used to obtain urea pH-biooptrode [
20].
This paper presents a possibility of using multi-walled carbon nanotubes to prepare disposable carbon-plastic potentiometric sensors, using the approach developed in our group earlier [
13]. In this work, the key interest was to prepare a simple methodology allowing for potentiometric readout. The sensors prepared were either ammonium- or hydrogen ion-selective. The ion-selective membranes were further modified with urease enzyme-immobilized with acetate cellulose on the top of ion-selective membranes.
3. Results and Discussion
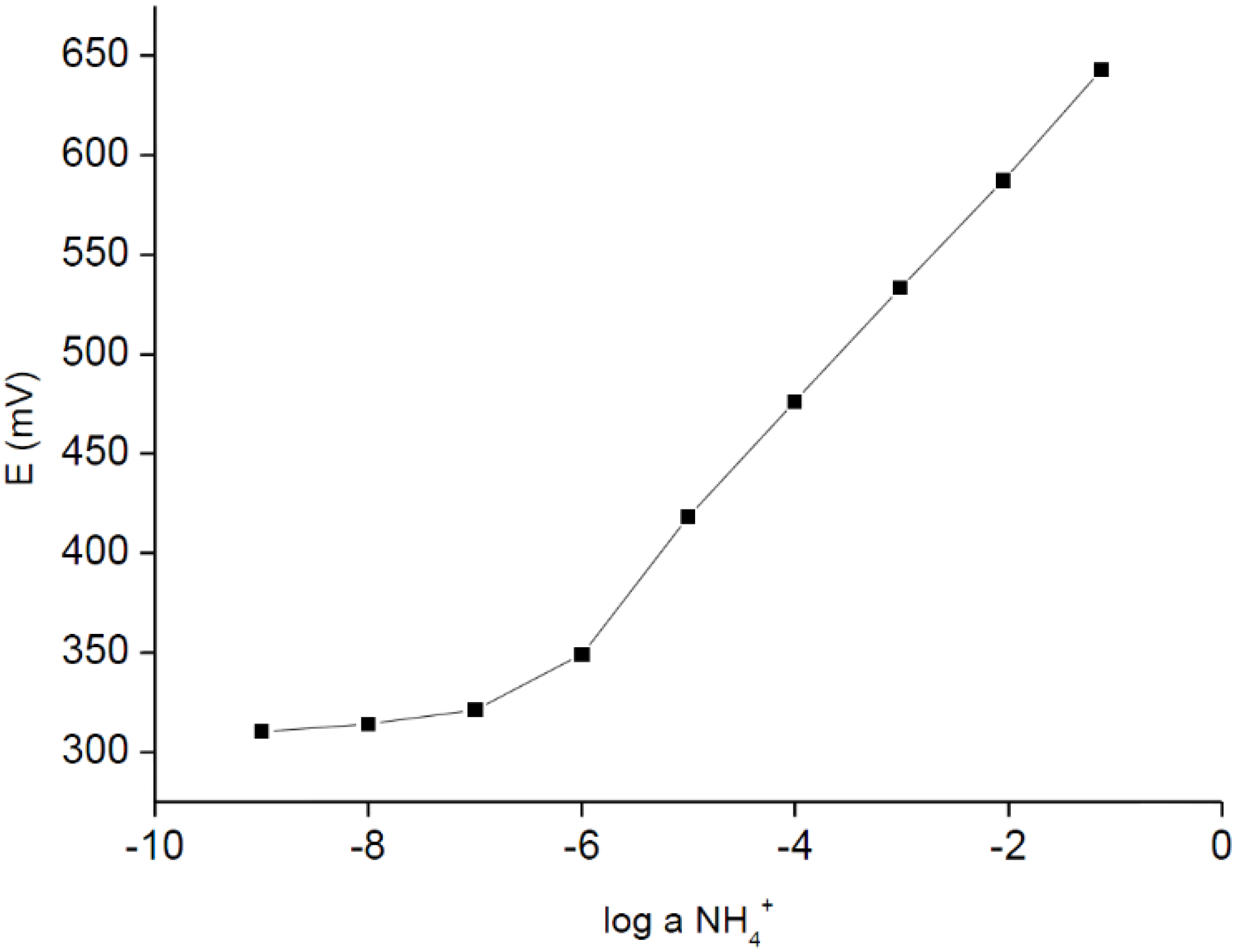
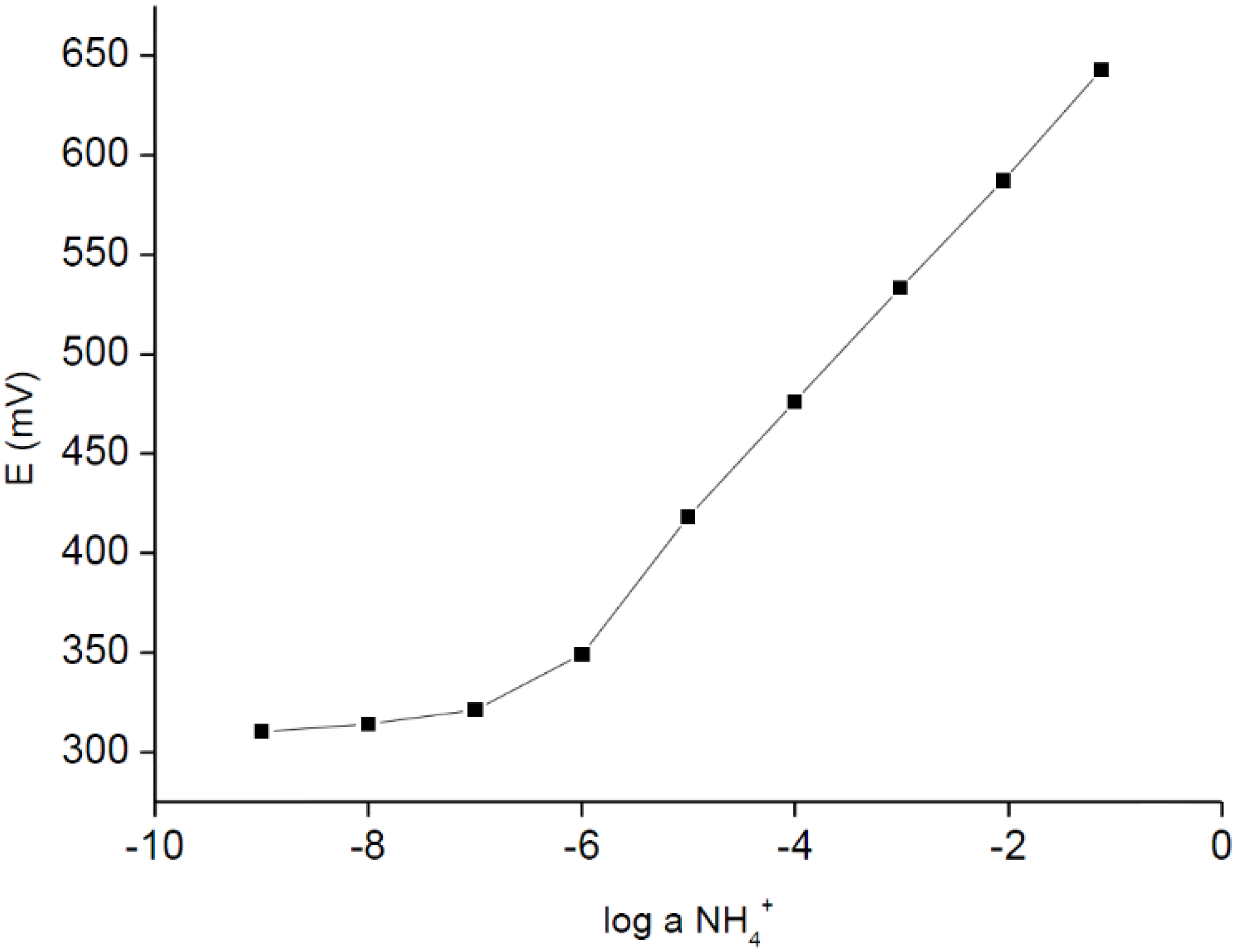
Figure 1 presents typical potentiometric responses obtained for miniature, disposable, plastic-carbon ammonium ion-selective electrodes (without urease). As it can be seen in
Figure 1, the sensors were characterized with linear responses within the broad activity range from 10
−1 to 10
−6 M, with a detection limit close to 10
−6.6 M and close to the Nernstian slope, equal to 59.4 ± 0.9 mV/dec (R
2 = 0.999). The logarithms of selectivity coefficients for the ammonium ion-selective electrode (calculated using the Nernstian slope for activities from 10
−1 M to 10
−4 M, separate solution method) were equal to −4.44 ± 0.5, −4.73 ± 0.4, −0.95 ± 0.04, −2.99 ± 0.01 for Mg
2+, Ca
2+, K
+, Na
+ cations, respectively. The above given analytical parameters clearly confirm that these miniature, nominally-disposable sensors were successfully obtained.
Figure 1.
Potentiometric response of ammonium ion-selective carbon-plastic sensor recorded in NH4Cl solutions.
Figure 1.
Potentiometric response of ammonium ion-selective carbon-plastic sensor recorded in NH4Cl solutions.
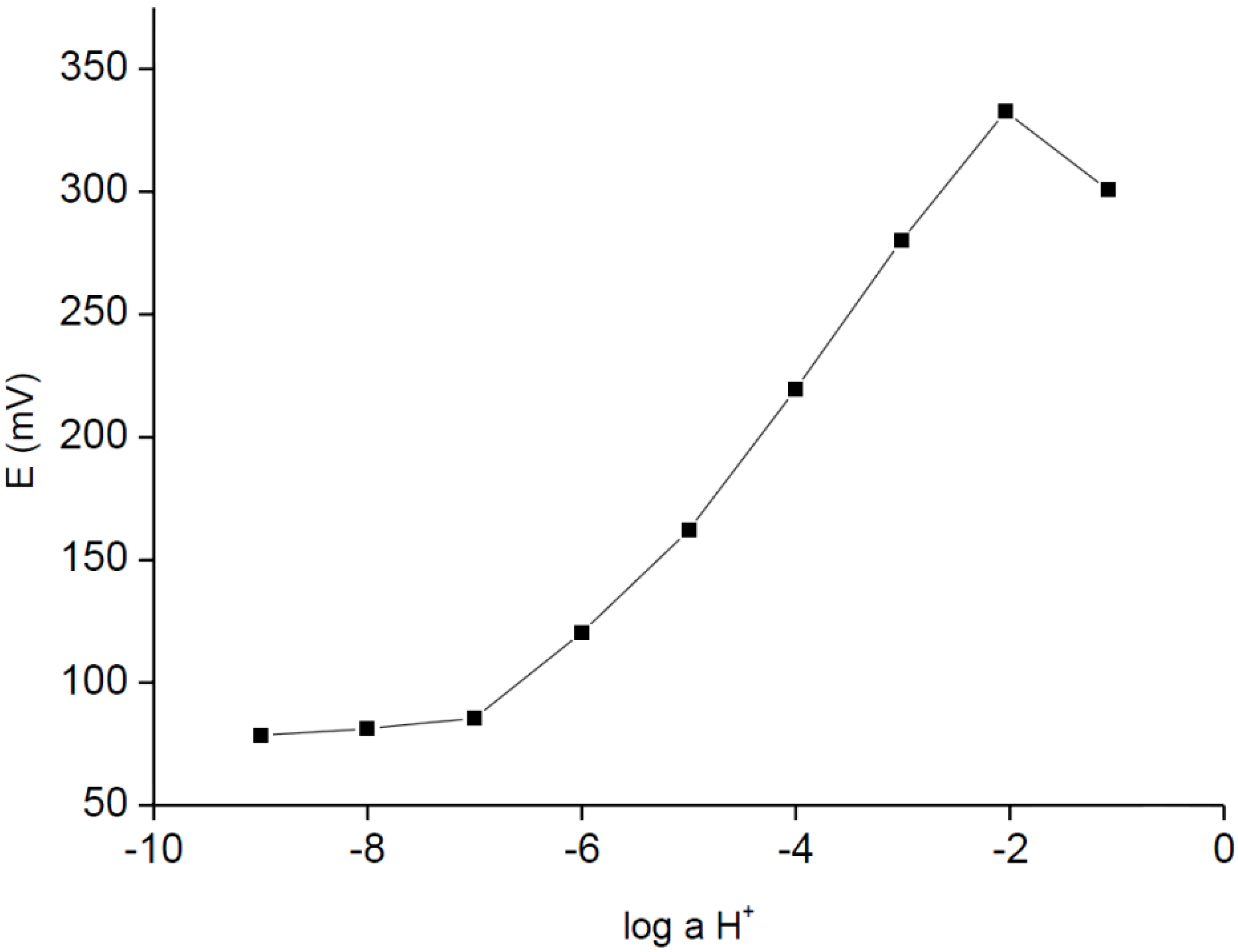
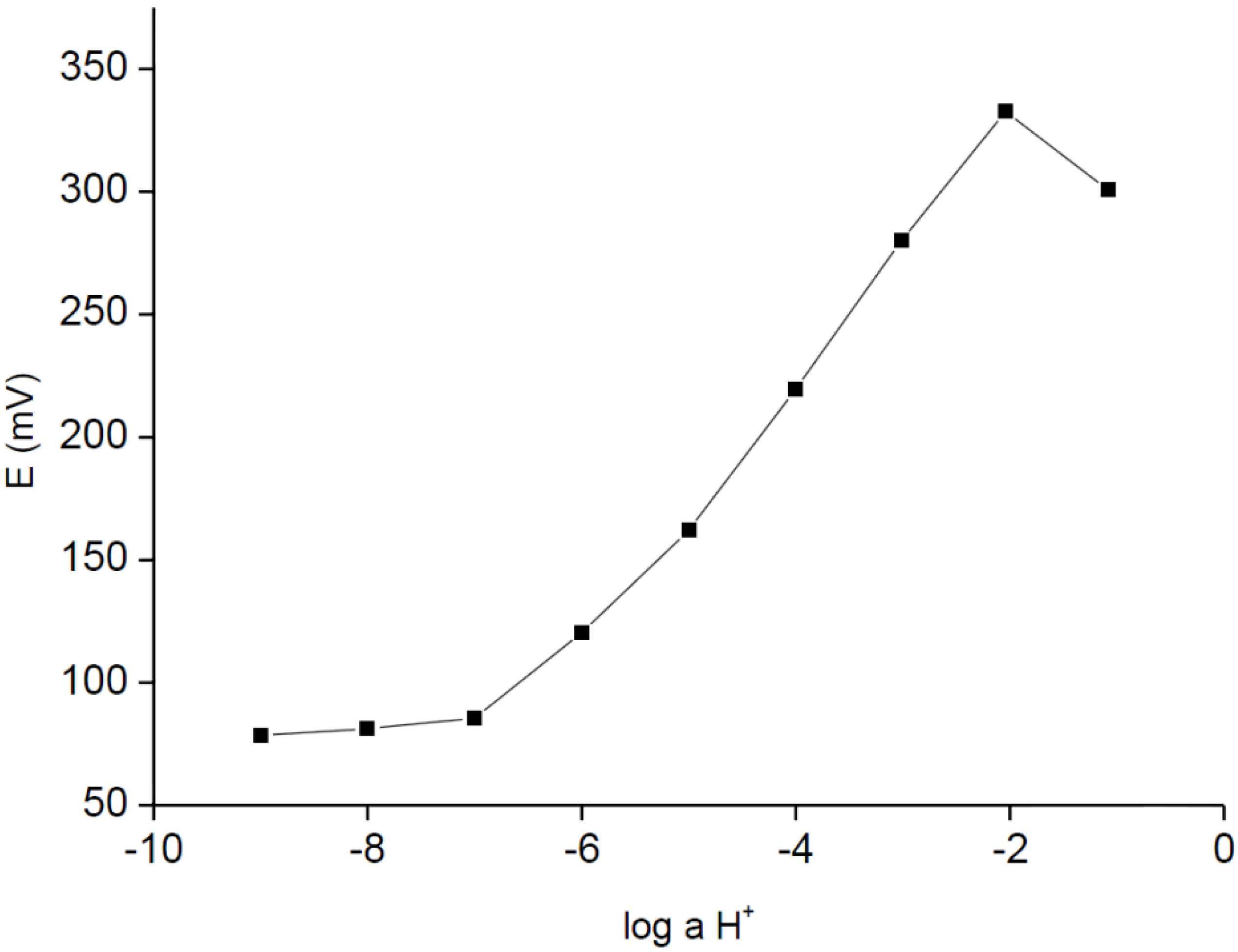
The H
+-selective electrode tested in parallel (without urease) were also characterized with potentiometric responses typical for its full-size, solid-contact or internal solution counterparts (
Figure 2). As it can be seen in
Figure 2, the sensor was characterized with linear responses within the broad activity range from 10
−2 M to 10
−6 M, with a detection limit close to 10
−6 M; the slope of dependence was close to Nernstian, within the range of experimental error, and was equal to 57.7 ± 0.7 mV/dec (R
2 = 0.999). The logarithms of selectivity coefficients (determined using theoretical slope, separate solution method, within the concentration range from 10
−1 M to 10
−4 M) were equal to −4.93 ± 0.5, −4.48 ± 0.7, −3.28 ± 0.1, −3.98 ± 0.4 for Mg
2+, Ca
2+, K
+ and Na
+ cations, respectively.
Figure 2.
Potentiometric response of H+-ISE carbon-plastic electrodes recorded in HCl/buffer solutions.
Figure 2.
Potentiometric response of H+-ISE carbon-plastic electrodes recorded in HCl/buffer solutions.
When the PVC-based ion-selective membranes were covered with urease in the cellulose acetate layer, some alteration of the potentiometric response to the H+ or NH4+ ions was observed, for H+-ISE or NH4+-ISE, respectively, in the absence of urea in solution. The slope of the dependence obtained for H+-ISE electrodes with the PVC surface modified with urease in cellulose acetate was close to 32.0 mV/dec, in the range from 10−2 M to 10−4 M (R2 = 0.998). Further, a decrease of solution H+ ion activity resulted in a decrease of potential; however, the slope of dependence further decreased. Similarly, for the NH4+-ISE surface modified with urease in cellulose acetate, the slope of potentiometric dependence was lower than Nernstian and was close to 38.0 mV/dec, in the range from 10−1 M to 10−5 M (R2 = 0.996).
The lower slope of the dependence recorded in the presence of cellulose acetate and the urease layer on the surface of the ion-selective membrane can be related to limitation in transport/ion-exchange at the ion-selective membrane/solution interface and to the pH buffering effect of this layer. The effect of the presence of the outer layer on the ion-selective membrane was more pronounced for H+-ISE, which is probably related to the possibility of protonation/deprotonation of enzyme proteins in the cellulose acetate layer. As a result, the changes of H+ ions directly at the membrane surface, i.e., where the signal is formed, are different from those occurring in the solution bulk. The effect of the surface enzyme-containing layer presence on the performance of NH4+-ISE was less pronounced, as in this case the effect is primarily attributed to analyte transport limitation.
The above-described effects should not affect the performance of the biosensors, in which the changes of pH due to enzymatic reaction are occurring in the ion-selective membrane modifying layer—that is at the ion-selective membrane interface.
For application of ion-selective electrodes with enzyme immobilized on the surface of ion-selective membranes for urea detection, the choice of background buffer and solution pH is vitally important. The choice of optimal pH is dependent on the enzyme-optimal activity, as well as on the analyte to be detected potentiometrically.
For the NH
4+-selective membrane the potentiometric detection requires rather low pH to favor the presence of the ammonium ions close to the membrane surface and prevent its spontaneous transformation to yield ammonia. Conversely, the optimal pH of the urease activity is close to 7.4 [
24]. Thus, clearly the measurement should be performed after adjustment of the sample pH, post enzymatic reaction. It should be stressed that the decrease of pH is expected to affect, mainly, the detection limit, as the amount of NH
4+ ions present in the solution of constant total ammonium ion concentration is pH-dependent.
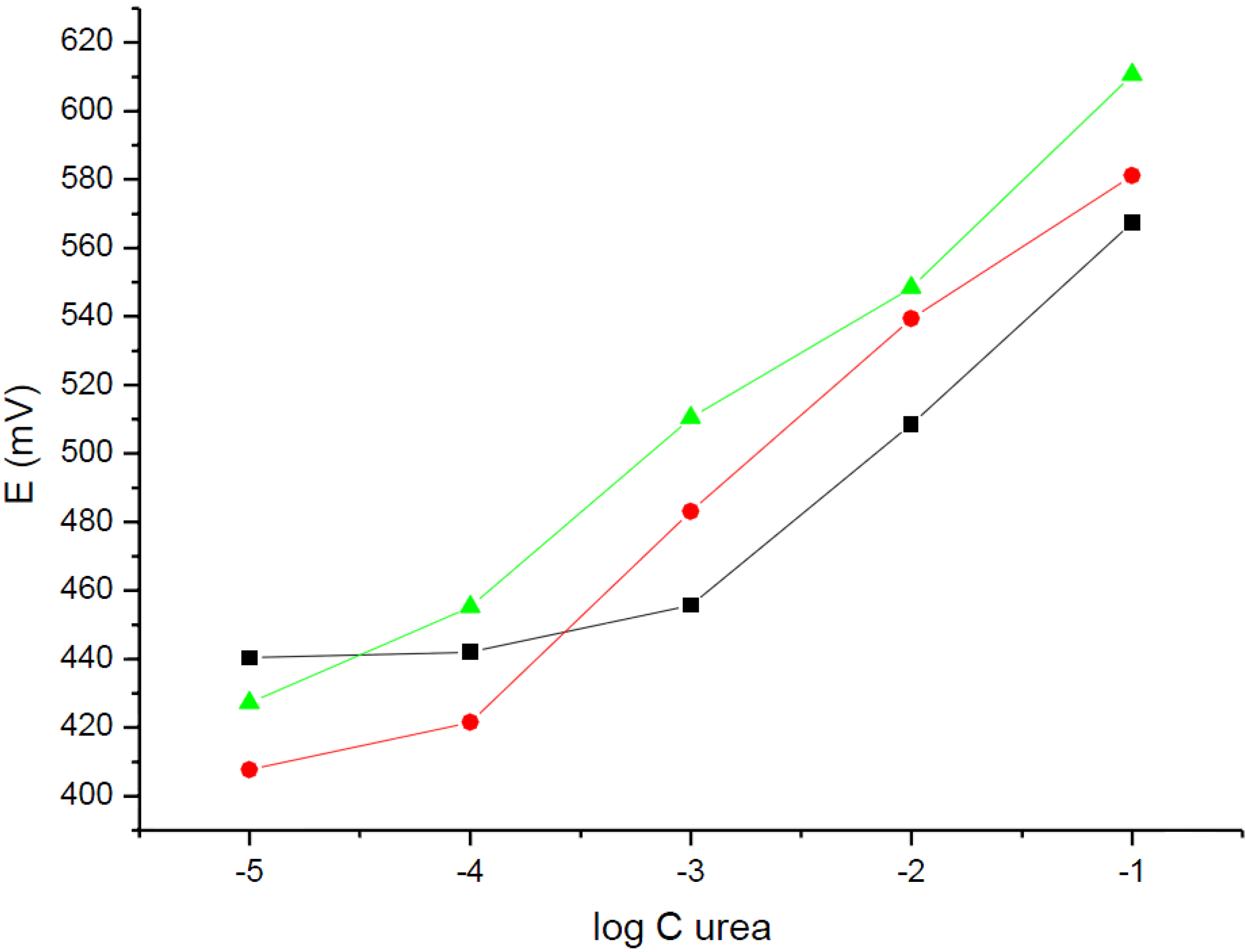
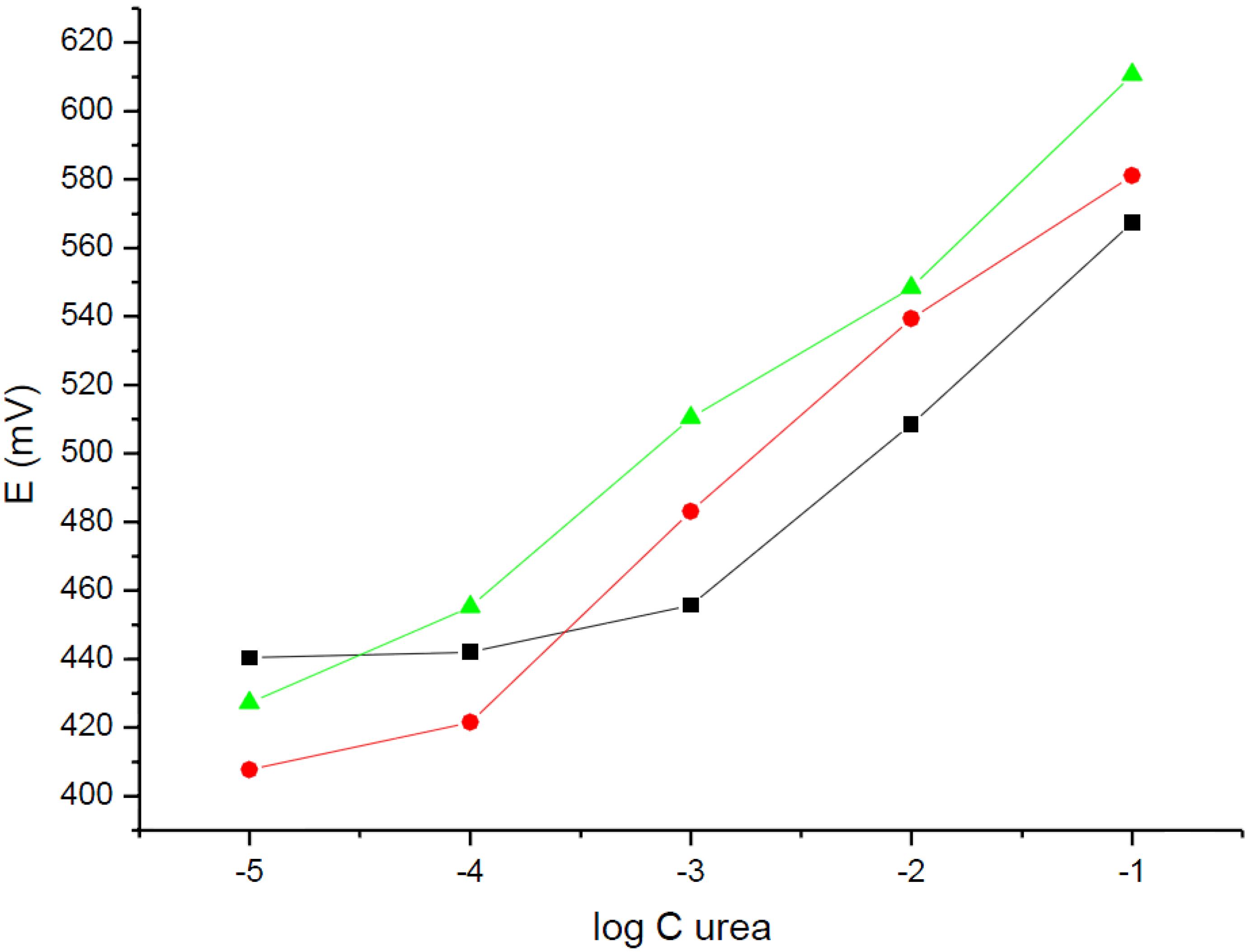
Figure 3 presents potentiometric responses of urea biosensors (urease-NH
4+-ISE) for different concentrations of urea in the solution, recorded for different working pH values. As it can be seen from
Figure 3, the detection limit of urease-NH
4+-ISE significantly depends on the working solution pH. The highest detection limit was obtained for a working solution pH equal to 7.19 and was equal to 10
−3 M of urea in solution. The slope of potentiometric dependence was equal to 55.9 ± 1.7 mV/dec (R
2 = 0.999).
Figure 3.
Potentiometric response of urea biosensors based on the ammonium ion-selective carbon-plastic electrodes in (■) 10−4 M Tris pH = 7.19, () H2O, and () 0.1 M acetate buffer pH = 5.35 solutions.
Figure 3.
Potentiometric response of urea biosensors based on the ammonium ion-selective carbon-plastic electrodes in (■) 10−4 M Tris pH = 7.19, () H2O, and () 0.1 M acetate buffer pH = 5.35 solutions.
If the determination was performed in the absence of a buffer, just in deionized water of pH close to 6.5, the linear dependence of potential on urea concentration in solution ranged from 10−1 M to 10−4 M, with a detection limit close 10−4 M. The slope of the dependence was within the range of experimental error, equal to that obtained in a Tris buffer of pH = 7.19 and was equal to 53.5 ± 3.2 mV/dec (R2 = 0.993).
For the measurements performed in an acetic buffer of pH = 5.35 (first the sensor was in contact with urea water solution for 5 min, then the solution pH was adjusted by addition of an acetic buffer, for the potentiometric measurement) the detection limit of urea obtained was equal to 10−5 M. The linear dependence within the range from 0.1 to 10−5 M was characterized with a slope of 50.0 ± 5.3 mV/dec. (R2 = 0.968). Thus, the slope of the dependence was within the limit of experimental error, comparable to above given values.
The observed detection limit dependence on the pH of the working solution results from the simple pH dependence of NH
4+ ion concentration for the total ammonium ion concentration, C, in solution:
Taking into account that K
a of NH
4+ is close to 10
−9.2, it can be expected that, for a solution of pH = 5.35 (acetic buffer used), practically all NH
4+ ions generated as a result of urea enzymatic hydrolysis are present in solution as ammonium ions,
i.e., C is close to [NH
4+]. Conversely, for slightly alkaline solution of pH = 7.19 (Tris buffer used), about 1% of total ammonium ion concentration is present in the solution as NH
3. However, from the dependence presented in
Figure 3, it is clear that the shift in the detection limit of potentiometric biosensor tested for the change of working solution pH from 5.35 to 7.19 is close to two orders of magnitude. This effect clearly points to a local effect of amplification (for pH = 5.35) or attenuation (for pH = 7.19) of the analytical signal obtained by simple solution equilibrium means.
Furthermore, for the urea detection based on the change of pH of the sample, due to alkalization of the solution in the course of enzymatic hydrolysis of urea, to maximize the sensitivity the pH of the working solution should be kept rather high to prevent spontaneous conversion of ammonia generated to ammonium ions.
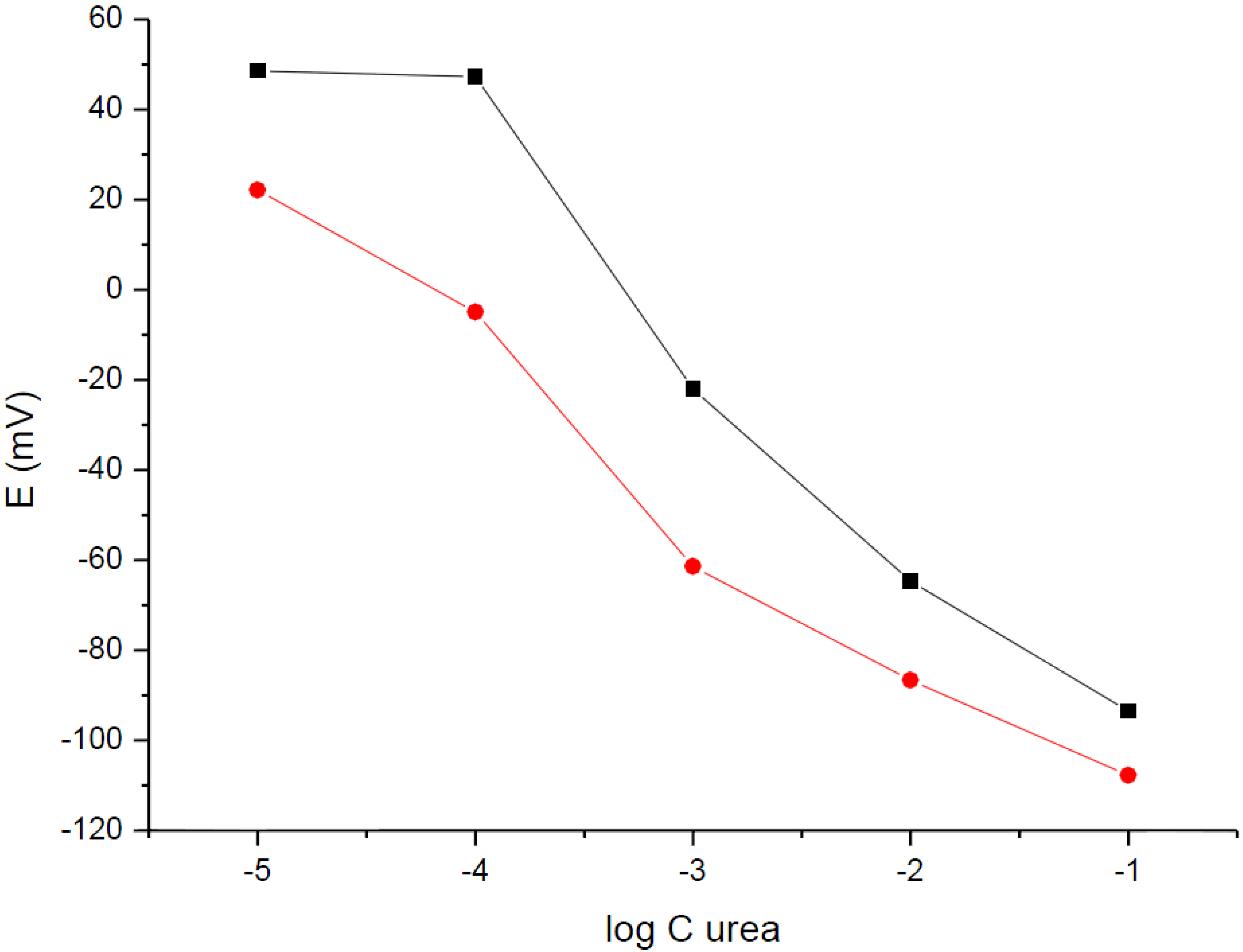
To check the effect of pH on the responses of urease-H
+-ISE, the dependences of potential recorded on the change of urea concentration was recorded in a solution of Tris buffer (pH = 7.19) and in deionized water (
Figure 4). Similarly, as in the case of urease-NH
4+-ISE, pH change has a profound effect on the obtained responses. The decrease of potential recorded in the 10
−4 M Tris buffer (pH = 7.19) for the change in the urea concentration in solution was observed within the range form 10
−4 M to 0.1 M. The dependence recorded was not linear and higher sensitivity was observed for lower concentrations of urea in solution. This effect can be attributed to the low buffering capacity of the buffer used. The low capacity was chosen to minimize the buffering effect, to lower the interfering ions influence (
i.e., ions present in the buffer), and to allow manifestation of the sample pH by a change of the electrode potential. Nevertheless, the effect of preventing spontaneous protonation of ammonia to form NH
4+ was achieved only at the low urea concentration tested.
Figure 4.
Potentiometric response of urea biosensors using H+-ISE carbon-plastic electrodes recorded in (■) 10−4 M Tris, pH = 7.19 and () H2O.
Figure 4.
Potentiometric response of urea biosensors using H+-ISE carbon-plastic electrodes recorded in (■) 10−4 M Tris, pH = 7.19 and () H2O.
Working in deionized water (in the absence of buffer) has the advantage of being a relatively sensitive system. Indeed, in the absence of a buffer, the significant potential change was observed when the urea concentration in the sample was changed from 10−5 to 10−4 M (detection limit close to 10−5 M). For the higher urea concentrations, similarly as in the case of the Tris buffer-containing samples described above, the potential dependence was observed; however, this relation was not linear. It should be stressed that for low concentration ranges (where the dependence of potential on the logarithm of concentration of urea is close to linear) the sensitivity, defined as a change of the potential per order of magnitude urea concentration, the change was lower for measurements performed in deionized water. The reproducibility of responses obtained for different, but nominally the same, sensors was in the range of single millivolts, but it can be probably further optimized if the sensor production method is automated.
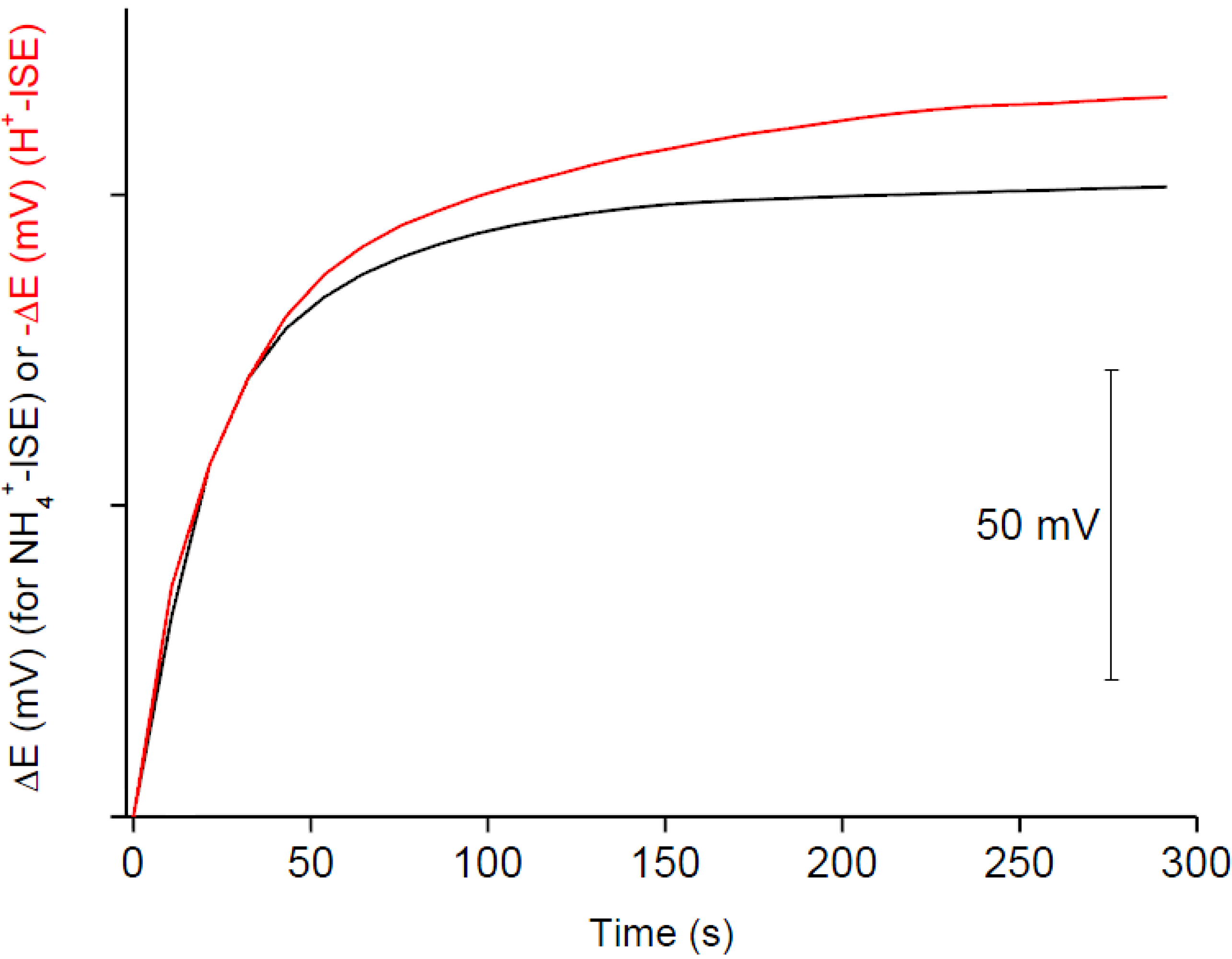
To compare the response time of H
+- and NH
4+-selective ISEs, with ion-selective membranes modified with urease in cellulose acetate layer, the potential accompanying change of the urea concentration (10
−3 M) in solution was followed for both sensors, while working in DI water (
Figure 5). As it can be seen from
Figure 5, both sensors quite rapidly respond to the introduction of urea to the solution. The response stabilizes faster for NH
4+-ISE; after about 100 s a stable potential is recorded. Conversely, for H
+-ISE, after the abrupt potential change directly after introduction of urea to the sample, a small potential change is observed even for the longer times tested. This, in our opinion, can be related to participation of the biosensing layer in protolitic equilibrium.
Figure 5.
The change of the potential recorded for biosensors containing urease in a cellulose acetate layer for H+-ISE (red line) and for NH4+-ISE (black line), following introduction 10−3 M urea to the unbuffered (i.e., DI water-containing) sample. The lines were shifted to give the same starting potentials; for H+-ISE, when the decrease of potential is recorded, the negative potential change was plotted.
Figure 5.
The change of the potential recorded for biosensors containing urease in a cellulose acetate layer for H+-ISE (red line) and for NH4+-ISE (black line), following introduction 10−3 M urea to the unbuffered (i.e., DI water-containing) sample. The lines were shifted to give the same starting potentials; for H+-ISE, when the decrease of potential is recorded, the negative potential change was plotted.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}