Is Five Percent Too Small? Analysis of the Overlaps between Disorder, Coiled Coil and Collagen Predictions in Complete Proteomes

Abstract
:1. Introduction
2. Experimental

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomic group | Number of proteomes |
|---|---|
| Archaea | 18 |
| Bacteria | 69 |
| Embryophyta | 10 |
| Eukaryota (except Metazoa & Embryophyta) | 49 |
| Metazoa | 57 |
| Viruses | 105 |
| All organisms | 308 |
3. Results and Discussion
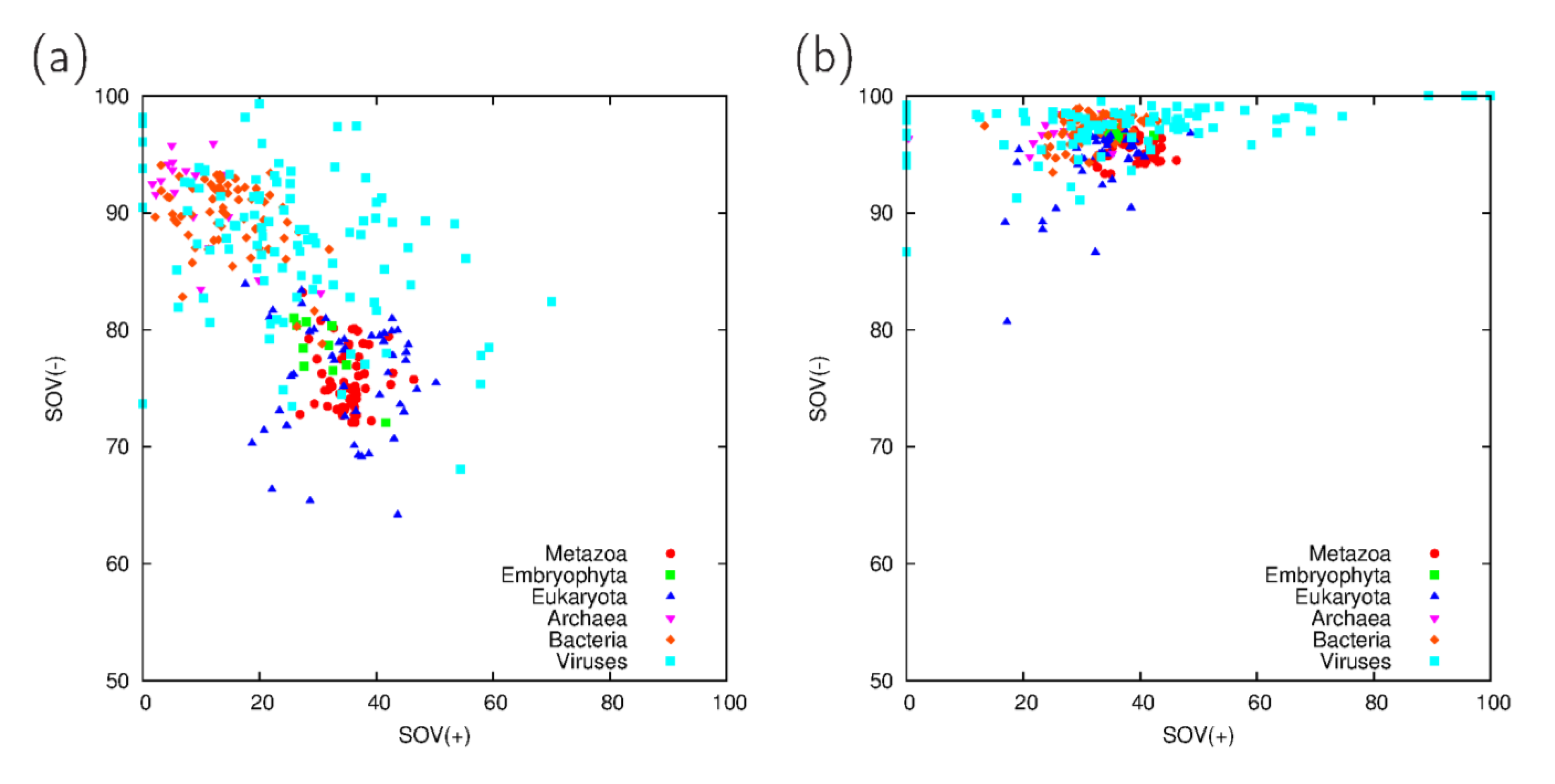
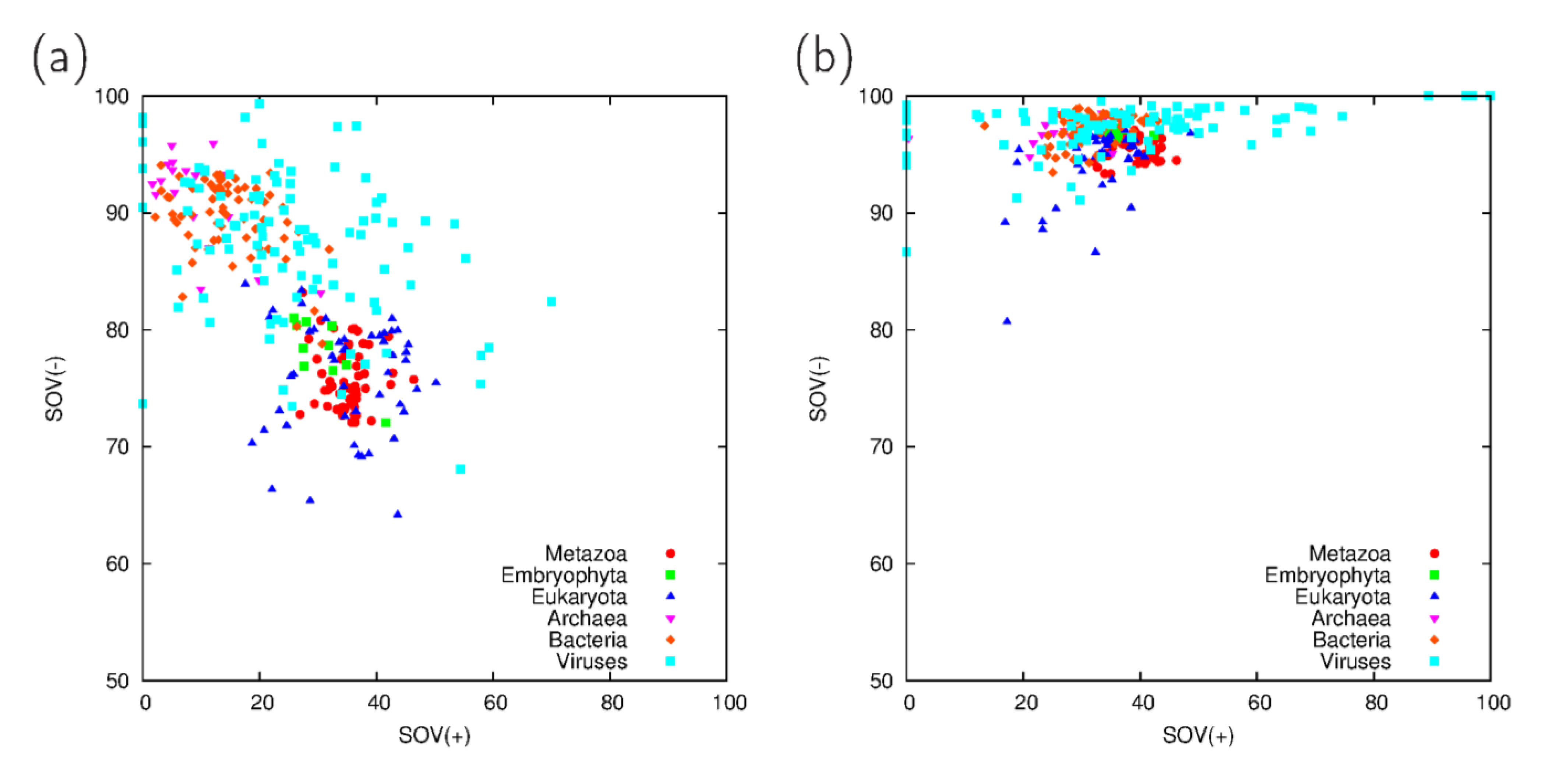
3.1. Comparison of Predictions on Full Proteomes
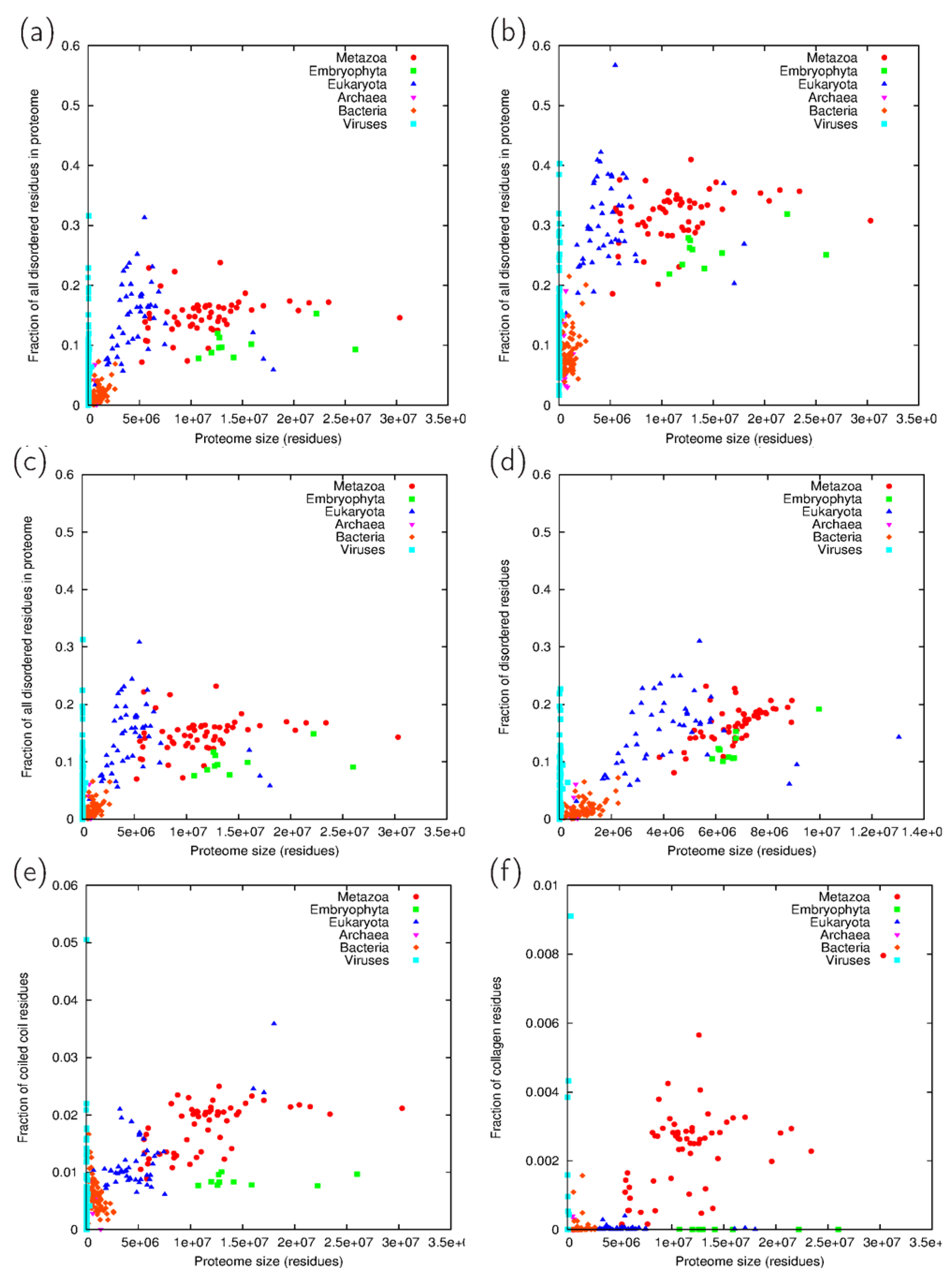
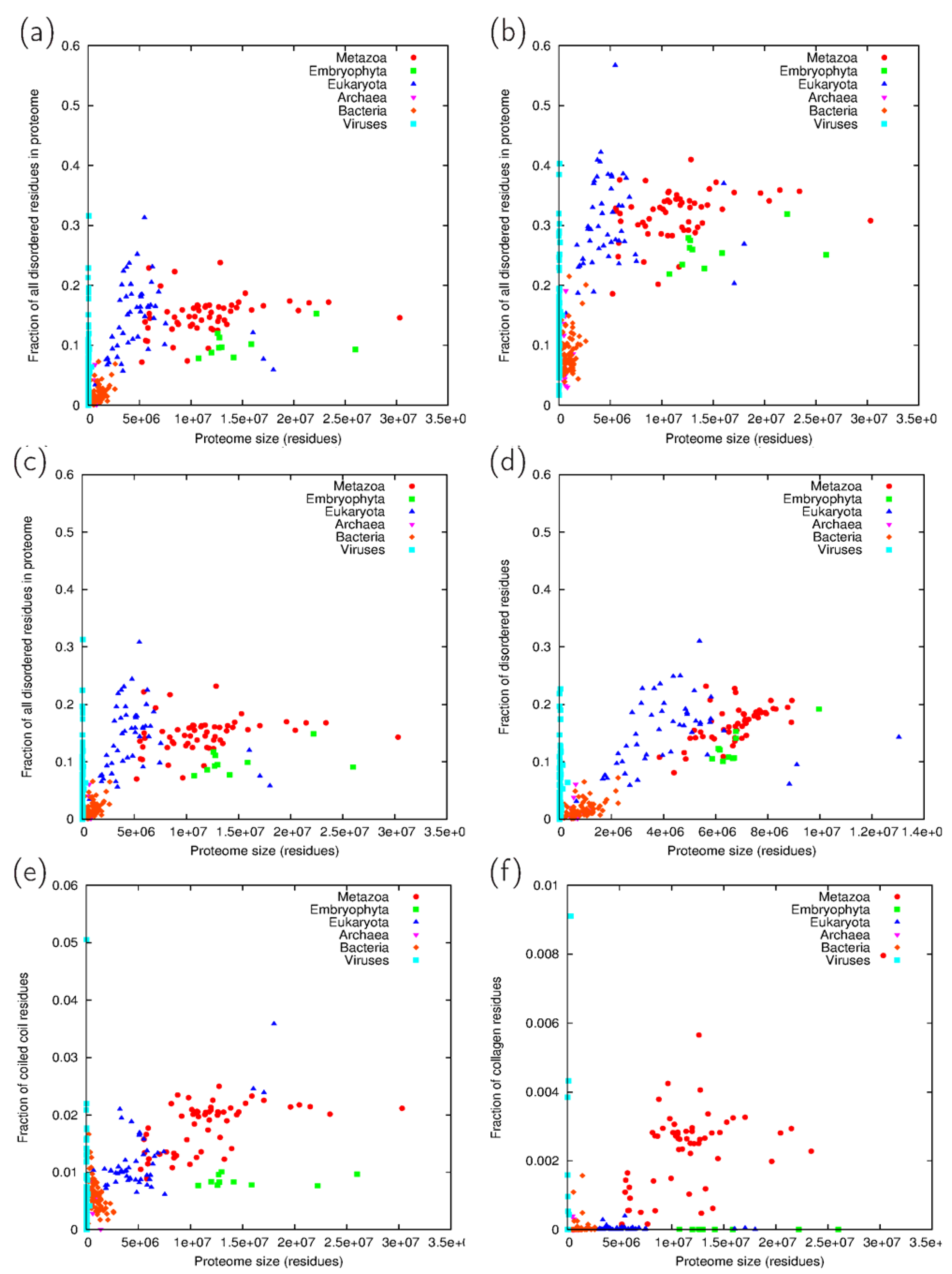
3.2. Fraction Residues Predicted to Lie in Disordered, Coiled Coil and Collagen Segments
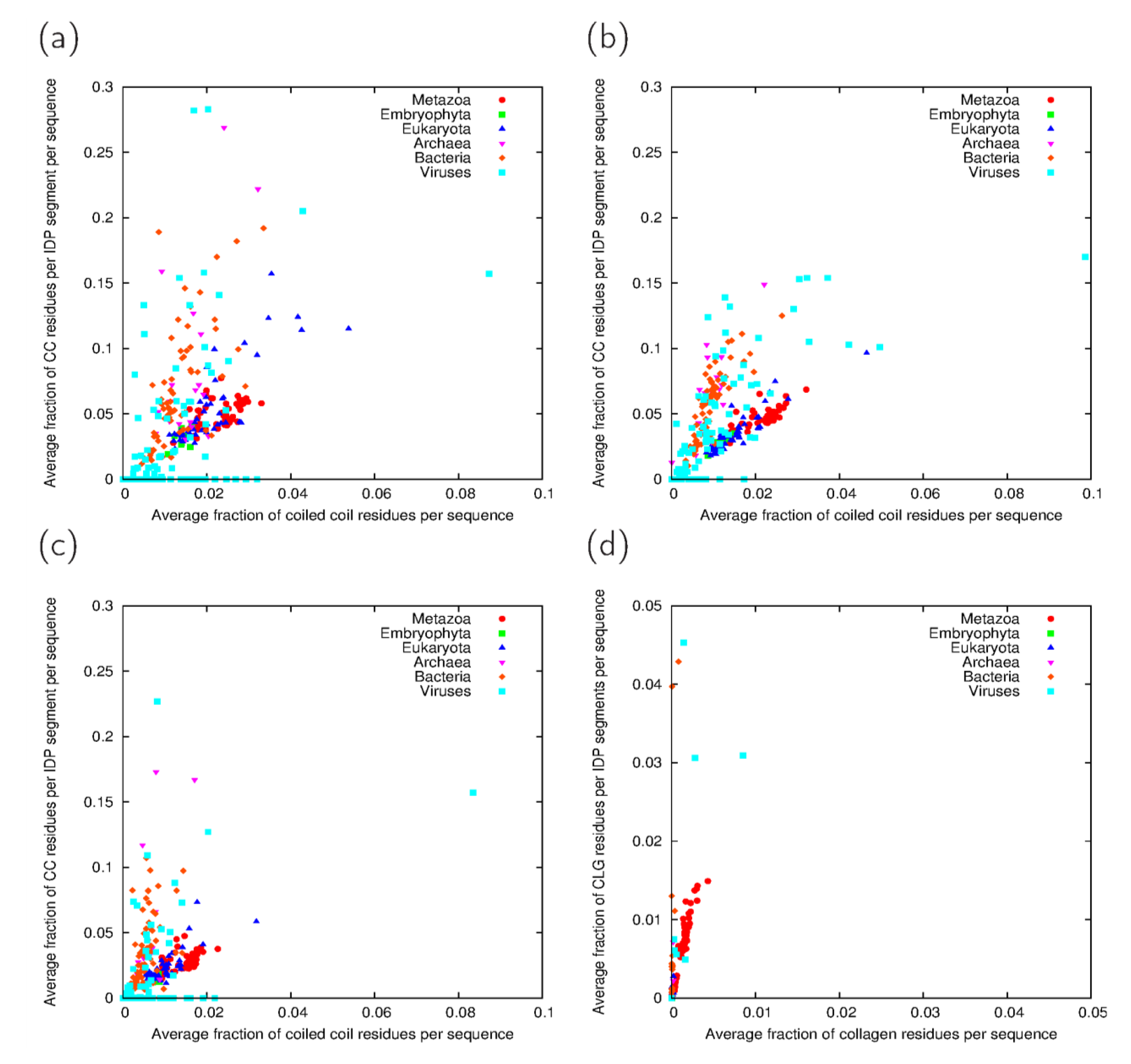
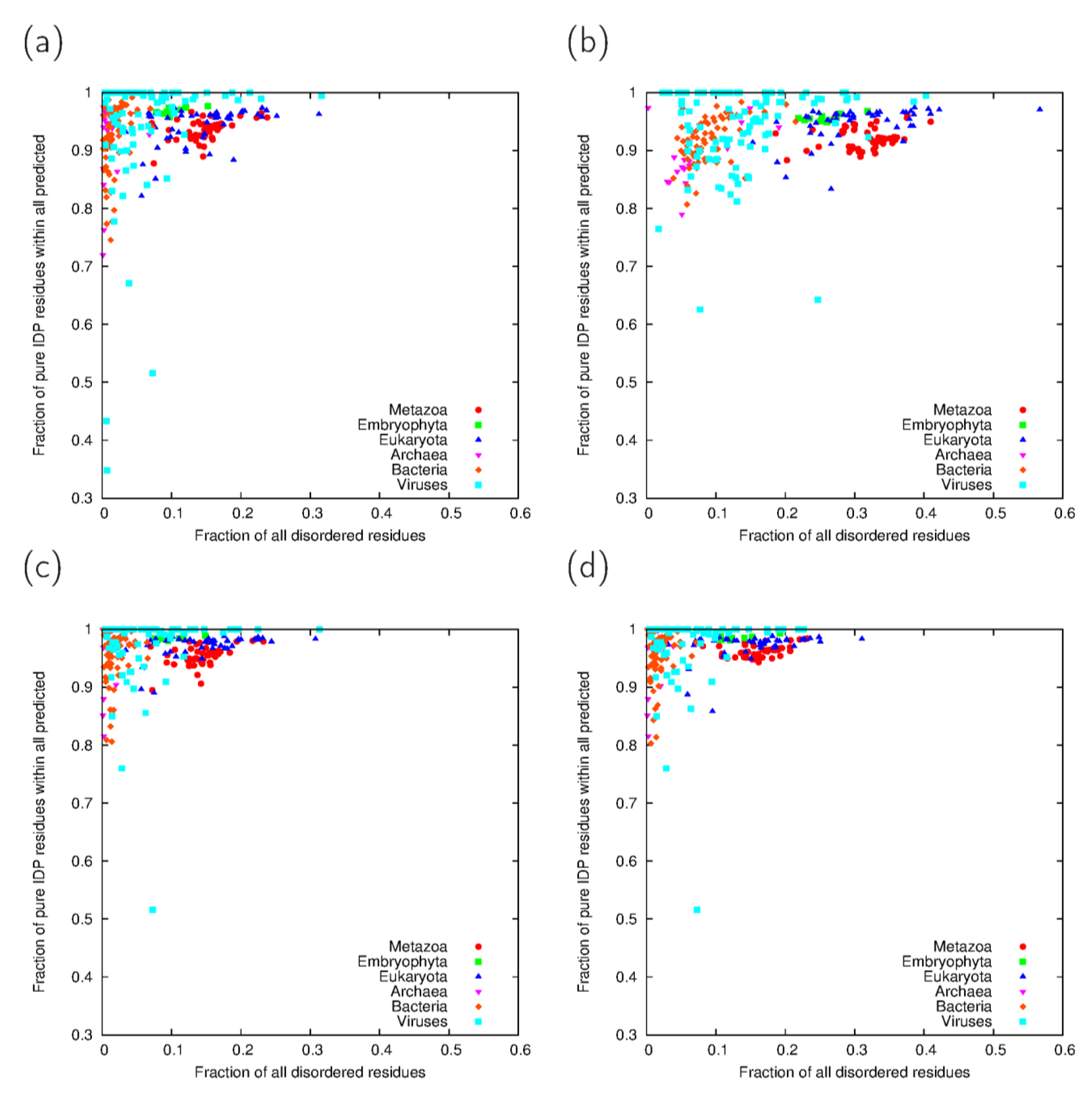
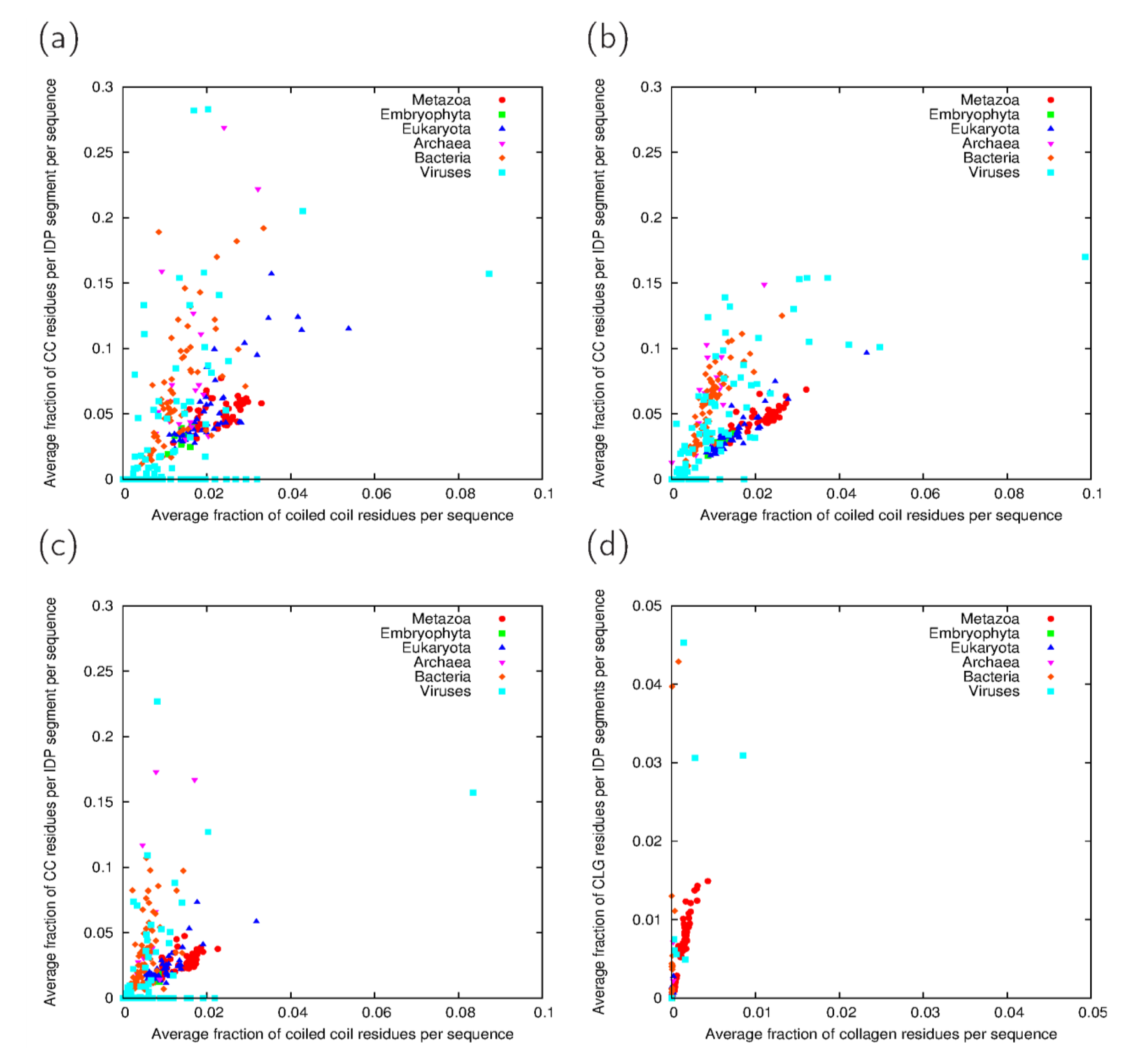
3.3. Analysis of Cross-Predicted Residues
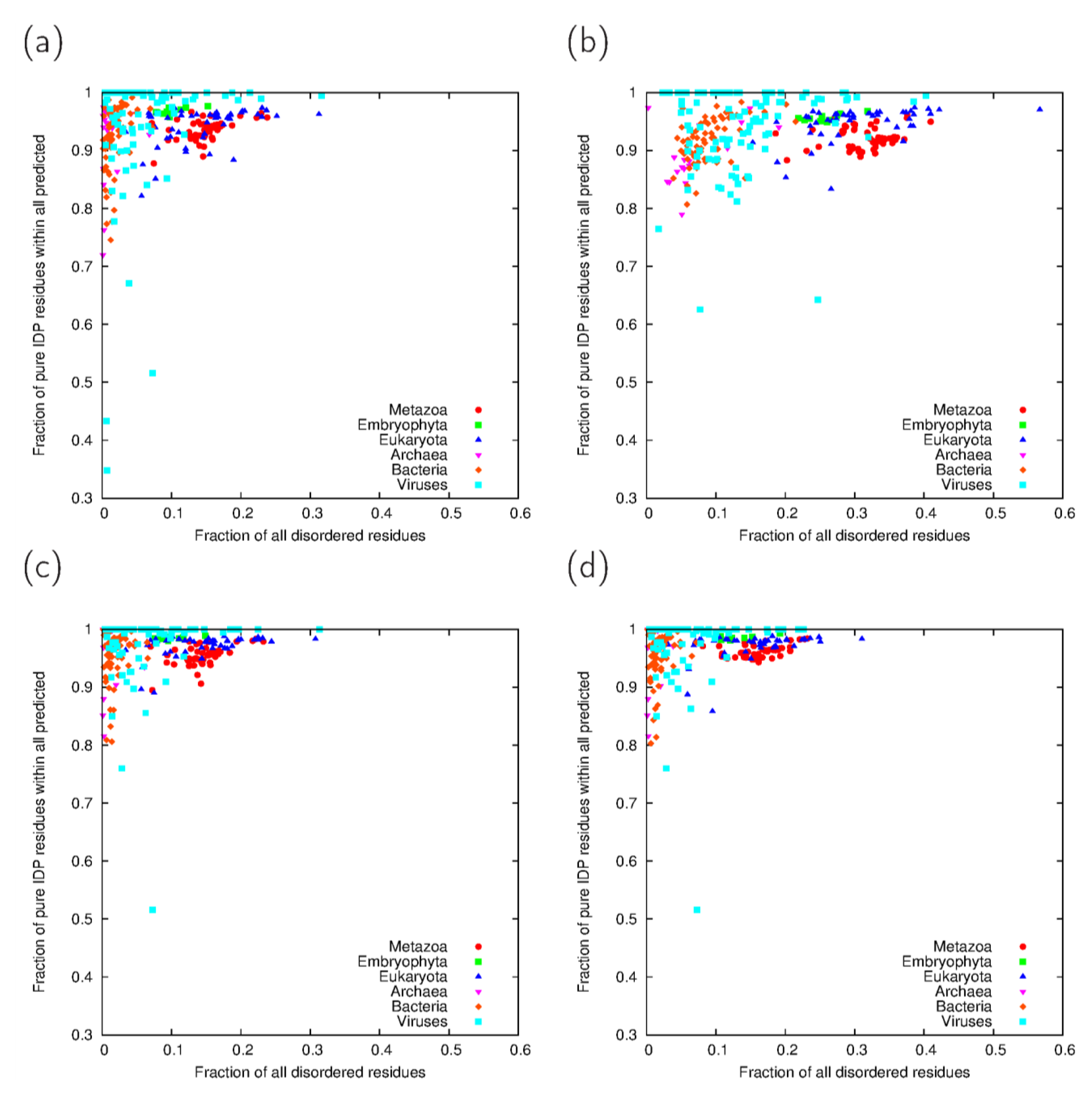
| Taxonomic group | IUPred-ncoils | IUPred-Paircoil2 | VSL2B-ncoils | VSL2B-Paircoil2 | Consensus | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Full | 40% | Full | 40% | Full | 40% | Full | 40% | Full | 40% | |
| Archaea | 1.000 | 1.000 | 1.000 | 1.000 | 0.998 | 0.998 | 0.999 | 0.999 | 1.000 | 1.000 |
| Bacteria | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 0.999 | 1.000 | 1.000 | 1.000 | 1.000 |
| Embryophyta | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Eukaryota | 0.997 | 0.998 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 |
| Metazoa | 0.999 | 0.999 | 1.000 | 0.999 | 1.000 | 0.999 | 1.000 | 1.000 | 1.000 | 0.999 |
| Viruses | 0.999 | 0.999 | 0.999 | 0,999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 | 0.999 |
| Taxonomic group | IUPred-ncoils | IUPred-Paircoil2 | VSL2B-ncoils | VSL2B-Paircoil2 | Consensus | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Full | 40% | Full | 40% | Full | 40% | Full | 40% | Full | 40% | |
| Archaea | 0.917 | 0.910 | 0.907 | 0.912 | 0.546 | 0.518 | 0.652 | 0.642 | 0.894 | 0.896 |
| Bacteria | 0.926 | 0.923 | 0.976 | 0.977 | 0.646 | 0.693 | 0.842 | 0.857 | 0.917 | 0.914 |
| Embryophyta | 0.991 | 0.946 | 0.995 | 0.932 | 0.879 | 0.063 | 0.911 | 0.053 | 0.990 | 0.916 |
| Eukaryota | 0.985 | 0.976 | 0.995 | 0.992 | 0.957 | 0.926 | 0.981 | 0.965 | 0.991 | 0.986 |
| Metazoa | 0.987 | 0.949 | 0.995 | 0.973 | 0.941 | 0.763 | 0.977 | 0.885 | 0.989 | 0.963 |
3.4. Relevance of Cross-Predictions
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Schad, E.; Tompa, P.; Hegyi, H. The relationship between proteome size, structural disorder and organism complexity. Genome Biol. 2011, 12, R120. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 130, 137–149. [Google Scholar]
- Szappanos, B.; Süveges, D.; Nyitray, L.; Perczel, A.; Gáspári, Z. Folded-unfolded cross-predictions ad protein evolution: the case study of coiled coils. FEBS Lett. 2010, 584, 1623–1627. [Google Scholar] [CrossRef]
- Mészáros, B.; Dosztányi, Z.; Magyar, C.; Simon, I. Bioinformatical approaches to unstructured/ disordered proteinsand their interactions. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes, Springer Series in Bio-Neuroinformatics; Liwo, A., Ed.; Springer: New York, NY, USA, 2014; Volume 1, pp. 525–556. [Google Scholar]
- Romero, P.; Obradovic, P.; Li, X.; Garner, E.C.; Brown, J.C.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Varadi, M.; Kosol, S.; Lebrun, P.; Valentini, E.; Blackledge, M.; Dunker, A.K.; Felli, I.C.; Forman-Kay, J.D.; Kriwacki, R.W.; Pierattelli, R.; et al. pE-DB: A database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucleic Acids Res. 2014, 42, D326–D235. [Google Scholar]
- Moran, L.B.; Schneider, J.P.; Kentsis, A.; Reddy, G.A.; Sosnick, T.R. Transition state heterogenity in GCN4 coiled coil folding studied by using multisite mutations and crosslinking. Proc. Natl. Acad. Sci. USA 1999, 96, 10699–10704. [Google Scholar] [CrossRef]
- Uversky, V. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef]
- McAlinden, A.; Smith, T.A.; Sandell, L.J.; Ficheux, D.; Parry, D.A.D.; Hulmes, D.J.S. Alpha-helical coiled-coil oligomerization domains are most ubiquitous in the collagen superfamily. J. Biol. Chem. 2003, 278, 42200–42207. [Google Scholar]
- Burkhard, P.; Stetefeld, J.; Strelkov, S.V. Coiled coils: A highly versatile protein folding motif. Trends Cell Biol. 2001, 11, 82–88. [Google Scholar] [CrossRef]
- Gáspári, Z.; Nyitray, L. Coiled coils as possible models of protein structure evolution. Biomol. Concepts 2011, 2, 199–210. [Google Scholar]
- Rose, A.; Schraegle, S.J.; Stahlberg, E.A.; Meier, I. Coiled-coil protein composition of 22 proteomes—Differences and common themes in subcellular infrastructure and traffic control. BMC Evol. Biol. 2005, 5, e66. [Google Scholar] [CrossRef]
- Exposito, J.-Y.; Lethias, C. Invertebrate and vertebrate collagens. In Evolution of Extracellular Matrix; Keeley, F.W., Mecham, R.P., Eds.; Springer: New York, NY, USA, 2013; pp. 39–72. [Google Scholar]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clusterin g and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Lupas, A.; van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 2005, 61, 176–182. [Google Scholar]
- McDonnell, A.V.; Jiang, T.; Keating, A.E.; Berger, B. Paircoil2: Improved prediction of coiled coils from sequence. Bioinformatics 2006, 22, 356–358. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C.; Schneider, R. Redefining the goals of protein secondary structure prediction. J. Mol. Biol. 1994, 235, 13–26. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Pfam database, family: Collagen. Available online: http://pfam.sanger.ac.uk/family/PF01391 (accessed on 15 July 2013).
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef]
- Minezaki, Y.; Hooma, K.; Nishikawa, K. Intrinsically disordered regions of human plasma membrane proteins preferentially occur in the cytoplasmic segment. J. Mol. Biol. 2007, 368, 302–913. [Google Scholar]
- Süveges, D.; Gáspári, Z.; Tóth, G.; Nyitray, L. Charged single alpha-helix: A versatile protein structural motif. Proteins 2009, 74, 905–916. [Google Scholar] [CrossRef]
- Gáspári, Z.; Süveges, D.; Perczel, A.; Nyitray, L.; Tóth, G. Charged single alpha-helices in proteomes revealed by a consensus prediction approach. Biochem. Biophys. Acta 2012, 1824, 637–646. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gáspári, Z. Is Five Percent Too Small? Analysis of the Overlaps between Disorder, Coiled Coil and Collagen Predictions in Complete Proteomes. Proteomes 2014, 2, 72-83. https://doi.org/10.3390/proteomes2010072
Gáspári Z. Is Five Percent Too Small? Analysis of the Overlaps between Disorder, Coiled Coil and Collagen Predictions in Complete Proteomes. Proteomes. 2014; 2(1):72-83. https://doi.org/10.3390/proteomes2010072
Chicago/Turabian StyleGáspári, Zoltán. 2014. "Is Five Percent Too Small? Analysis of the Overlaps between Disorder, Coiled Coil and Collagen Predictions in Complete Proteomes" Proteomes 2, no. 1: 72-83. https://doi.org/10.3390/proteomes2010072