Pathological, Morphological, Cytogenomic, Biochemical and Molecular Data Support the Distinction between Colletotrichum cigarro comb. et stat. nov. and Colletotrichum kahawae

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Pathogenicity Tests
2.2. Induction of Perithecia
2.3. Substrate Use
2.4. Morphology
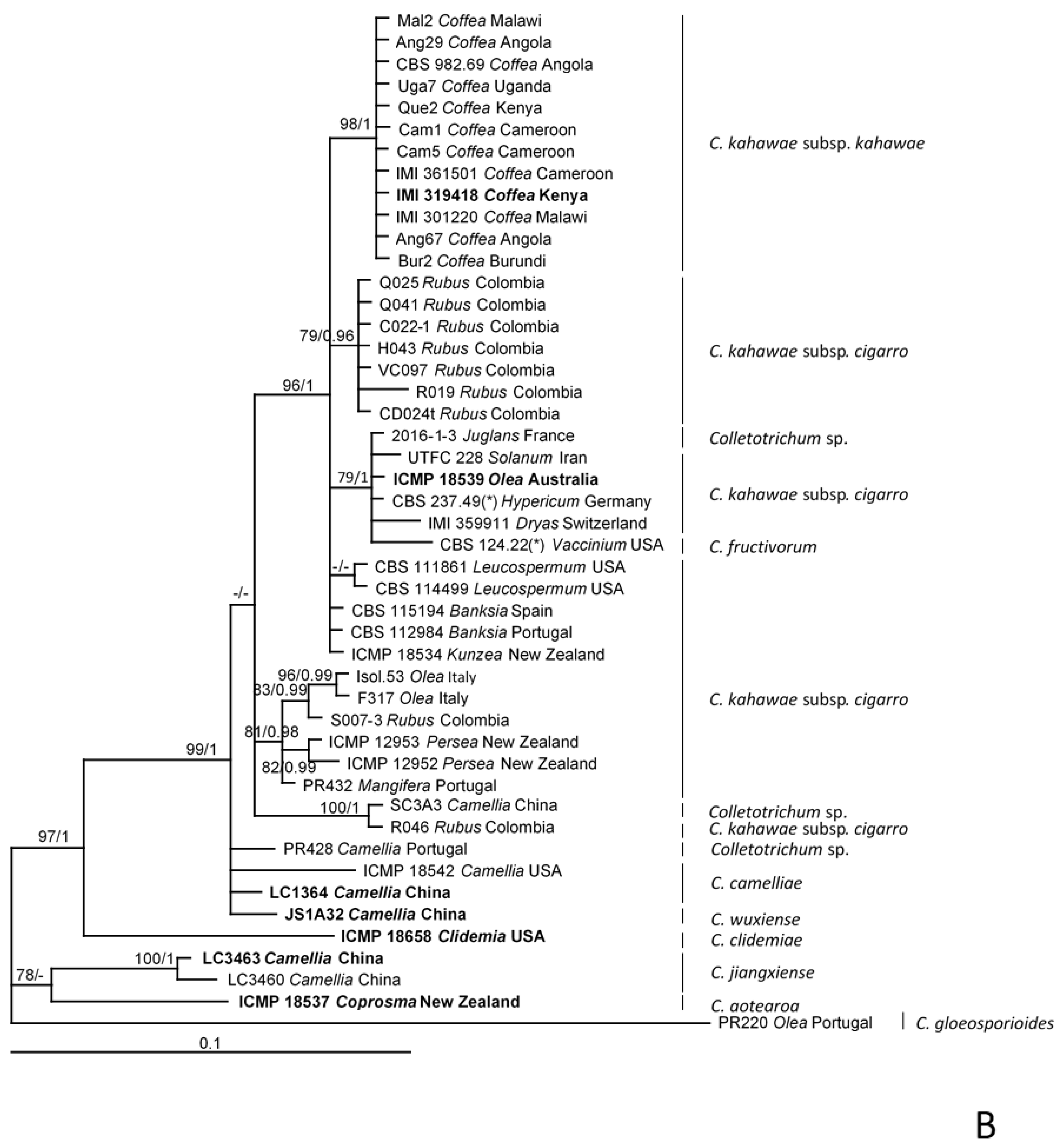
2.5. Phylogenetic Analyses
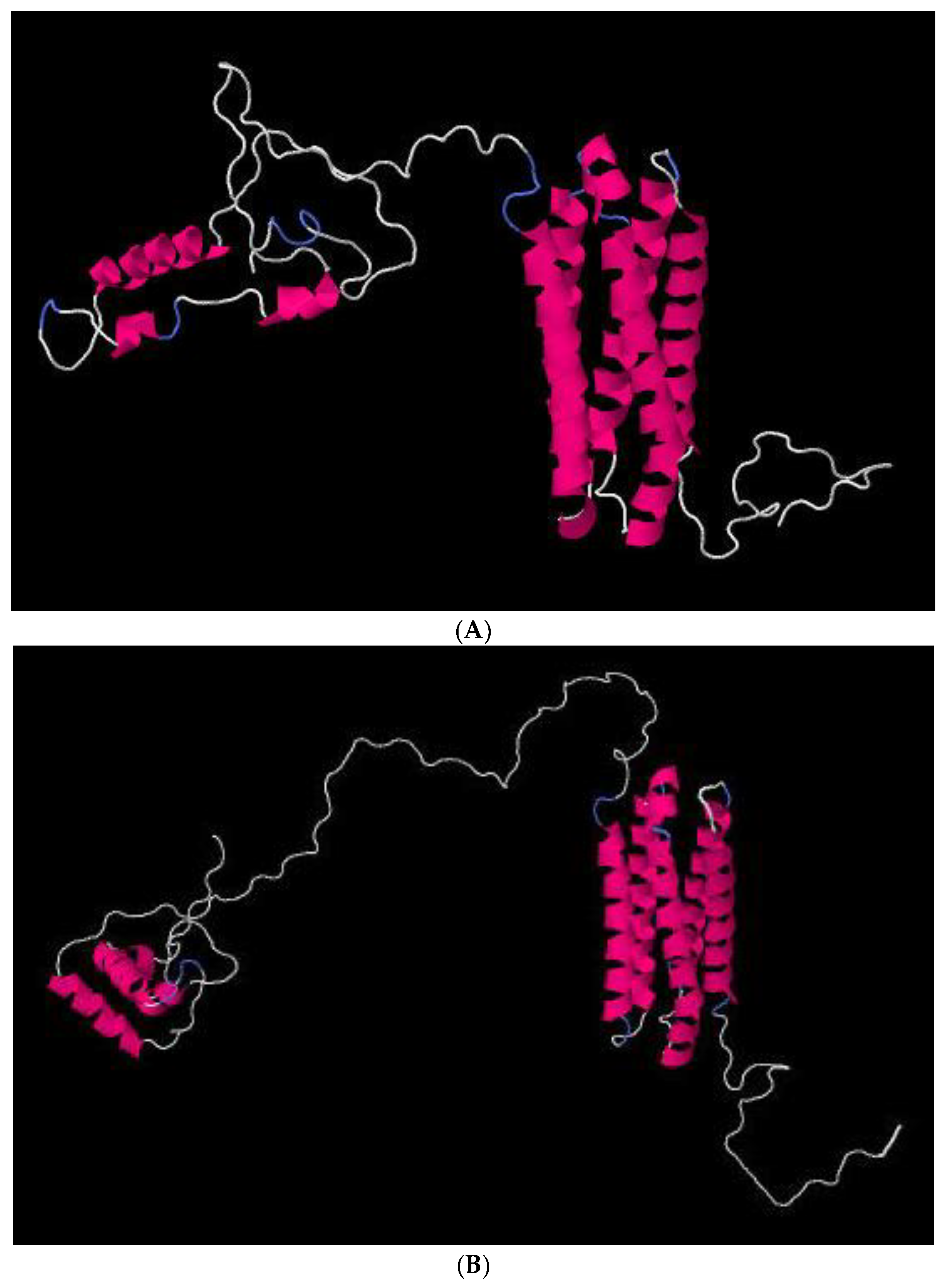
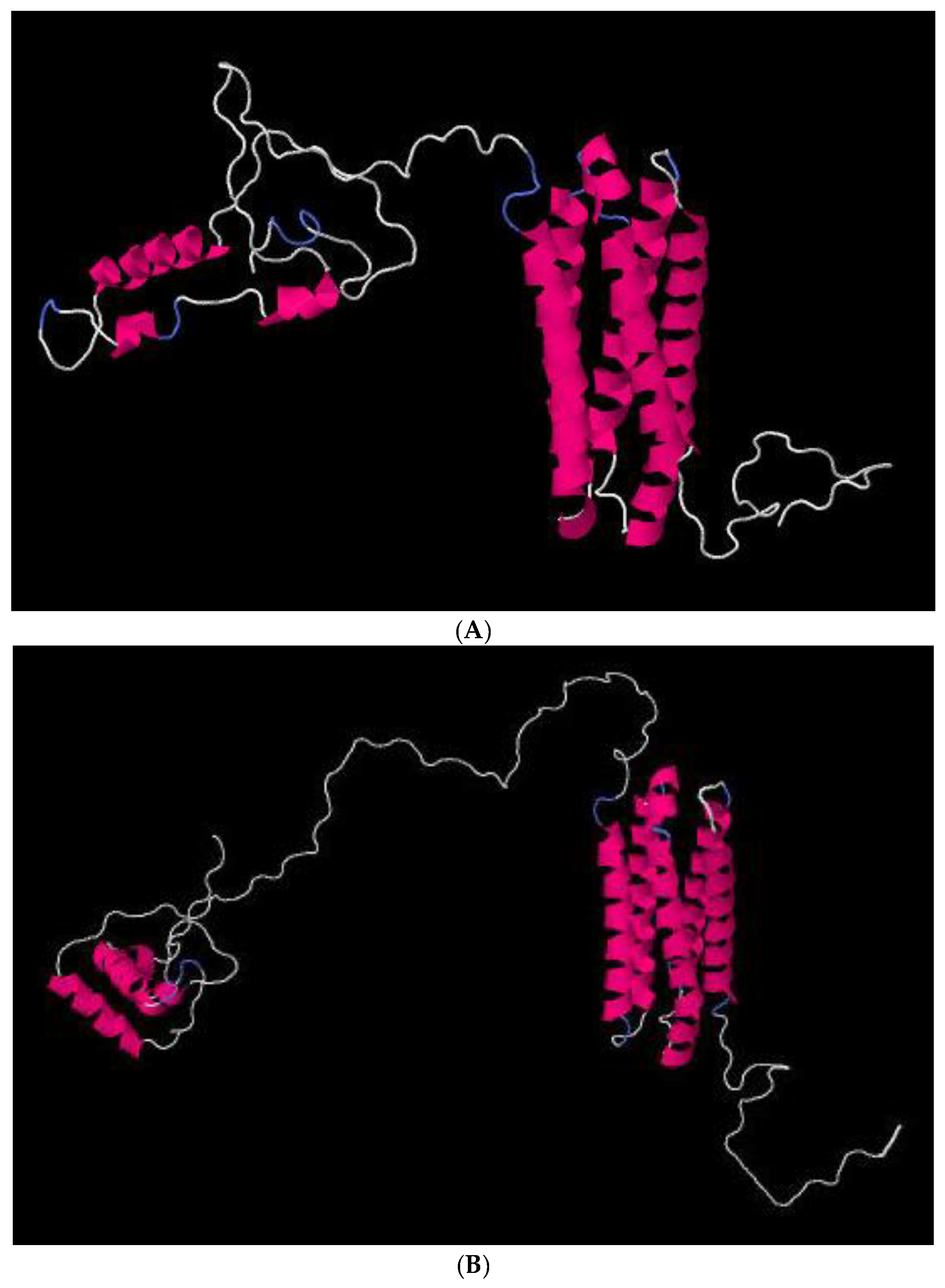
2.6. In Silico Analyses of Proteins
2.7. Genome Size
2.8. Taxonomy
3. Discussion
4. Materials and Methods
4.1. Fungal Material
4.2. Pathogenicity Tests
4.3. Induction of Perithecia
4.4. Phenotypic Characters
4.5. Substrate Use
4.6. DNA Extraction, PCR Amplification, and Sequencing
4.7. Phylogenetic Analyses
4.8. Bioinformatic Prediction Tools
4.9. Genome Size
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- von Arx, J.A. Die arten der gattung Colletotrichum Cda. Phytopathol. Z. 1957, 29, 413–468. [Google Scholar]
- Sutton, B.C. The Coelomycetes. Fungi Imperfecti with Pycnidia, Acervuli and Stromata; CABI: Kew, UK, 1980. [Google Scholar]
- Cannon, P.F.; Damm, U.; Johnston, P.R.; Weir, B.S. Colletotrichum—Current status and future directions. Stud. Mycol. 2012, 73, 181–213. [Google Scholar] [CrossRef] [Green Version]
- Jayawardena, R.S.; Hyde, K.D.; Damm, U.; Cai, L.; Liu, M.; Li, X.H.; Zhang, W.; Zhao, W.S.; Yan, J.Y. Notes on currently accepted species of Collet. Mycosphere 2016, 7, 1192–1260. [Google Scholar] [CrossRef]
- Baroncelli, R.; Talhinhas, P.; Pensec, F.; Sukno, S.A.; Le Floch, G.; Thon, M.R. The Colletotrichum acutatum species complex as a model system to study evolution and host specialization in plant pathogens. Front. Microbiol. 2017, 8, 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Felix, Y.; Groenewald, J.Z.; Cai, L.; Chen, Q.; Marincowitz, S.; Barnes, I.; Bensch, K.; Braun, U.; Camporesi, E.; Damm, U.; et al. Genera of phytopathogenic fungi: GOPHY 1. Stud. Mycol. 2017, 86, 99–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.; Johnston, P.R.; Damm, U. The Colletotrichum gloeosporioides species complex. Stud. Mycol. 2012, 73, 115–180. [Google Scholar] [CrossRef] [Green Version]
- Sreenivasaprasad, S.; Brown, A.E.; Mills, P.R. Coffee berry disease pathogen in Africa: Genetic structure and relationship to the group species Colletotrichum gloeosporioides. Mycol. Res. 1993, 97, 995–1000. [Google Scholar] [CrossRef]
- Waller, J.M.; Bridge, P.D.; Black, R.; Hakiza, G. Characterization of the coffee berry disease pathogen, Colletotrichum kahawae sp. nov. Mycol. Res. 1993, 97, 989–994. [Google Scholar] [CrossRef]
- van der Vossen, H.A.M.; Walyaro, D.J. Additional evidence for oligogenic inheritance of durable host resistance to coffee berry disease (Colletotrichum kahawae) in Arabica coffee (Coffea arabica L.). Euphytica 2009, 165, 105–111. [Google Scholar] [CrossRef]
- Loureiro, A.; Nicole, M.R.; Várzea, V.; Moncada, P.; Bertrand, B.; Silva, M.C. Coffee resistance to Colletotrichum kahawae is associated with lignification, accumulation of phenols and cell death at infection sites. Physiol. Mol. Plant Pathol. 2012, 77, 23–32. [Google Scholar] [CrossRef]
- de Silva, D.D.; Crous, P.W.; Ades, P.K.; Hyde, K.D.; Taylor, P.W.J. Life styles of Colletotrichum species and implications for plant biosecurity. Fungal Biol. Rev. 2017, 31, 155–168. [Google Scholar] [CrossRef]
- Batista, D.; Silva, D.N.; Vieira, A.; Cabral, A.; Pires, A.S.; Loureiro, A.; Guerra-Guimarães, L.; Pereira, A.P.; Azinheira, H.; Talhinhas, P.; et al. Legitimacy and implications of reducing Colletotrichum kahawae to subspecies in plant pathology. Front. Plant Sci. 2017, 7, 2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, A.; Silva, D.N.; Várzea, V.; Paulo, O.S.; Batista, D. Novel insights on colonization routes and evolutionary potential of Colletotrichum kahawae, a severe pathogen of Coffea arabica. Mol. Plant Pathol. 2018, 19, 2488–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Australia Group Common Control List Handbook—Volume II: Biological Weapons-Related Common Control Lists. Available online: https://australiagroup.net/en/documents/Australia-Group-Common-Control-List-Handbook-Volume-II.pdf (accessed on 6 March 2020).
- Hindorf, H. Colletotrichum spp. isolated from Coffea arabica L. in Kenya. Z. PflKrankh. PflSchutz. 1970, 77, 328–331. [Google Scholar]
- Prihastuti, H.; Cai, L.; Chen, H.; McKenzie, E.H.C.; Hyde, K.D. Characterisation of Colletotrichum species associated with coffee berries in northern Thailand. Fungal Divers. 2009, 39, 89–109. [Google Scholar]
- Gibbs, J.N. Inoculum sources for coffee berry disease. Ann. Appl. Biol. 1969, 64, 515–522. [Google Scholar] [CrossRef]
- Hindorf, H. Colletotrichum-Arten aus dem Kaffeeanbaugebiet von Kiambu in Kenia. Z. PflKrankh. PflSchutz. 1974, 81, 108–113. [Google Scholar]
- Silva, D.N.; Talhinhas, P.; Várzea, V.; Cai, L.; Paulo, O.S.; Batista, D. Application of the Apn2/MAT locus to improve the systematics of the Colletotrichum gloeosporioides complex: An example from coffee (Coffea spp.) hosts. Mycologia 2012, 104, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Várzea, V.; Rodrigues, J.C.; Lewis, B. Distinguishing characteristics and vegetative compatibility of Colletotrichum kahawae in comparison with other related species from coffee. Plant Pathol. 2002, 51, 202–207. [Google Scholar]
- Doyle, V.P.; Oudemans, P.V.; Rehner, S.A.; Litt, A. Habitat and host indicate lineage identity in Colletotrichum gloeosporioides s.l. from wild and agricultural landscapes in North America. PLoS ONE 2013, 8, e62394. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Weir, B.S.; Damm, U.; Crous, P.W.; Wang, Y.; Liu, B.; Wang, M.; Zhang, M.; Cai, L. Unravelling Colletotrichum species associated with Camellia: Employing ApMat and GS loci to resolve species in the C. gloeosporioides complex. Persoonia 2015, 35, 63–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-C.; Hao, X.-Y.; Wang, L.; Xiao, B.; Wang, X.-C.; Yang, Y.-J. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 2016, 6, 35287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnaccia, V.; Groenewald, J.Z.; Polizzi, G.; Crous, P.W. High species diversity in Colletotrichum associated with citrus diseases in Europe. Persoonia 2017, 39, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Giraud, T.; Gladieux, P.; Gavrilets, S. Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol. Evol. 2010, 25, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, D.N.; Talhinhas, P.; Cai, L.; Manuel, L.; Gichuru, E.K.; Loureiro, A.; Várzea, V.; Paulo, O.S.; Batista, D. Host-jump drives rapid and recent ecological speciation of the emergent fungal pathogen Colletotrichum kahawae. Mol. Ecol. 2012, 21, 2655–2670. [Google Scholar] [CrossRef]
- Crouch, J.A.; Tredway, L.; Clarke, B.; Hillman, B. Phylogenetic and population genetic divergence correspond with habitat for the pathogen Colletotrichum cereale and allied taxa across diverse grass communities. Mol. Ecol. 2009, 18, 123–135. [Google Scholar] [CrossRef]
- Lardner, R.; Johnston, P.R.; Plummer, K.M.; Pearson, M.N. Morphological and molecular analysis of Colletotrichum acutatum sensu lato. Mycol. Res. 1999, 103, 275–285. [Google Scholar] [CrossRef]
- Pires, A.S.; Azinheira, H.G.; Cabral, A.; Tavares, S.; Tavares, D.; Castro, M.; Várzea, V.; Silva, M.C.; Abranches, R.; Loureiro, J.; et al. Cytogenomic characterisation of Colletotrichum kahawae, the causal agent of Coffee Berry Disease, reveals diversity in minichromosome profiles and genome size expansion. Plant Pathol. 2016, 65, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://jmol.sourceforge.net/ (accessed on 1 October 2019).
- Garibaldi, A.; Gilardi, G.; Puglisi, I.; Cacciola, S.O.; Gullino, M.L. First report of leaf spot caused by Colletotrichum kahawae on cultivated rocket (Eruca sativa) in Italy. Plant Dis. 2016, 100, 1240. [Google Scholar] [CrossRef]
- Garibaldi, A.; Gilardi, G.; Franco-Ortega, S.; Gullino, M.L. First report of leaf spot caused by Colletotrichum kahawae on American Sweetgum (Liquidambar styraciflua) in Italy. J. Plant Pathol. 2016, 95, 36. [Google Scholar]
- Coronado-Ruiz, C.; Avendaño, R.; Escudero-Leyva, E.; Conejo-Barboza, G.; Chaverri, P.; Chavarría, M. Two new cellulolytic fungal species isolated from a 19th-century art collection. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Grammen, A.; Wenneker, M.; van Campenhout, J.; Pham, K.T.K.; van Hemelrijck, W.; Bylemans, D.; Geeraerd, A.; Keulemans, W. Identification and pathogenicity assessment of Colletotrichum isolates causing bitter rot of apple fruit in Belgium. Eur. J. Plant Pathol. 2019, 153, 253–269. [Google Scholar] [CrossRef]
- Sharma, G.; Shenoy, B. Colletotrichum systematics: Past, present and prospects. Mycosphere 2016, 7, 1093–1102. [Google Scholar] [CrossRef]
- Liang, X.; Wang, B.; Dong, Q.; Li, L.; Rollins, J.A.; Zhang, R.; Sun, G. Pathogenic adaptations of Colletotrichum fungi revealed by genome wide gene family evolutionary analyses. PLoS ONE 2018, 13, e0196303. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Y.L.; Gerke, J.; Park, H.-S.; Bayram, Ö.; Neumann, P.; Ni, M.; Dickmanns, A.; Kim, S.C.; Yu, J.-H.; Braus, G.H.; et al. The Velvet Family of Fungal Regulators Contains a DNA-Binding Domain Structurally Similar to NF-κB. PLoS Biol. 2013, 11, e1001750. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Yu, J.H. A novel regulator couples sporogenesis and trehalose biogenesis in Aspergillus nidulans. PLoS ONE 2007, 2, e970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 7, 310–322. [Google Scholar]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.; Narusaka, M.; Kumakura, N.; Tsushima, A.; Takano, Y.; Narusaka, Y.; Shirasu, K. Genus-wide comparative genome analyses of Colletotrichum species reveal specific gene family losses and gains during adaptation to specific infection lifestyles. Genome Biol. Evol. 2016, 8, 1467–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundin, D.; Poole, A.M.; Sjöberg, B.M.; Högbom, M. Use of structural phylogenetic networks for classification of the ferritin-like superfamily. J. Biol. Chem. 2012, 287, 20565–20575. [Google Scholar] [CrossRef] [Green Version]
- Spanu, P.D.; Abbott, J.C.; Amselem, J.; Burgis, T.A.; Soanes, D.M.; Stüber, K.; van Themaat, E.V.L.; Brown, J.K.M.; Butcher, S.A.; Gurr, S.J.; et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 2010, 330, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Tavares, S.; Ramos, A.P.; Pires, A.S.; Azinheira, H.G.; Caldeirinha, P.; Link, T.; Abranches, R.; Silva, M.C.; Voegele, R.T.; Loureiro, J.; et al. Genome size analyses of Pucciniales reveal the largest fungal genomes. Front. Plant Sci. 2014, 5, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhinhas, P.; Neves-Martins, J.; Oliveira, H.; Sreenivasaprasad, S. The distinctive population structure of Colletotrichum species associated with olive anthracnose in the Algarve region of Portugal reflects a host-pathogen diversity hot spot. FEMS Microbiol. Lett. 2009, 296, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, A.; Guerra-Guimarães, L.; Lidon, F.C.; Bertrand, B.; Silva, M.C.; Várzea, V. Isoenzymatic characterization of Colletotrichum kahawae isolates with different levels of aggressiveness. Trop. Plant Pathol. 2011, 36, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Guerber, J.C.; Correll, J.C. Morphological description of Glomerella acutata, the teleomorph of Colletotrichum acutatum. Mycologia 2001, 93, 216–229. [Google Scholar] [CrossRef]
- Nirenberg, H. Untersuchungen über die morphologische und biologische differenzierung in der Fusarium-Sektion Liseola. Mitt. Biol. Bundesanst. Land Forstwirtsch. Berl Dahl. 1976, 169, 1–117. [Google Scholar]
- Cenis, J.L. Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res. 1992, 20, 2380. [Google Scholar] [CrossRef]
- Lehr, N.A.; Wang, Z.; Li, N.; Hewitt, D.A.; López-Giráldez, F.; Trail, F.; Townsend, J.P. Differences among Three Neurospora Species Reveal Genes Required for Sexual Reproduction in Neurospora crassa. PLoS ONE 2014, 9, e110398. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, G.; Lohman, D.J.; Meier, R. Sequence Matrix: Concatenation software for the fast assembly of multigene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayers, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large modelspace. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; Institute of Electrical and Electronics Engineers: Piscataway, NJ, USA, 2010. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Schoffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinf. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2018, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [CrossRef] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W665. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Gupta, R. Prediction of Glycosylation Sites in Proteomes: From Post-Translational Modifications to Protein Function. Ph.D. Thesis, Technical University of Denmark (DTU), Lyngby, Denmark, 2001. [Google Scholar]
- Xie, Y.; Zheng, Y.; Li, H.; Luo, X.; He, Z.; Cao, S.; Shi, Y.; Zhao, Q.; Xue, Y.; Zuo, Z.; et al. GPS-Lipid: A robust tool for the prediction of multiple lipid modification sites. Sci. Rep. 2016, 6, 28249. [Google Scholar] [CrossRef]
- Haas, J.; Roth, S.; Arnold, K.; Kiefer, F.; Schmidt, T.; Bordoli, L.; Schwede, T. The Protein Model Portal—A comprehensive resource for protein structure and model information. Database 2013, 2013, bat031. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Bourne, E.C.; Mina, D.; Gonçalves, S.C.; Loureiro, J.; Freitas, H.; Muller, L.A.H. Large and variable genome size unrelated to serpentine adaptation but supportive of cryptic sexuality in Cenococcum geophilum. Mycorrhiza 2014, 24, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Talhinhas, P.; Tavares, D.; Ramos, A.P.; Gonçalves, S.; Loureiro, J. Validation of Standards Suitable for Genome Size Estimation of Fungi. J. Microbiol. Methods 2017, 142, 76–78. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Slippers, B.; Wingfield, M.J. Global food and fibre security threatened by current inefficiencies in fungal identification. Phil Trans. R. Soc. B 2016, 371, 20160024. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loci 1 | Taxa | Nucleotide Substitution Models | Chars | Constant | Parsimony-Informative | Parsimony-Uninformative | ML-ln L 2 | BI-ln L 3 |
|---|---|---|---|---|---|---|---|---|
| apMAT | 64 | K80 | 727 | 595 | 20 | 110 | −1707.61 | −1826.67 |
| gs | 47 | HKY + G | 877 | 751 | 39 | 68 | −1941.59 | −2021.35 |
| tub2 | 96 | HKY + G | 599 | 541 | 19 | 31 | −1180.52 | −1289.96 |
| apMAT | 17 | K80 | 725 | 614 | 12 | 98 | −1528.87 | −1588.01 |
| apn25L | 17 | GTR | 837 | 749 | 17 | 71 | −1625.35 | −1693.53 |
| cas1 | 17 | GTR + G | 845 | 760 | 25 | 59 | −1647.75 | −1737.89 |
| cellwall | 17 | GTR + I | 1239 | 1112 | 50 | 73 | −2481.99 | −2521.34 |
| gs | 17 | HKY + G | 859 | 785 | 18 | 53 | −1601.82 | −1640.14 |
| mat1-2-1 | 17 | GTR + G | 843 | 786 | 10 | 47 | −1465.25 | −1512.12 |
| siRNA | 17 | HKY + G | 1292 | 1112 | 52 | 125 | −2839.28 | −2880.50 |
| tub2 | 17 | HKY | 597 | 548 | 8 | 33 | −1067.93 | −1100.61 |
| vosA | 17 | HKY + G | 1345 | 1190 | 33 | 121 | −2723.89 | −2760.43 |
| combined | 17 | 8582 | 7656 | 225 | 680 | −17098.40 | −17350.83 |
| Identity | Isolate | Protein/Amino Acid Position in the Protein Alignment | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cas1 | siRNA | vosA | Cellwall | |||||||||||||||||||||||||||||||||||||||||
| 176 | 178 | 71 | 85 | 101 | 146 | 194 | 227 | 236 | 281 | 306 | 322 | 49 | 94 | 128 | 203 | 309 | 42 | 79 | 181 | 182 | 183 | 208 | 209 | 210 | 211 | 212 | 213 | 214 | 215 | 216 | 217 | 218 | 219 | 220 | 221 | 222 | 223 | 224 | 225 | 226 | 227 | 243 | ||
| CBD | Ang29 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P |
| Ang67 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Bur2 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Cam1 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Cam5 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Mal2 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Que2 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| Uga7 | D | I | E | V | D | F | G | E | D | Y | S | D | M | V | R | P | L | L | K | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | P | |
| non-CBD | ICMP 12953 | N | V | D | V | E | F | A | D | E | K | S | D | K | V | G | A | H | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L |
| ICMP 18534 | N | V | E | I | D | L | A | E | D | Y | S | D | K | V | G | A | L | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L | |
| ICMP 18539 | N | V | D | V | E | F | A | E | E | K | S | G | K | V | G | A | L | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L | |
| CBS 237.49 (*) | N | V | E | I | D | L | A | E | D | Y | S | D | K | A | G | A | L | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L | |
| PR432 | N | V | D | V | E | F | A | E | E | K | P | G | K | V | G | A | L | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L | |
| PR428 | N | V | D | V | E | F | A | D | E | K | S | D | K | V | G | A | L | Q | Q | A | T | P | P | A | T | P | K | T | P | A | A | A | P | A | A | P | K | T | P | A | A | A | L | |
| Isolate | Species | Host | Country, Region |
|---|---|---|---|
| Ang6 | C. kahawae | Coffea arabica | Angola, Chianga |
| Ang29 | C. kahawae | C. arabica | Angola, Ganda |
| Ang30 | C. kahawae | C. arabica | Angola, Ganda |
| Ang67 | C. kahawae | C. arabica | Angola, Ganda |
| Ang81 | C. kahawae | C. arabica | Angola, Huambo |
| Bur2 | C. kahawae | C. arabica | Burundi |
| Cam1 | C. kahawae | C. arabica | Cameroon, Babadjou |
| Cam2 | C. kahawae | C. arabica | Cameroon, Santa |
| Cam5 | C. kahawae | C. arabica | Cameroon, Baham |
| Cam8 | C. kahawae | C. arabica | Cameroon, Kumbo |
| Eti3 | C. kahawae | C. arabica | Ethiopia, Sidamo |
| Eti9 | C. kahawae | C. arabica | Ethiopia, Sidamo |
| Eti20 | C. kahawae | C. arabica | Ethiopia |
| Mal2 | C. kahawae | C. arabica | Malawi |
| Que2 | C. kahawae | C. arabica | Kenya |
| Que42 | C. kahawae | C. arabica | Kenya |
| Que48 | C. kahawae | C. arabica | Kenya, Taita Taveta |
| Que72 | C. kahawae | C. arabica | Kenya, Ruiru |
| Que82 | C. kahawae | C. arabica | Kenya, Kitale |
| Que84 | C. kahawae | C. arabica | Kenya, Mgumguri |
| Rua1 | C. kahawae | C. arabica | Rwanda, Gicumbo |
| Tan2 | C. kahawae | C. arabica | Tanzania, Mbinga |
| Tan13 | C. kahawae | C. arabica | Tanzania, Mbinga |
| Uga2 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Uga3 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Uga5 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Uga6 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Uga7 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Uga9 | C. kahawae | C. arabica | Uganda, Kapchorwa |
| Zim1 | C. kahawae | C. arabica | Zimbabwe, Hiton |
| Zim12 | C. kahawae | C. arabica | Zimbabwe |
| Zim14 | C. kahawae | C. arabica | Zimbabwe |
| CBS 237.49 (*), ICMP 17922, C1275.8 | C. cigarro (syn. Glomerella cingulata var. migrans) | Hypericum perforatum | Germany |
| ICMP 12953, C1206.3 | C. cigarro | Persea americana | New Zealand |
| ICMP 18534, C1252.12 | C. cigarro | Kunzea ericoides | New Zealand |
| ICMP 18539, C1262.12 | C. cigarro | Olea europaea | Australia |
| ICMP 18542, C1291, CG02g | C. camelliae | Camellia sp. | USA |
| PR432 | C. cigarro | Mangifera indica | Portugal, Lisbon |
| PR428 | Colletotrichum sp. | Camellia japonica | Portugal, Lisbon |
| PR220 | C. gloeosporioides s.s. | O. europaea | Portugal, Tondela |
| PR808 | C. gloeosporioides s.s. | O. europaea | Portugal, Silves |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, A.; Azinheira, H.G.; Talhinhas, P.; Batista, D.; Ramos, A.P.; Silva, M.d.C.; Oliveira, H.; Várzea, V. Pathological, Morphological, Cytogenomic, Biochemical and Molecular Data Support the Distinction between Colletotrichum cigarro comb. et stat. nov. and Colletotrichum kahawae. Plants 2020, 9, 502. https://doi.org/10.3390/plants9040502
Cabral A, Azinheira HG, Talhinhas P, Batista D, Ramos AP, Silva MdC, Oliveira H, Várzea V. Pathological, Morphological, Cytogenomic, Biochemical and Molecular Data Support the Distinction between Colletotrichum cigarro comb. et stat. nov. and Colletotrichum kahawae. Plants. 2020; 9(4):502. https://doi.org/10.3390/plants9040502
Chicago/Turabian StyleCabral, Ana, Helena G. Azinheira, Pedro Talhinhas, Dora Batista, Ana Paula Ramos, Maria do Céu Silva, Helena Oliveira, and Vítor Várzea. 2020. "Pathological, Morphological, Cytogenomic, Biochemical and Molecular Data Support the Distinction between Colletotrichum cigarro comb. et stat. nov. and Colletotrichum kahawae" Plants 9, no. 4: 502. https://doi.org/10.3390/plants9040502