Role of Prokineticin Receptor-1 in Epicardial Progenitor Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Prokineticins and Cognate Receptors
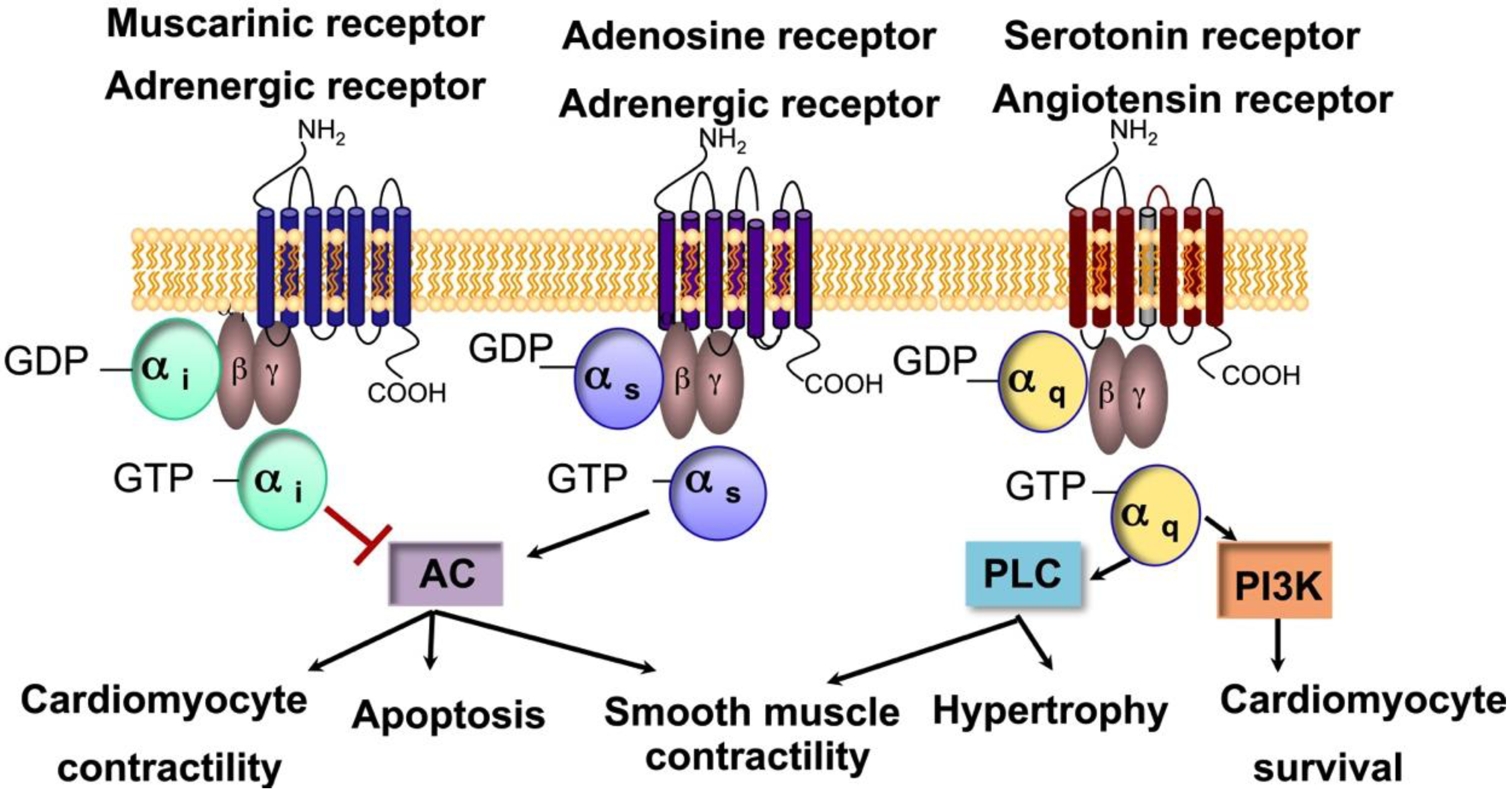
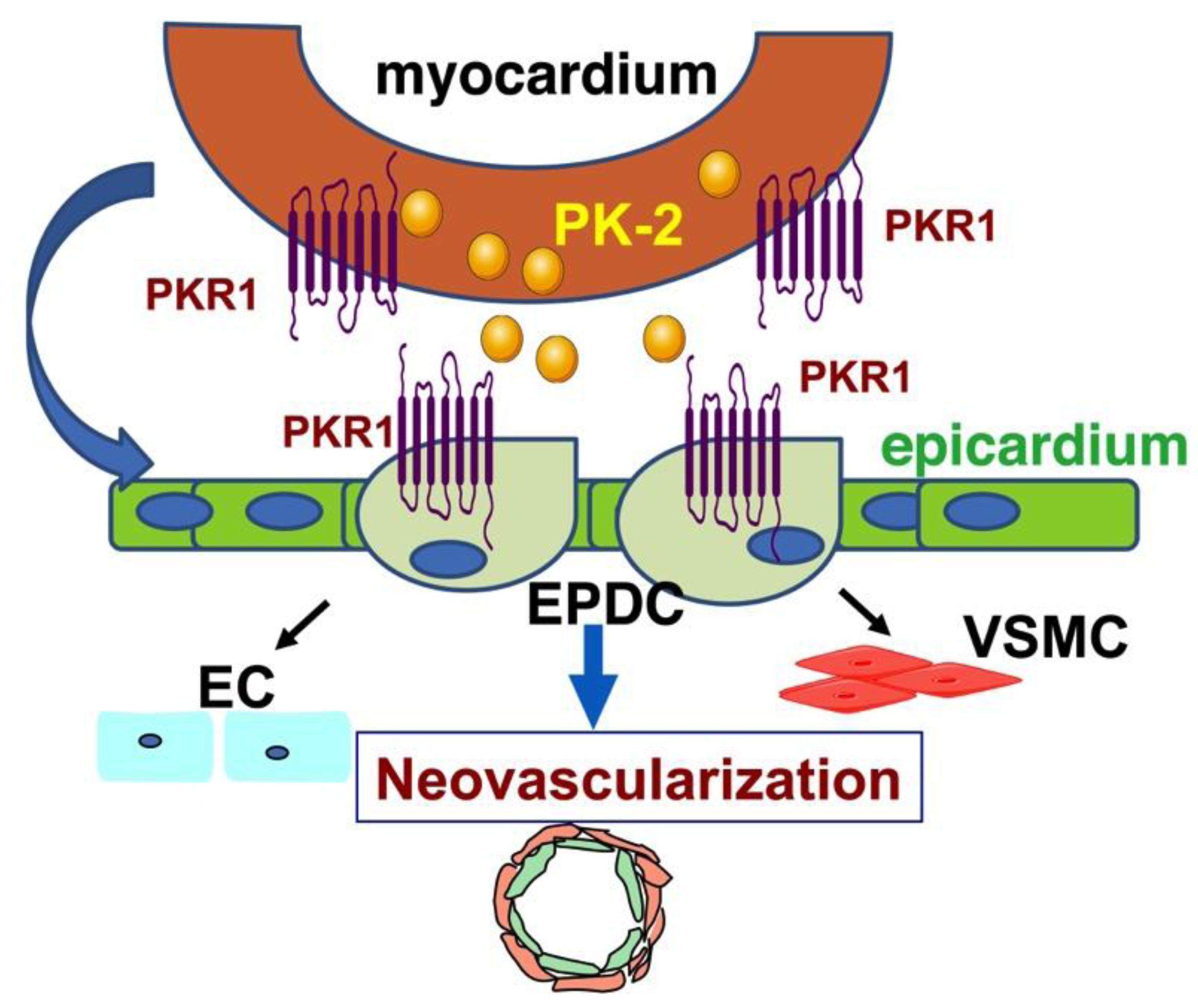
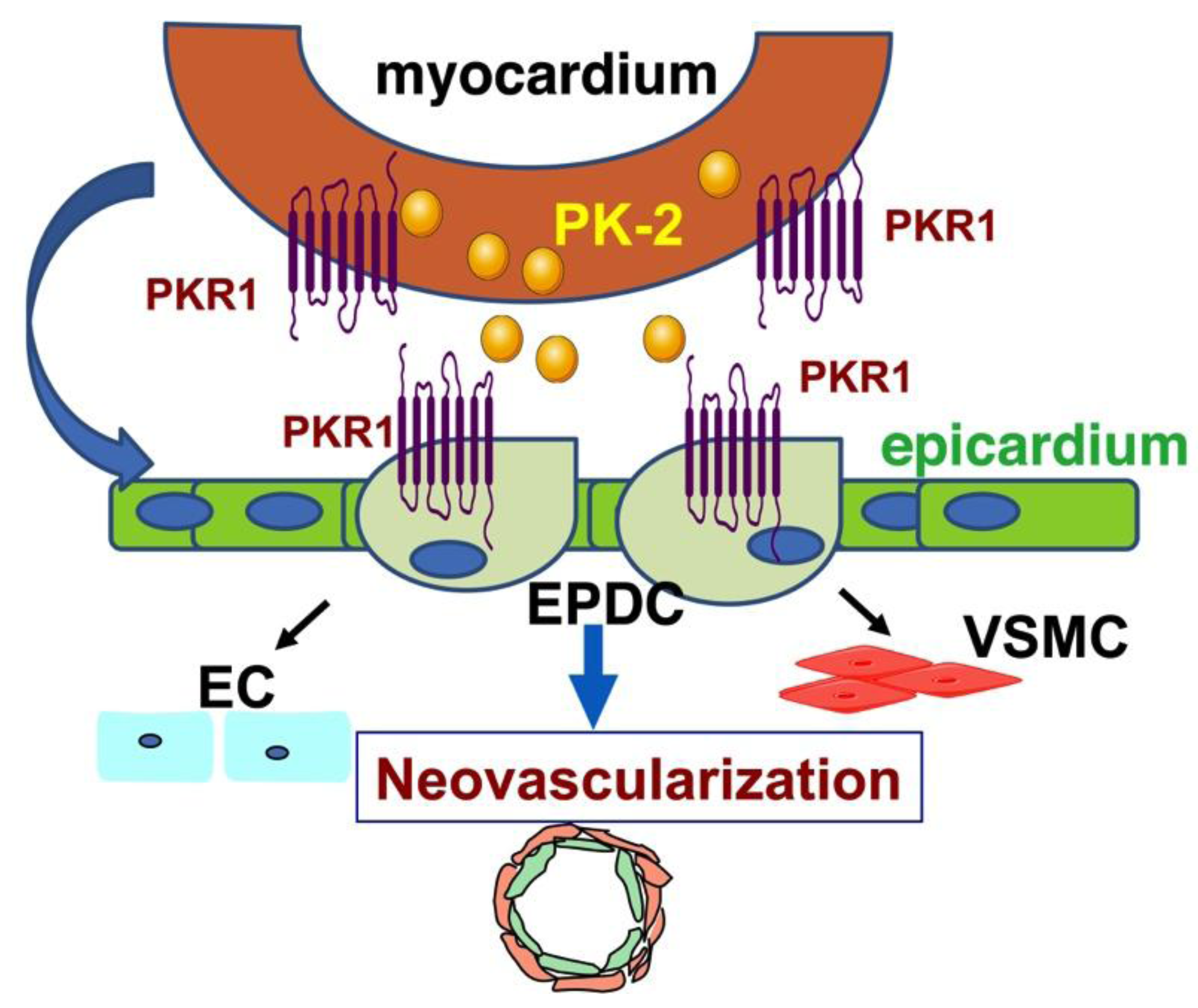
3. Prokineticin Signaling in Myocardium and Epicardium Interaction
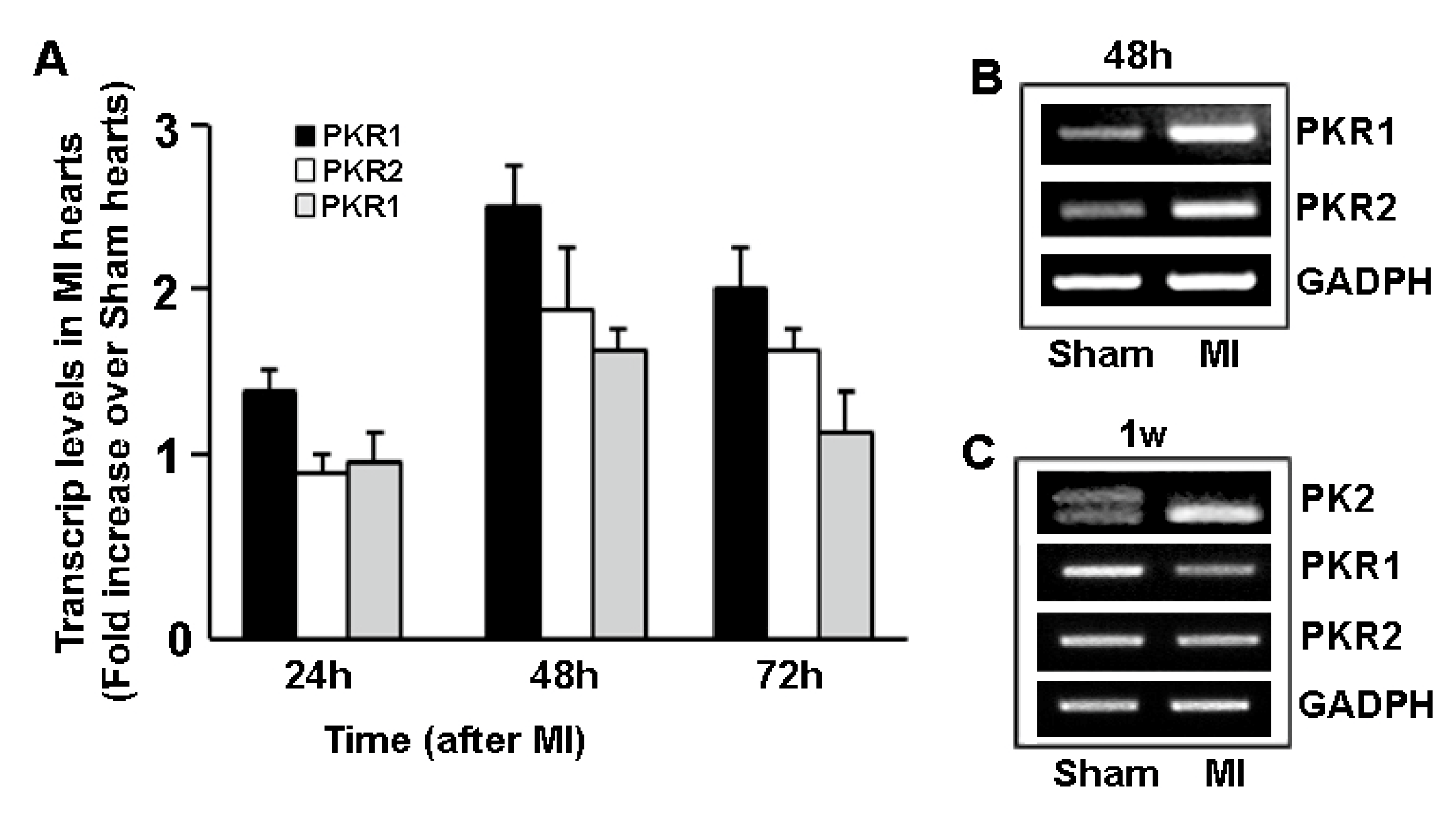
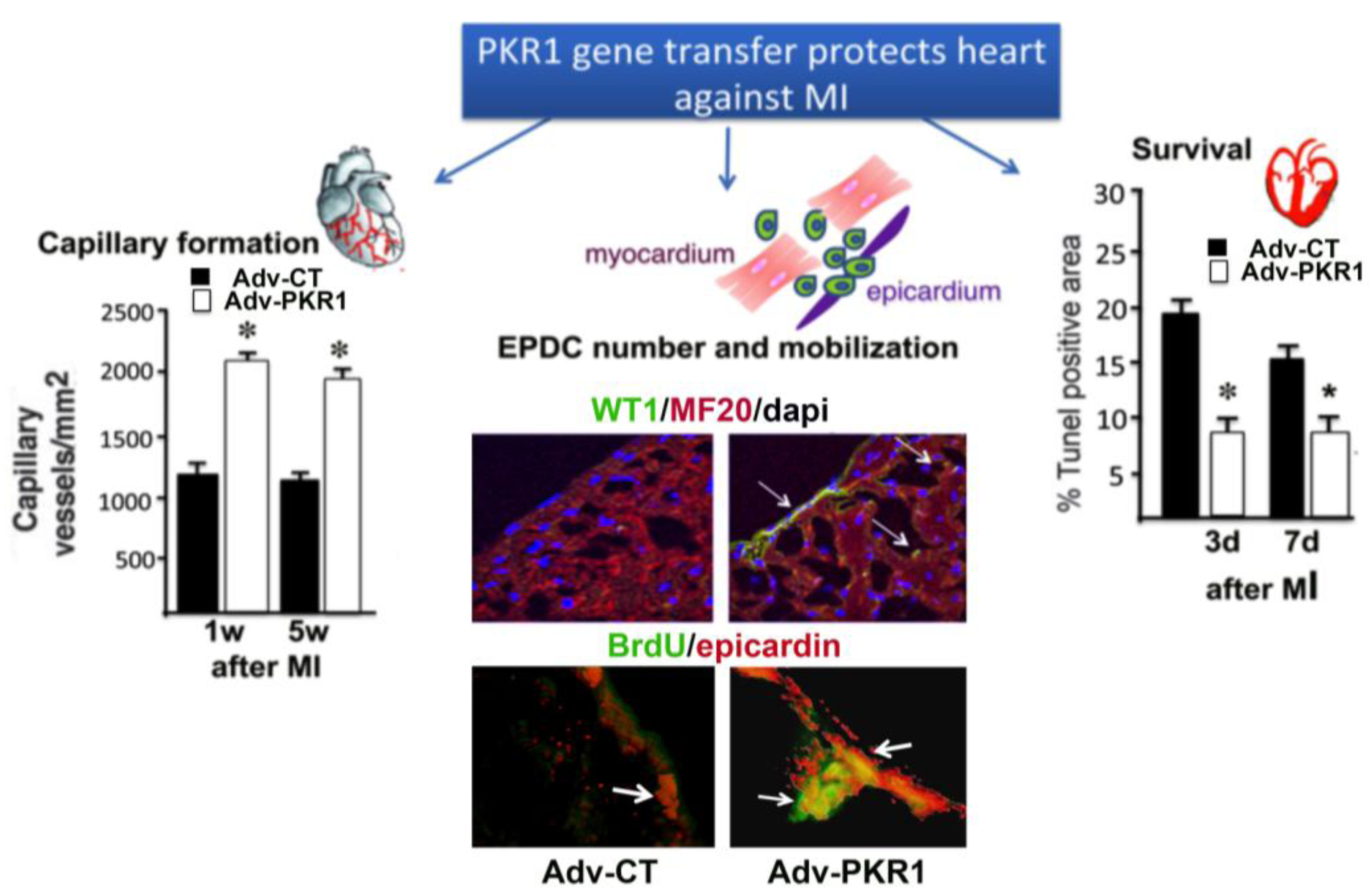
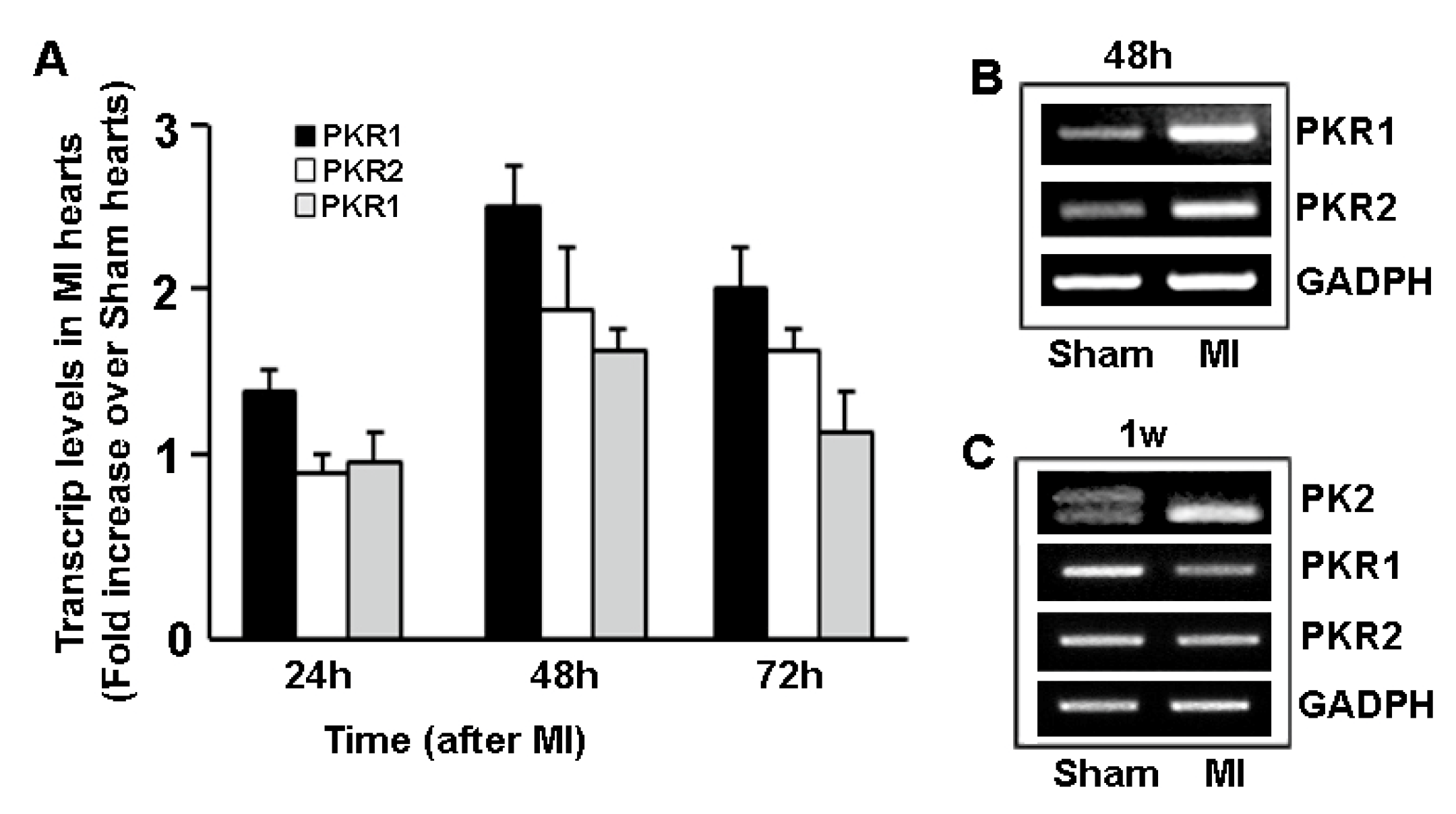
4. Prokineticin Signaling in Myocardial Infarction (MI)

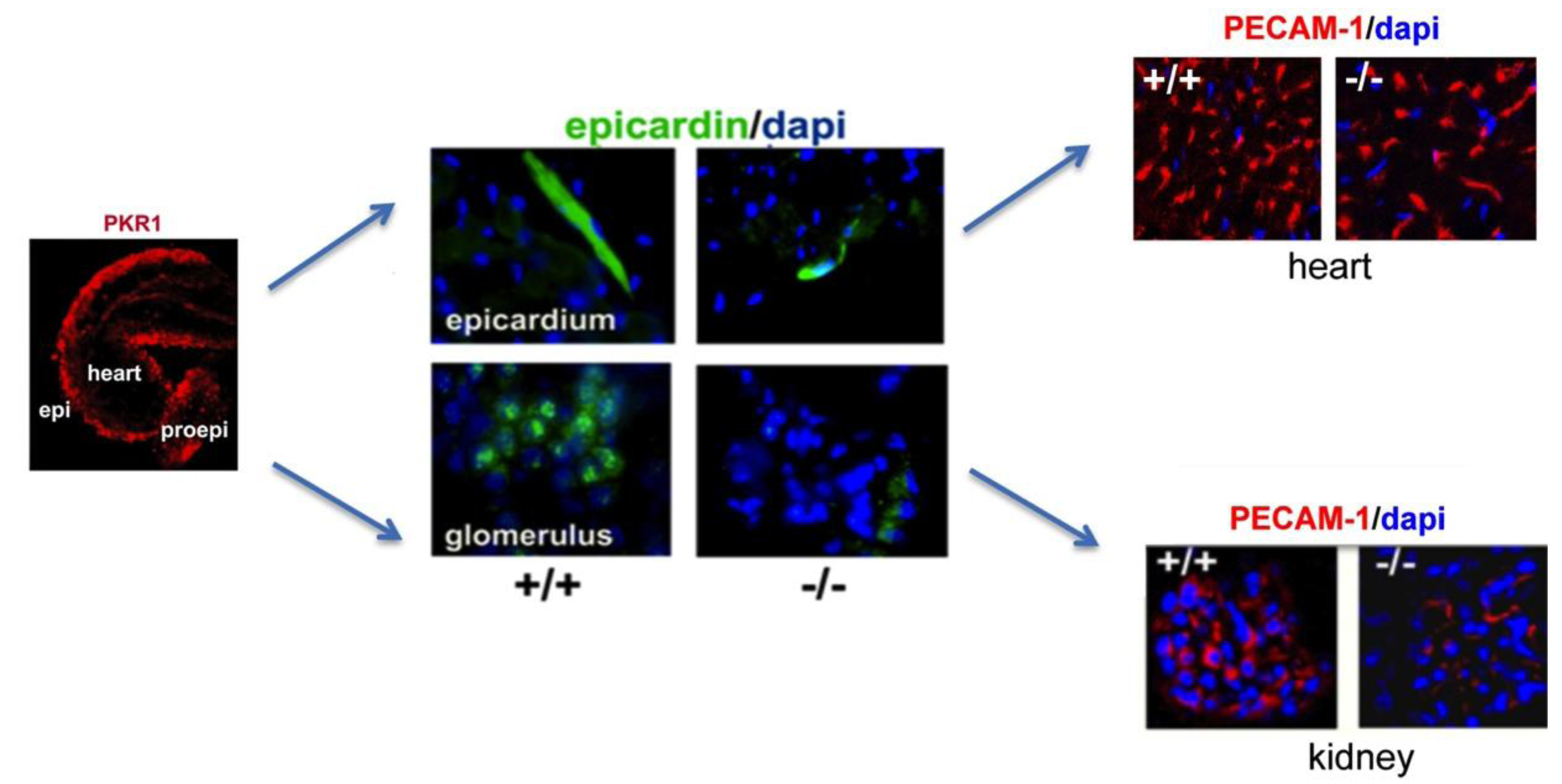
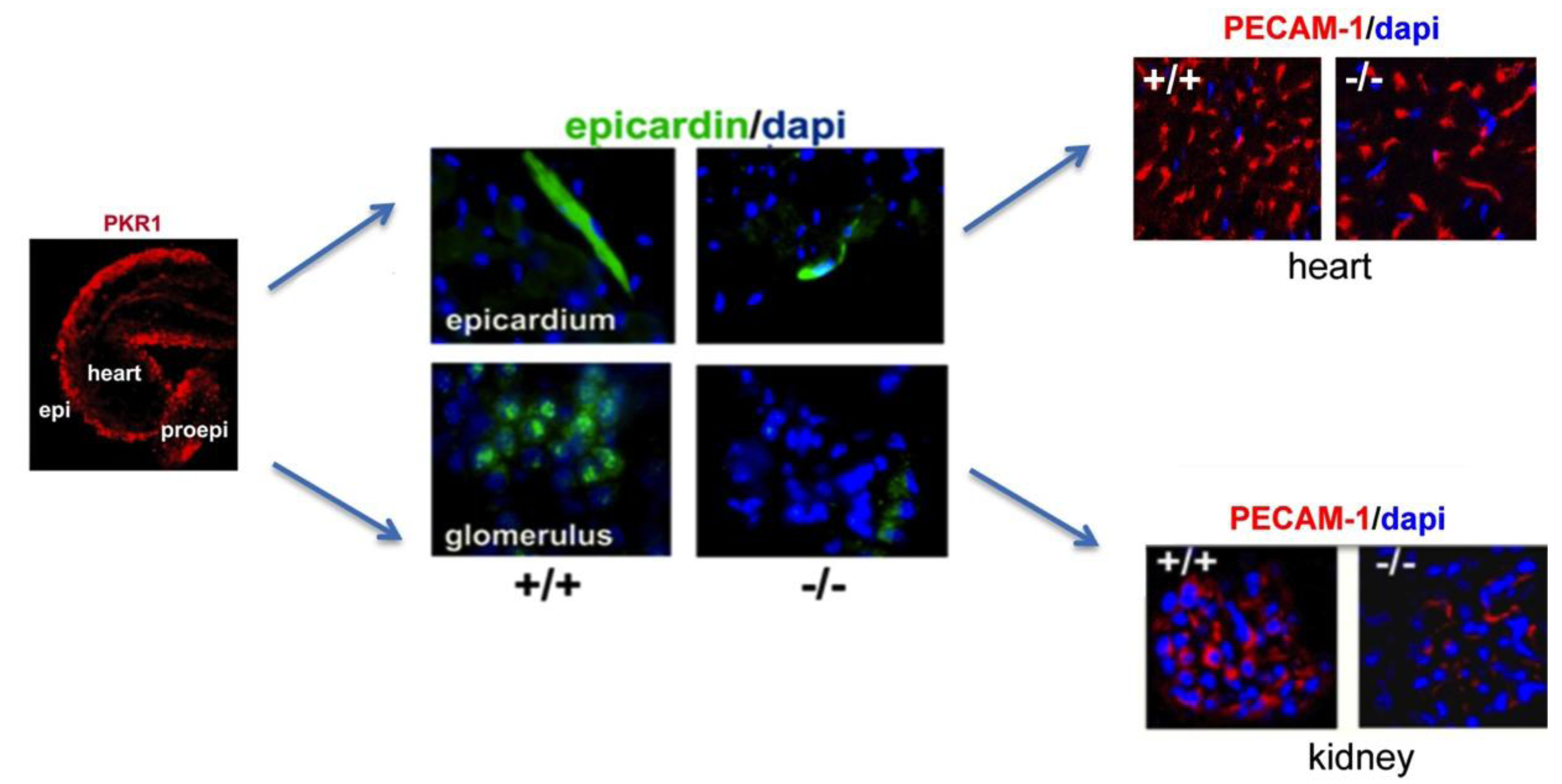
5. Prokineticin Signaling in the Epicardium-Kidney Axis
6. Concluding Remarks
Acknowledgments
Conflict of Interest
References and Notes
- Zimmermann, W.H.; Eschenhagen, T. Questioning the relevance of circulating cardiac progenitor cells in cardiac regeneration. Cardiovasc. Res. 2005, 68, 344–346. [Google Scholar] [CrossRef]
- Gonzales, C.; Pedrazzini, T. Progenitor cell therapy for heart disease. Exp. Cell. Res. 2009, 315, 3077–3085. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; Pu, W.T.; Riley, P.R. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef]
- Limana, F.; Capogrossi, M.C.; Germani, A. The epicardium in cardiac repair: from the stem cell view. Pharmacol. Ther. 2011, 129, 82–96. [Google Scholar] [CrossRef]
- Limana, F.; Zacheo, A.; Mocini, D.; Mangoni, A.; Borsellino, G.; Diamantini, A.; De Mori, R.; Battistini, L.; Vigna, E.; Santini, M.; Loiaconi, V.; Pompilio, G.; Germani, A.; Capogrossi, M.C. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ. Res. 2007, 101, 1255–1265. [Google Scholar]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar]
- Rockman, H.A.; Lefkowitz, R.J. Introduction to the series on novel aspects of cardiovascular G protein-coupled receptor signaling. Circ. Res. 2011, 109, 202–204. [Google Scholar] [CrossRef]
- Tang, C.M.; Insel, P.A. GPCR expression in the heart; "new" receptors in myocytes and fibroblasts. Trends Cardiovasc. Med. 2004, 14, 94–99. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. G protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Wettschureck, N.; Rutten, H.; Zywietz, A.; Gehring, D.; Wilkie, T.M.; Chen, J.; Chien, K.R.; Offermanns, S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 2001, 7, 1236–1240. [Google Scholar] [CrossRef]
- Nebigil, C.G.; Jaffre, F.; Messaddeq, N.; Hickel, P.; Monassier, L.; Launay, J.M.; Maroteaux, L. Overexpression of the serotonin 5-HT2B receptor in heart leads to abnormal mitochondrial function and cardiac hypertrophy. Circulation 2003, 107, 3223–3229. [Google Scholar] [CrossRef]
- Howes, A.L.; Miyamoto, S.; Adams, J.W.; Woodcock, E.A.; Brown, J.H. Galphaq expression activates EGFR and induces Akt mediated cardiomyocyte survival: dissociation from Galphaq mediated hypertrophy. J. Mol. Cell. Cardiol. 2006, 40, 597–604. [Google Scholar] [CrossRef]
- Kaplan, D.D.; Meigs, T.E.; Casey, P.J. Distinct regions of the cadherin cytoplasmic domain are essential for functional interaction with Galpha 12 and beta-catenin. J. Biol. Chem. 2001, 276, 44037–44043. [Google Scholar] [CrossRef]
- Offermanns, S.; Mancino, V.; Revel, J.P.; Simon, M.I. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 1997, 275, 533–536. [Google Scholar] [CrossRef]
- Gaudin, C.; Ishikawa, Y.; Wight, D.C.; Mahdavi, V.; Nadal-Ginard, B.; Wagner, T.E.; Vatner, D.E.; Homcy, C.J. Overexpression of Gs alpha protein in the hearts of transgenic mice. J. Clin. Invest. 1995, 95, 1676–1683. [Google Scholar] [CrossRef]
- Smart, N.; Risebro, C.A.; Melville, A.A.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef]
- Urayama, K.; Guilini, C.; Turkeri, G.; Takir, S.; Kurose, H.; Messaddeq, N.; Dierich, A.; Nebigil, C.G. Prokineticin receptor-1 induces neovascularization and epicardial-derived progenitor cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 841–849. [Google Scholar] [CrossRef]
- Kaser, A.; Winklmayr, M.; Lepperdinger, G.; Kreil, G. The AVIT protein family. Secreted cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. 2003, 4, 469–473. [Google Scholar] [CrossRef]
- Nebigil, C.G. Prokineticin receptors in cardiovascular function: foe or friend? Trends Cardiovasc. Med. 2009, 19, 55–60. [Google Scholar]
- Li, M.; Bullock, C.M.; Knauer, D.J.; Ehlert, F.J.; Zhou, Q.Y. Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle. Mol. Pharmacol. 2001, 59, 692–698. [Google Scholar]
- Masuda, Y.; Takatsu, Y.; Terao, Y.; Kumano, S.; Ishibashi, Y.; Suenaga, M.; Abe, M.; Fukusumi, S.; Watanabe, T.; Shintani, Y.; Yamada, T.; Hinuma, S.; Inatomi, N.; Ohtaki, T.; Onda, H.; Fujino, M. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G protein-coupled receptors. Biochem. Biophys. Res. Commun. 2002, 293, 396–402. [Google Scholar] [CrossRef]
- LeCouter, J.; Lin, R.; Ferrara, N. The role of EG-VEGF in the regulation of angiogenesis in endocrine glands. Cold Spring Harb Symp. Quant. Biol. 2002, 67, 217–221. [Google Scholar] [CrossRef]
- Chen, J.; Kuei, C.; Sutton, S.; Wilson, S.; Yu, J.; Kamme, F.; Mazur, C.; Lovenberg, T.; Liu, C. Identification and pharmacological characterization of prokineticin 2 beta as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 2005, 67, 2070–2076. [Google Scholar] [CrossRef]
- Soga, T.; Matsumoto, S.; Oda, T.; Saito, T.; Hiyama, H.; Takasaki, J.; Kamohara, M.; Ohishi, T.; Matsushime, H.; Furuichi, K. Molecular cloning and characterization of prokineticin receptors. Biochim. Biophys. Acta 2002, 1579, 173–179. [Google Scholar] [CrossRef]
- Negri, L.; Lattanzi, R.; Giannini, E.; Colucci, M.; Margheriti, F.; Melchiorri, P.; Vellani, V.; Tian, H.; De Felice, M.; Porreca, F. Impaired nociception and inflammatory pain sensation in mice lacking the prokineticin receptor PKR1: focus on interaction between PKR1 and the capsaicin receptor TRPV1 in pain behavior. J. Neurosci. 2006, 26, 6716–6727. [Google Scholar] [CrossRef]
- Li, J.D.; Hu, W.P.; Boehmer, L.; Cheng, M.Y.; Lee, A.G.; Jilek, A.; Siegel, J.M.; Zhou, Q.Y. Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J. Neurosci. 2006, 26, 11615–11623. [Google Scholar] [CrossRef]
- Hu, W.P.; Li, J.D.; Zhang, C.; Boehmer, L.; Siegel, J.M.; Zhou, Q.Y. Altered circadian and homeostatic sleep regulation in prokineticin 2-deficient mice. Sleep 2007, 30, 247–256. [Google Scholar]
- LeCouter, J.; Ferrara, N. EG-VEGF and the concept of tissue-specific angiogenic growth factors. Semin. Cell. Dev. Biol. 2002, 13, 3–8. [Google Scholar] [CrossRef]
- Ng, K.L.; Li, J.D.; Cheng, M.Y.; Leslie, F.M.; Lee, A.G.; Zhou, Q.Y. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 2005, 308, 1923–1927. [Google Scholar] [CrossRef]
- LeCouter, J.; Zlot, C.; Tejada, M.; Peale, F.; Ferrara, N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc. Natl. Acad. Sci. USA 2004, 101, 16813–16818. [Google Scholar]
- Negri, L.; Lattanzi, R.; Giannini, E.; De Felice, M.; Colucci, A.; Melchiorri, P. Bv8, the amphibian homologue of the mammalian prokineticins, modulates ingestive behaviour in rats. Br. J. Pharmacol. 2004, 142, 181–191. [Google Scholar] [CrossRef]
- Li, J.D.; Hu, W.P.; Zhou, Q.Y. Disruption of the circadian output molecule prokineticin 2 results in anxiolytic and antidepressant-like effects in mice. Neuropsychopharmacology 2009, 34, 367–373. [Google Scholar] [CrossRef]
- Guilini, C.; Urayama, K.; Turkeri, G.; Dedeoglu, D.B.; Kurose, H.; Messaddeq, N.; Nebigil, C.G. Divergent roles of prokineticin receptors in the endothelial cells: Angiogenesis and fenestration. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H844–H852. [Google Scholar] [CrossRef]
- Ngan, E.S.; Lee, K.Y.; Sit, F.Y.; Poon, H.C.; Chan, J.K.; Sham, M.H.; Lui, V.C.; Tam, P.K. Prokineticin-1 modulates proliferation and differentiation of enteric neural crest cells. Biochim. Biophys. Acta 2007, 1773, 536–545. [Google Scholar] [CrossRef]
- Giannini, E.; Lattanzi, R.; Nicotra, A.; Campese, A.F.; Grazioli, P.; Screpanti, I.; Balboni, G.; Salvadori, S.; Sacerdote, P.; Negri, L. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc. Natl. Acad. Sci. USA 2009, 106, 14646–14651. [Google Scholar] [CrossRef]
- Dorsch, M.; Qiu, Y.; Soler, D.; Frank, N.; Duong, T.; Goodearl, A.; O'Neil, S.; Lora, J.; Fraser, C.C. PK1/EG-VEGF induces monocyte differentiation and activation. J. Leukoc. Biol. 2005, 78, 426–434. [Google Scholar] [CrossRef]
- Ngan, E.S.; Tam, P.K. Prokineticin-signaling pathway. Int. J. Biochem. Cell Biol. 2008, 40, 1679–1684. [Google Scholar] [CrossRef]
- Urayama, K.; Guilini, C.; Messaddeq, N.; Hu, K.; Steenman, M.; Kurose, H.; Ert, G.; Nebigil, C.G. The prokineticin receptor-1 (GPR73) promotes cardiomyocyte survival and angiogenesis. FASEB J. 2007, 21, 2980–2993. [Google Scholar] [CrossRef]
- Martucci, C.; Franchi, S.; Giannini, E.; Tian, H.; Melchiorri, P.; Negri, L.; Sacerdote, P. Bv8, the amphibian homologue of the mammalian prokineticins, induces a proinflammatory phenotype of mouse macrophages. Br. J. Pharmacol. 2006, 147, 225–234. [Google Scholar] [CrossRef]
- Essafi, A.; Webb, A.; Berry, R.L.; Slight, J.; Burn, S.F.; Spraggon, L.; Velecela, V.; Martinez-Estrada, O.M.; Wiltshire, J.H.; Roberts, S.G.; Brownstein, D.; Davies, J.A.; Hastie, N.D.; Hohenstein, P. A wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell 2011, 21, 559–574. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Banfi, S.; Eskiocak, B.; Tallquist, M.D. Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis 2011, 49, 870–877. [Google Scholar] [CrossRef]
- Robb, L.; Mifsud, L.; Hartley, L.; Biben, C.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Harvey, R.P. Epicardin: A novel basic helix-loop-helix transcription factor gene expressed in epicardium, branchial arch myoblasts, and mesenchyme of developing lung, gut, kidney, and gonads. Dev. Dyn. 1998, 213, 105–113. [Google Scholar] [CrossRef]
- Wagner, N.; Morrison, H.; Pagnotta, S.; Michiels, J.F.; Schwab, Y.; Tryggvason, K.; Schedl, A.; Wagner, K.D. The podocyte protein nephrin is required for cardiac vessel formation. Hum. Mol. Genet. 2011, 20, 2182–2194. [Google Scholar] [CrossRef]
- Airik, R.; Bussen, M.; Singh, M.K.; Petry, M.; Kispert, A. Tbx18 regulates the development of the ureteral mesenchyme. J. Clin. Invest. 2006, 116, 663–674. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9; Discussion E9–E10. [Google Scholar] [CrossRef]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; Hosen, N.; Hill, R.E.; Munoz-Chapuli, R.; Hastie, N.D. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar]
- Hohenstein, B.; Hausknecht, B.; Boehmer, K.; Riess, R.; Brekken, R.A.; Hugo, C.P. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006, 69, 1654–1661. [Google Scholar] [CrossRef]
- Suri, M.; Kelehan, P.; O'Neill, D.; Vadeyar, S.; Grant, J.; Ahmed, S.F.; Tolmie, J.; McCann, E.; Lam, W.; Smith, S.; Fitzpatrick, D.; Hastie, N.D.; Reardon, W. WT1 mutations in Meacham syndrome suggest a coelomic mesothelial origin of the cardiac and diaphragmatic malformations. Am. J. Med. Genet. A 2007, 143A, 2312–2320. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Carmona, R.; Gonzalez-Iriarte, M.; Atencia, G.; Wessels, A.; Munoz-Chapuli, R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int J. Dev. Biol. 2002, 46, 1005–1013. [Google Scholar]
- Pombal, M.A.; Carmona, R.; Megias, M.; Ruiz, A.; Perez-Pomares, J.M.; Munoz-Chapuli, R. Epicardial development in lamprey supports an evolutionary origin of the vertebrate epicardium from an ancestral pronephric external glomerulus. Evol. Dev. 2008, 10, 210–216. [Google Scholar] [CrossRef]
- Boulberdaa, M.; Turkeri, G.; Urayama, K.; Dormishian, M.; Szatkowski, C.; Zimmer, L.; Messaddeq, N.; Laugel, V.; Dolle, P.; Nebigil, C.G. Genetic inactivation of prokineticin receptor-1 leads to heart and kidney disorders. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 842–850. [Google Scholar] [CrossRef]
- Boulberdaa, M.; Urayama, K.; Nebigil, C.G. Prokineticin receptor 1 (PKR1) signalling in cardiovascular and kidney functions. Cardiovasc. Res. 2011, 92, 191–198. [Google Scholar] [CrossRef]
- Noguchi, H.; Ueda, M.; Matsumoto, S.; Kobayashi, N.; Hayashi, S. BETA2/NeuroD protein transduction requires cell surface heparan sulfate proteoglycans. Hum. Gene Ther 2007, 18, 10–17. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; Pu, W.T. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; Stallcup, W.B.; Denton, C.P.; McCulloch, A.; Chen, J.; Evans, S.M. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; Olson, E.N.; Tallquist, M.D. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell. 2012, 22, 639–650. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nguyen, T.L.; Gasser, A.; Nebigil, C.G. Role of Prokineticin Receptor-1 in Epicardial Progenitor Cells. J. Dev. Biol. 2013, 1, 20-31. https://doi.org/10.3390/jdb1010020
Nguyen TL, Gasser A, Nebigil CG. Role of Prokineticin Receptor-1 in Epicardial Progenitor Cells. Journal of Developmental Biology. 2013; 1(1):20-31. https://doi.org/10.3390/jdb1010020
Chicago/Turabian StyleNguyen, Thu Lan, Adelin Gasser, and Canan G. Nebigil. 2013. "Role of Prokineticin Receptor-1 in Epicardial Progenitor Cells" Journal of Developmental Biology 1, no. 1: 20-31. https://doi.org/10.3390/jdb1010020