
Microstructure and Photoluminescence of ZrTiO4:Eu3+ Phosphors: Host-Sensitized Energy Transfer and Optical Thermometry

 ,
,
Abstract
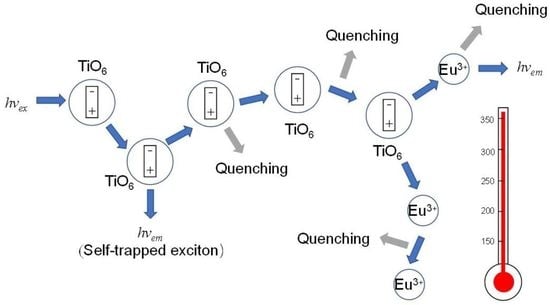
:
1. Introduction
2. Experimental
2.1. Chemicals and Materials Preparation
2.2. Characterization and Measurements
3. Results and Discussion
3.1. Composition, Crystalline Structure and Morphology
3.2. Electronic Band Structure of Zr1−xEuxTiO4
3.3. Luminescence Properties of ZrTiO4
3.4. Luminescence Properties of Zr1−xEuxTiO4
3.5. Optical Thermometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Diaz, A.L. Progress in understanding host-sensitized excitation processes in luminescent materials. ECS J. Solid State Sci. Technol. 2019, 8, R14–R26. [Google Scholar] [CrossRef]
- Marciniak, L.; Kniec, K.; Elżbieciak-Piecka, K.; Trejgis, K.; Stefanska, J.; Dramićanin, M. Luminescence thermometry with transition metal ions. A review. Coordin. Chem. Rev. 2022, 469, 214671–214702. [Google Scholar] [CrossRef]
- Bednarkiewicz, A.; Drabik, J.; Trejgis, K.; Jaque, D.; Ximendes, E.; Marciniak, L. Luminescence based temperature bio-imaging: Status, challenges, and perspectives. Appl. Phys. Rev. 2021, 8, 011317–011371. [Google Scholar] [CrossRef]
- Jaque, D.; Vetrone, F. Luminescence nanothermometry. Nanoscale 2012, 4, 4301–4326. [Google Scholar] [CrossRef]
- Zheng, B.; Fan, J.; Chen, B.; Qin, X.; Wang, J.; Wang, F.; Deng, R.; Liu, X. Rare-earth doping in nanostructured inorganic materials. Chem. Rev. 2022, 122, 5519–5603. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Gusmano, G.; Freer, R.; Smith, P. Zirconium titanate microwave dielectrics prepared via polymeric precursor route. J. Eur. Ceram. Soc. 1999, 19, 959–963. [Google Scholar] [CrossRef]
- Polliotto, V.; Albanese, E.; Livraghi, S.; Indyka, P.; Sojka, Z.; Pacchioni, G.; Giamello, E. Fifty–Fifty Zr–Ti solid solution with a TiO2-Type structure: Electronic structure and photochemical properties of zirconium titanate ZrTiO4. J. Phys. Chem. C 2017, 121, 5487–5497. [Google Scholar] [CrossRef]
- Cosentino, I.C.; Muccillo, E.N.S.; Muccillo, R. Development of zirconia-titania porous ceramics for humidity sensors. Sens. Actuat. B-Chem. 2003, 96, 677–683. [Google Scholar] [CrossRef]
- Park, Y. Zr0.98Sn0.02TiO4 single crystal in a low electric field: Birefringence, dielectric, and synchrotron X-ray studies. Phys. Rev. B 2000, 62, 8794–8801. [Google Scholar] [CrossRef]
- Lynch, R.W.; Morosin, B. Thermal expansion, compressibility, and polymorphism in hafnium and zirconium titanates. J. Am. Ceram. Soc. 1972, 55, 409–413. [Google Scholar] [CrossRef]
- Bordet, P.; Mchale, A.; Santoro, A.; Roth, R. Powder neutron diffraction study of ZrTiO4, Zr5Ti7O24, and FeNb2O6. J. Solid State Chem. 1986, 64, 30–46. [Google Scholar] [CrossRef]
- Bayer, G.; Hofmann, M.; Gauckler, L.J. Effect of ionic substitution on the thermal expansion of ZrTiO4. J. Am. Ceram. Soc. 1991, 74, 2205–2208. [Google Scholar] [CrossRef]
- Mchale, A.; Roth, R. Investigation of the phase-transition in ZrTiO4 and ZrTiO4-SnO2 solid-solutions. J. Am. Ceram. Soc. 1983, 66, C18–C20. [Google Scholar] [CrossRef]
- Oanh, L.M.; Do, D.B.; Hung, N.M.; Thang, D.V.; Phuong, D.T.; Ha, D.T.; Van Minh, N. Formation of Crystal structure of zirconium titanate ZrTiO4 powders prepared by Sol–Gel method. J. Electron. Mater. 2016, 45, 2553–2558. [Google Scholar] [CrossRef]
- de Lucena, P.R.; Roberto, L.E.; Pontes, F.M.; Longo, E.; Pizani, P.S.; Arana, V.J. Photoluminescence: A probe for short, medium and long-range self-organization order in oxide. J. Solid State Chem. 2006, 179, 3997–4002. [Google Scholar] [CrossRef]
- Shi, Q.; You, F.; Huang, S.; Cui, J.; Huang, Y.; Tao, Y. Host sensitization of Tb3+ through Gd3+ in Na3Gd(BO3):Tb3+. J. Alloys Compd. 2016, 654, 441–444. [Google Scholar] [CrossRef]
- Eempicki, A. The physics of inorganic scintillators. J. Appl. Spectrosc. 1995, 62, 787–802. [Google Scholar] [CrossRef]
- Richard, C.P.; Blasse, G. Energy transfer in concentrated systems. In Luminescence and Energy Transfer; Springer: Berlin/Heidelberg, Germany, 1980; Volume 42, pp. 43–96. [Google Scholar]
- Blasse, G. Luminescence of inorganic solids: From isolated centres to concentrated systems. Prog. Solid State Chem. 1988, 18, 79–171. [Google Scholar] [CrossRef]
- Alarcon, J.; Blasse, G. Luminescence of Eu3+-doped lanthanum titanate (La2TiO5), a system with one-dimensional energy migration. J. Phys. Chem. Solids 1992, 53, 677–680. [Google Scholar] [CrossRef]
- Dehaart, L.; Devries, A.; Blasse, G. On the photoluminescence of semiconducting titanates applied in photoelectrochemical cells. J. Solid State Chem. 1985, 59, 291–300. [Google Scholar] [CrossRef]
- Pan, G.; Zhang, L.; Wu, H.; Qu, X.; Wu, H.; Hao, Z.; Zhang, L.; Zhang, X.; Zhang, J. On the luminescence of Ti4+ and Eu3+ in monoclinic ZrO2: High performance optical thermometry derived from energy transfer. J. Mater. Chem. C 2020, 8, 4518–4533. [Google Scholar] [CrossRef]
- Yu, D.; Li, H.; Zhang, D.; Zhang, Q.; Meijerink, A.; Suta, M. One ion to catch them all: Targeted high-precision Boltzmann thermometry over a wide temperature range with Gd3+. Light-Sci. Appl. 2021, 10, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Chen, F.; Wu, H.; Wu, H.; Zhang, L.; Pan, G.; Liu, F.; Wang, X.; Zhang, J. Afterglow-intensity-ratio-based temperature sensing using a persistent phosphor. J. Mater. Chem. C 2022, 10, 11884–11890. [Google Scholar] [CrossRef]
- Marciniak, L.; Bednarkiewicz, A.; Strek, W. Tuning of the up-conversion emission and sensitivity of luminescent thermometer in LiLaP4O12: Tm, Yb nanocrystals via Eu3+ dopants. J. Lumin. 2017, 184, 179–184. [Google Scholar] [CrossRef]
- Yu, H.; Su, W.; Chen, L.; Deng, D.; Xu, S. Excellent temperature sensing characteristics of europium ions self-reduction Sr3P4O13 phosphors for ratiometric luminescence thermometer. J. Alloys Compd. 2019, 806, 833–840. [Google Scholar] [CrossRef]
- Maciejewska, K.; Pozniak, B.P.; Tikhomirov, M.; Kobylin´ska, A.; Marciniak, L. Synthesis, cytotoxicity assessment and optical properties characterization of colloidal GdPO4: Mn2+, Eu3+ for high sensitivity luminescent nanothermometers operating in the physiological temperature range. Nanomaterials 2020, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Kniec, K.; Marciniak, L. Spectroscopic properties of LaGaO3: V, Nd3+ nanocrystals as a potential luminescent thermometer. Phys. Chem. Chem. Phys. 2018, 20, 21598–21606. [Google Scholar] [CrossRef]
- Kolesnikov, I.E.; Mamonova, D.V.; Kurochkin, M.A.; Kolesnikov, E.Y.; Lähderanta, E. Multimode luminescence thermometry based on emission and excitation spectra. J. Lumin. 2021, 231, 117828–117831. [Google Scholar] [CrossRef]
- Elzbieciak, K.; Bednarkiewicz, A.; Marciniak, L. Temperature sensitivity modulation through crystal field engineering in Ga3+ co-doped Gd3Al5-xGaxO12:Cr3+, Nd3+ nanothermometers. Sens. Actuat. B-Chem. 2018, 269, 96–102. [Google Scholar] [CrossRef]
- Mykhaylyk, V.; Kraus, H.; Zhydachevskyy, Y.; Tsiumra, V.; Luchechko, A.; Wagner, A.; Suchocki, A. Multimodal non-contact luminescence thermometry with Cr-doped oxides. Sensors 2020, 20, 5259. [Google Scholar] [CrossRef]
- Marciniak, L.; Bednarkiewicz, A.; Kowalska, D.; Strek, W. A new generation of highly sensitive luminescent thermometers operating in the optical window of biological tissues. J. Mater. Chem. C 2016, 4, 5559–5563. [Google Scholar] [CrossRef]
- Katsumata, T.; Nakayama, A.; Kano, Y.; Aizawa, H.; Komuro, S. IEEE Characteristics of Ti-doped sapphire for fluorescence thermo-sensor. In Proceedings of the 2007 International Conference on Control, Automation and Systems, Seoul, Republic of Korea, 17–20 October 2007; pp. 1025–1028. [Google Scholar]
- Drabik, J.; Cichy, B.; Marciniak, L. New type of nanocrystalline luminescent thermometers based on Ti3+/Ti4+ and Ti4+/Ln3+ (Ln3+ = Nd3+, Eu3+, Dy3+) luminescence intensity ratio. J. Phys. Chem. C 2018, 122, 14928–14936. [Google Scholar] [CrossRef]
- Wang, C.; Jin, Y.; Yuan, L.; Wu, H.; Ju, G.; Li, Z.; Liu, D.; Lv, Y.; Chen, L.; Hu, Y. A spatial/temporal dual-mode optical thermometry platform based on synergetic luminescence of Ti4+-Eu3+ embedded flexible 3D micro-rod arrays: High-sensitive temperature sensing and multi-dimensional high-level secure anti-counterfeiting. Chem. Eng. J. 2019, 374, 992–1004. [Google Scholar] [CrossRef]
- Dramićanin, M.D. Sensing temperature via downshifting emissions of lanthanide-doped metal oxides and salts. A review. Methods Appl. Fluoresc. 2016, 4, 042001–042024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, N.G.; Per-Anders, H.; Helmer, F.; Ola, N. Luminescent properties of europium titanium phosphate thin films deposited by atomic layer deposition. RSC Adv. 2017, 7, 8051–8059. [Google Scholar] [CrossRef] [Green Version]
- Veronica, P.; Marco, B.; Rosanna, R.; Asmaa El, K.; Manuela, R.; Giancarlo, D.V.; Francesco, C. Characterization and Luminescence of Eu3+- and Gd3+-Doped Hydroxyapatite Ca10(PO4)6(OH)2. Crystals 2020, 10, 806. [Google Scholar] [CrossRef]
- de Haart, L.G.J.; Boessenkoo, H.J.; Blasse, G. Photoelectrochemical properties of titanium niobate (TiNb2O7) and titanium tantalate (TiTa2O7). Mater. Chem. Phys. 1985, 13, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Blasse, G.; Bril, A. The influence of crystal structure on the fluorescence of oxidic niobates and related compounds. Z. Phys. Chem. 1968, 57, 187–202. [Google Scholar] [CrossRef]
- Blasse, G. Optical electron-transfer between metal-ions and its consequences. Struct. Bond. 1991, 76, 153–187. [Google Scholar]
- Haynes, W.M.; Bruno, T.J.; David, R. CRC Handbook of Chemistry and Physics, 96th ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Krebs, M.A.; Condrate, R.A. A Raman spectral characterization of various crystalline mixtures in the ZrO2-TiO2 and HfO2-TiO2 systems. J. Mater. Sci. Lett. 1988, 7, 1327–1330. [Google Scholar] [CrossRef]
- Nowak, M.; Kauch, B.; Szperlich, P. Determination of energy band gap of nanocrystalline SbSI using diffuse reflectance spectroscopy. Rev. Sci. Instrum. 2009, 80, 046107–046110. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Lin, P.; Tseng, T. Optical properties of ZrTiO4 films grown by radio-frequency magnetron sputtering. J. Appl. Phys. 1995, 77, 4445–4451. [Google Scholar] [CrossRef]
- Toyozawa, Y. Electrons, Holes and Excitons in Deformable Lattice. In Relaxation of Elementary Excitations; Ryogo, K., Eiichi, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1980; pp. 3–18. [Google Scholar]
- Blasse, G. The influence of crystal structure on the luminescence of tantalates and niobates. J. Solid State Chem. 1988, 72, 72–79. [Google Scholar] [CrossRef]
- Blasse, G.; Bril, A. On the Eu3+ Fluorescence in mixed metal oxides. III. Energy transfer in Eu3+-activated tungstates and molybdates of the type Ln2WO6 and Ln2MoO6. J. Chem. Phys. 1966, 45, 2350–2355. [Google Scholar] [CrossRef]
- Blasse, G.; Bril, A. Fluorescence of Eu3+-activated sodium lanthanide titanates (NaLn1–xEuxTiO4). J. Chem. Phys. 1968, 48, 3652–3656. [Google Scholar] [CrossRef]
- Ran, W.; Noh, H.; Park, S.; Lee, B.; Kim, J.; Jeong, J.; Shi, J. Er3+-activated NaLaMgWO6 double perovskite phosphors and their bifunctional application in solid-state lighting and non-contact optical thermometry. Dalton Trans. 2019, 48, 4405–4412. [Google Scholar] [CrossRef]
- Shi, R.; Lin, L.; Dorenbos, P.; Liang, H. Development of a potential optical thermometric material through photoluminescence of Pr3+ in La2MgTiO6. J. Mater. Chem. C 2017, 5, 10737–10745. [Google Scholar] [CrossRef]
- Glais, E.; Pellerin, M.; Castaing, V.; Alloyeau, D.; Touati, N.; Viana, B.; Chaneac, C. Luminescence properties of ZnGa2O4:Cr3+, Bi3+ nanophosphors for thermometry applications. RSC Adv. 2018, 8, 41767–41774. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, C.; Deng, D.; Yu, H.; Li, H.; Wang, L.; Shen, C.; Jing, X.; Xu, S. High-sensitivity based on Eu2+/Cr3+ co-doped BaAl12O19 phosphors for dual-mode optical thermometry. J. Lumin. 2021, 237, 118142–118150. [Google Scholar] [CrossRef]
- Li, L.; Tong, Y.; Chen, J.; Chen, Y.; Ghulam, A.A.; Chen, L.; Pang, T.; Guo, H. Up-conversion and temperature sensing properties of Na2GdMg2(VO4)3: Yb3+, Er3+ phosphors. J. Am. Ceram. Soc. 2021, 105, 384–391. [Google Scholar] [CrossRef]
- Piotrowski, W.M.; Ristic, Z.; Dramićanin, M.D.; Marciniak, Ł. Modification of the thermometric performance of the lifetime-based luminescent thermometer exploiting Ti3+ emission in SrTiO3 and CaTiO3 by doping with lanthanide ions. J. Alloys Compd. 2022, 906, 164398–164406. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Luminescent Ions for Sensing | Excitation Wavelength (nm) | Maximum Sa (K−1) | Maximum Sr (%K−1) | Temperature Range (K) | Ref. |
|---|---|---|---|---|---|---|
| LiLaP4O12 | Tm3+, Eu3+ | 975 | - | 1.8 | 123–473 | [25] |
| Sr3P4O13 | Eu2+, Eu3+ | 360 | 0.008 | 1.06 | 293–573 | [26] |
| GdPO4: Mn2+, Eu3+ | Mn2+, Eu3+ | 375 | - | 8.88 | 303–323 | [27] |
| NaLaMgWO6:Er3+ | Er3+ | 378 | 0.0223 | 1.04 | 303–483 | [50] |
| La2MgTiO6: Pr3+ | Pr3+ | 350 | 0.072 | 1.28 | 80–500 | [51] |
| ZnGa2O4:Cr3+, Bi3+ | Cr3+, Bi3+ | 430 | - | 1.93 | 293–473 | [52] |
| BaAl12O19:Eu2+, Cr3+ | Eu2+, Cr3+ | 325 | 0.0143 | 0.466 | 290–480 | [53] |
| Na2GdMg2(VO4)3 | Yb3+, Er3+ | 980 | 0.749 | 0.976 | 303–573 | [54] |
| CaTiO3:Ti3+, Yb3+ | Ti3+, Yb3+ | 266 | 0.311 | 3.55 | 77–480 | [55] |
| GaTi2O7:Eu3+ | Traps Eu3+ | - | - | 0.46 | 313–423 | [36] |
| LiTaO3:Ti4+, Eu3+ | Ti4+, Eu3+ | 270 | 0.671 | 3.395 | 303–443 | [35] |
| ZrO2: Ti4+, Eu3+ | Ti4+, Eu3+ | 280 | 0.414 | 3.84 | 303–413 | [22] |
| ZrTiO4: Eu3+ | Eu3+, Ti4+ | 340 | 0.084 | 1.11 | 153–313 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, A.; Pan, G.-H.; Wu, H.; Zhang, L.; Zhang, L.; Wu, H.; Zhang, J. Microstructure and Photoluminescence of ZrTiO4:Eu3+ Phosphors: Host-Sensitized Energy Transfer and Optical Thermometry. Chemosensors 2022, 10, 527. https://doi.org/10.3390/chemosensors10120527
Gu A, Pan G-H, Wu H, Zhang L, Zhang L, Wu H, Zhang J. Microstructure and Photoluminescence of ZrTiO4:Eu3+ Phosphors: Host-Sensitized Energy Transfer and Optical Thermometry. Chemosensors. 2022; 10(12):527. https://doi.org/10.3390/chemosensors10120527
Chicago/Turabian StyleGu, Anheng, Guo-Hui Pan, Huajun Wu, Liangliang Zhang, Ligong Zhang, Hao Wu, and Jiahua Zhang. 2022. "Microstructure and Photoluminescence of ZrTiO4:Eu3+ Phosphors: Host-Sensitized Energy Transfer and Optical Thermometry" Chemosensors 10, no. 12: 527. https://doi.org/10.3390/chemosensors10120527