New Thalidomide-Resembling Dicarboximides Target ABC50 Protein and Show Antileukemic and Immunomodulatory Activities

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
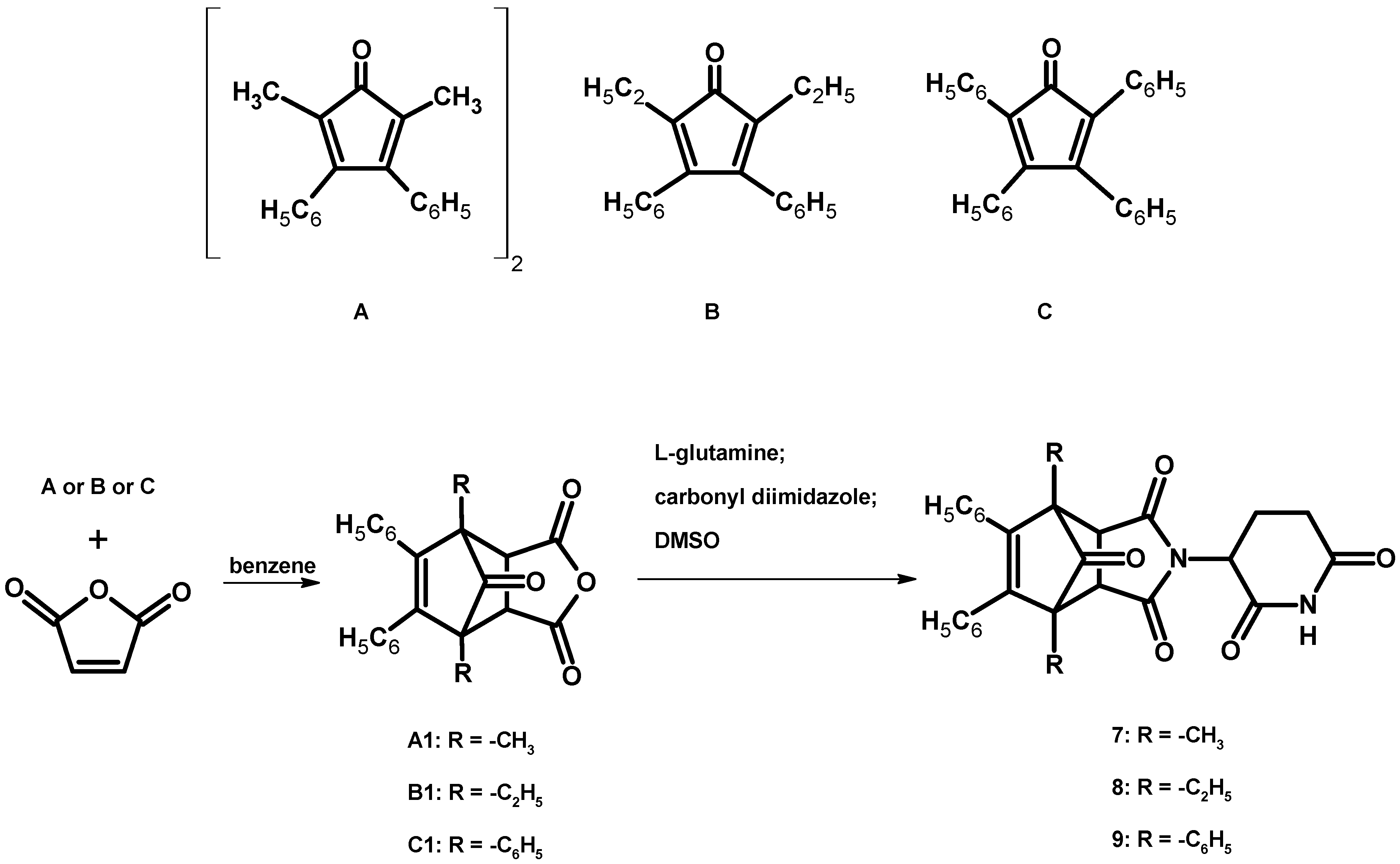
2.1.1. Synthesis of 1,7,8,9-tetraphenyl-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione (D)
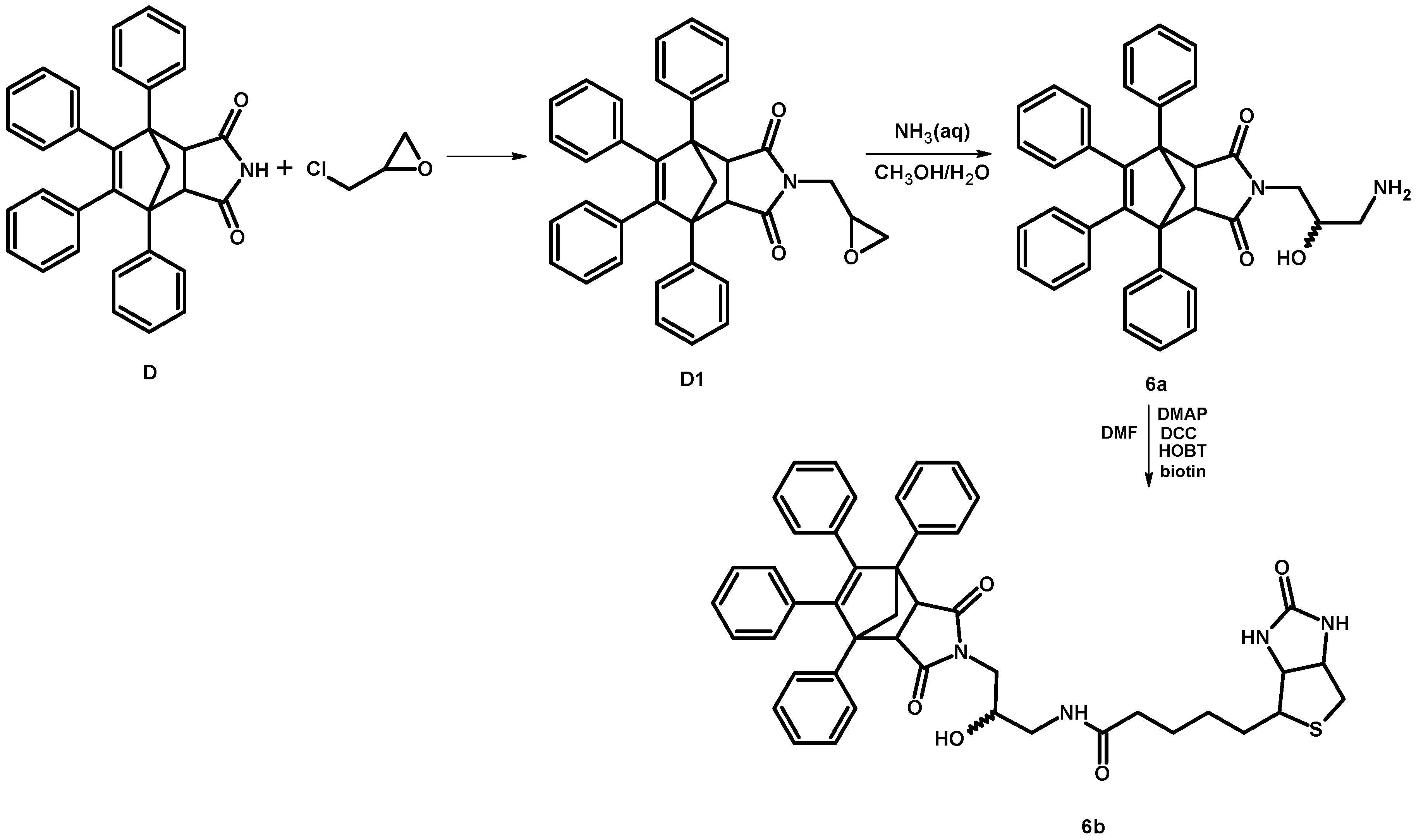
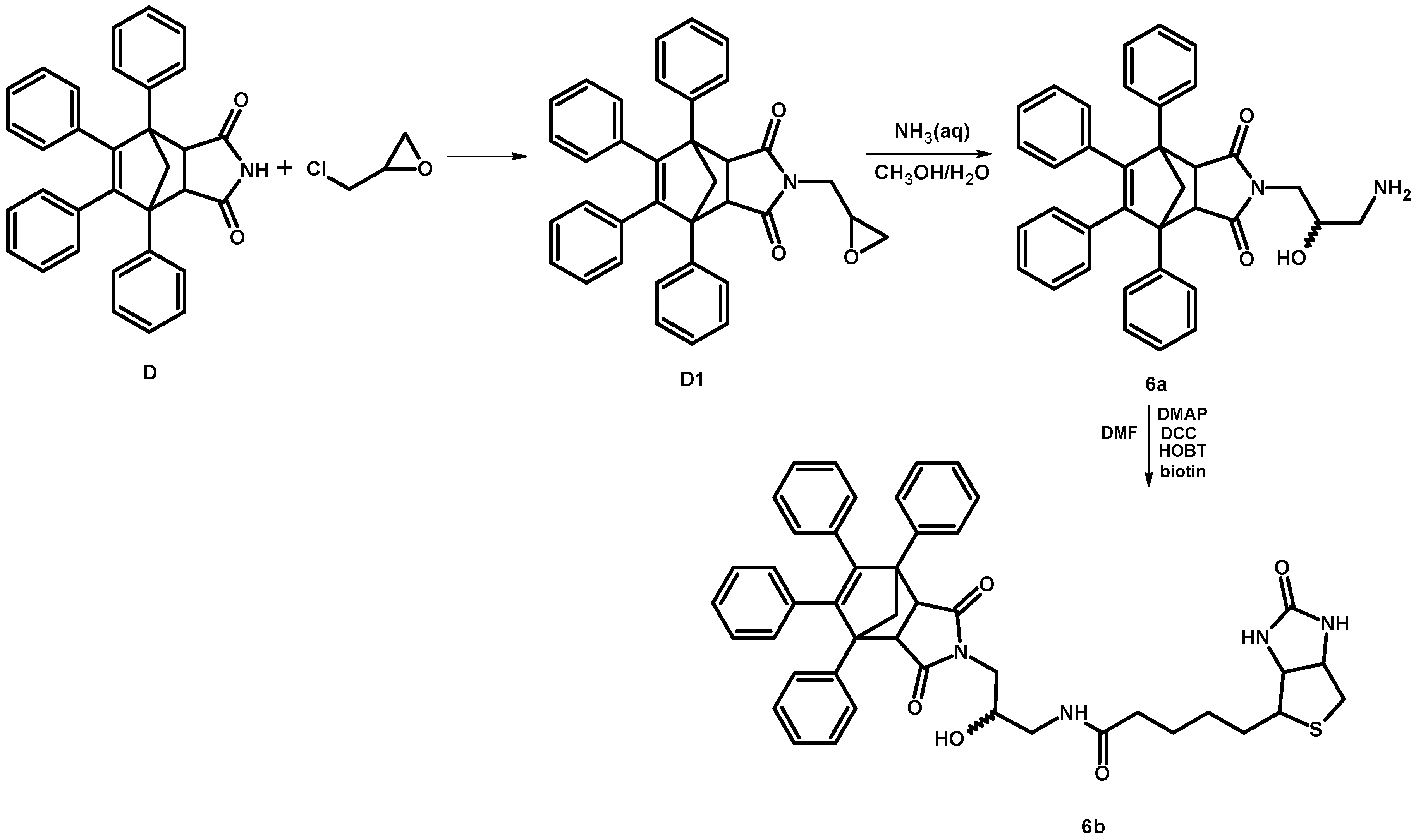
2.1.2. Synthesis of 4-[-3-(amino)-2-hydroxypropyl]-1,7,8,9-tetraphenyl-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione (6a)
2.1.3. Synthesis of Biotinylated Derivative 6b
2.1.4. Synthesis of Derivatives 7–9
Synthesis of Anhydrides A1, B1 and C1
- 2,5-dimethyl-3,4-diphenylcyclopentadienone as a dimer (0.002 mol of dimer)
- 2,5-diethyl-3,4-diphenylcyclopenta-2,4-dienone (0.007 mol)
- 1,2,3,4-tetraphenylcyclopenta-1,3-dienone (0.007 mol)
Synthesis of Derivatives 7–9
2.2. Cell Culturing and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cytotoxicity Assay
2.3. Activation of Caspase-3/7 and Caspase-8/9 Determined by Fluorescent and Luminescent Assays
2.4. Cleavage of Caspase 3/8/9 and Poly(ADP-Ribose)Polymerase (PARP)
2.5. Annexin V/Propidium Iodide (PI) Flow Cytometry
2.6. Annexin V/7-Aminoactinomycin D (7-AAD) Flow Cytometry
2.7. Real-Time Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. The Effect of Thalidomide, Lenalidomide and Test Dicarboximides on the IKZF1 and IKZF3 Level in Leukemia Cells
2.9. IKZF1, IKZF3 and ABC50 Immunoblotting
2.10. Identification of Cellular Proteins Targeted by Dicarboximides—Pull-Down Assay
2.11. Analysis of ABC50 Protein: Cell Culture, siRNA, Transfections
2.12. Statistical Analysis
3. Results
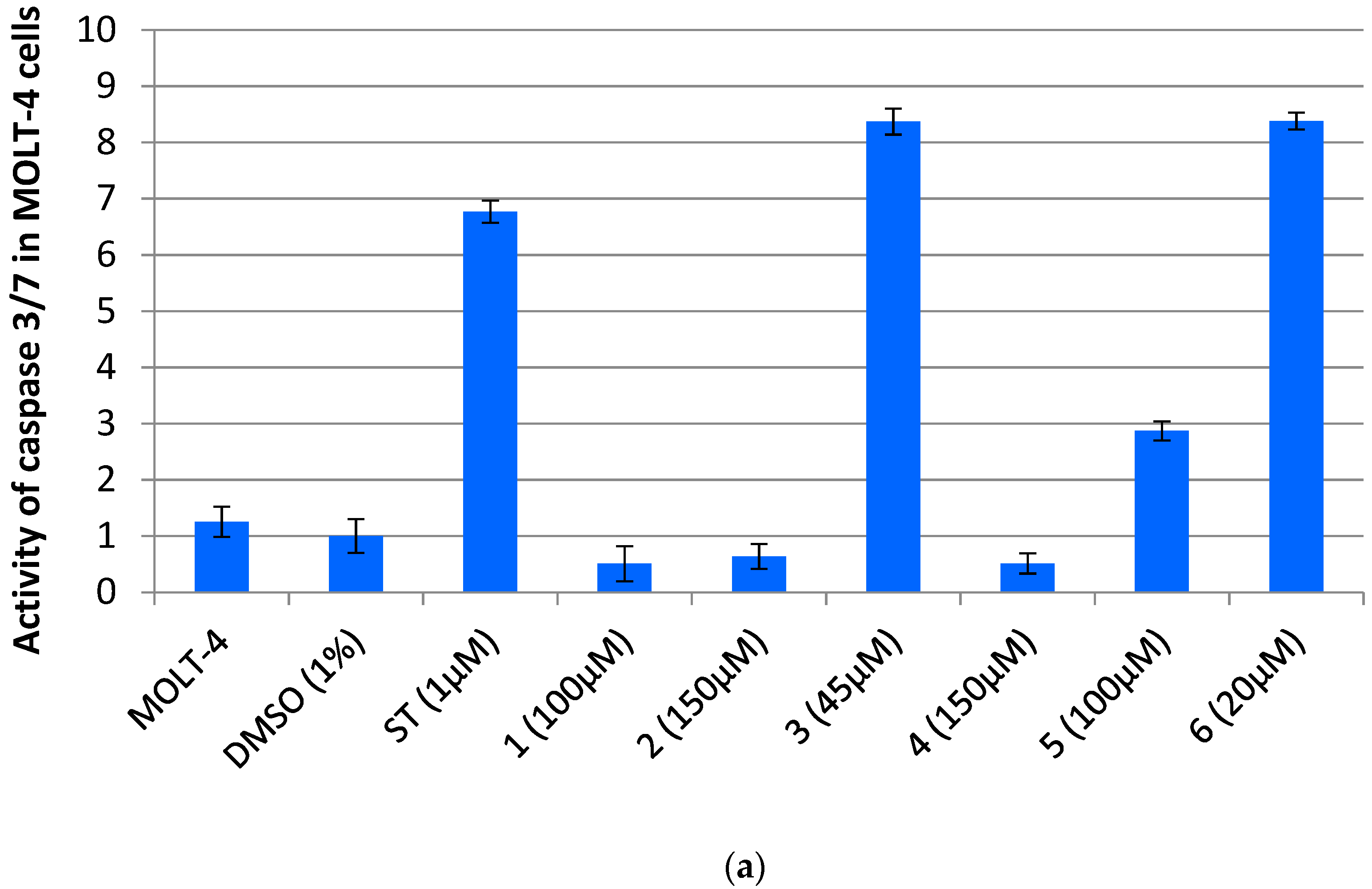
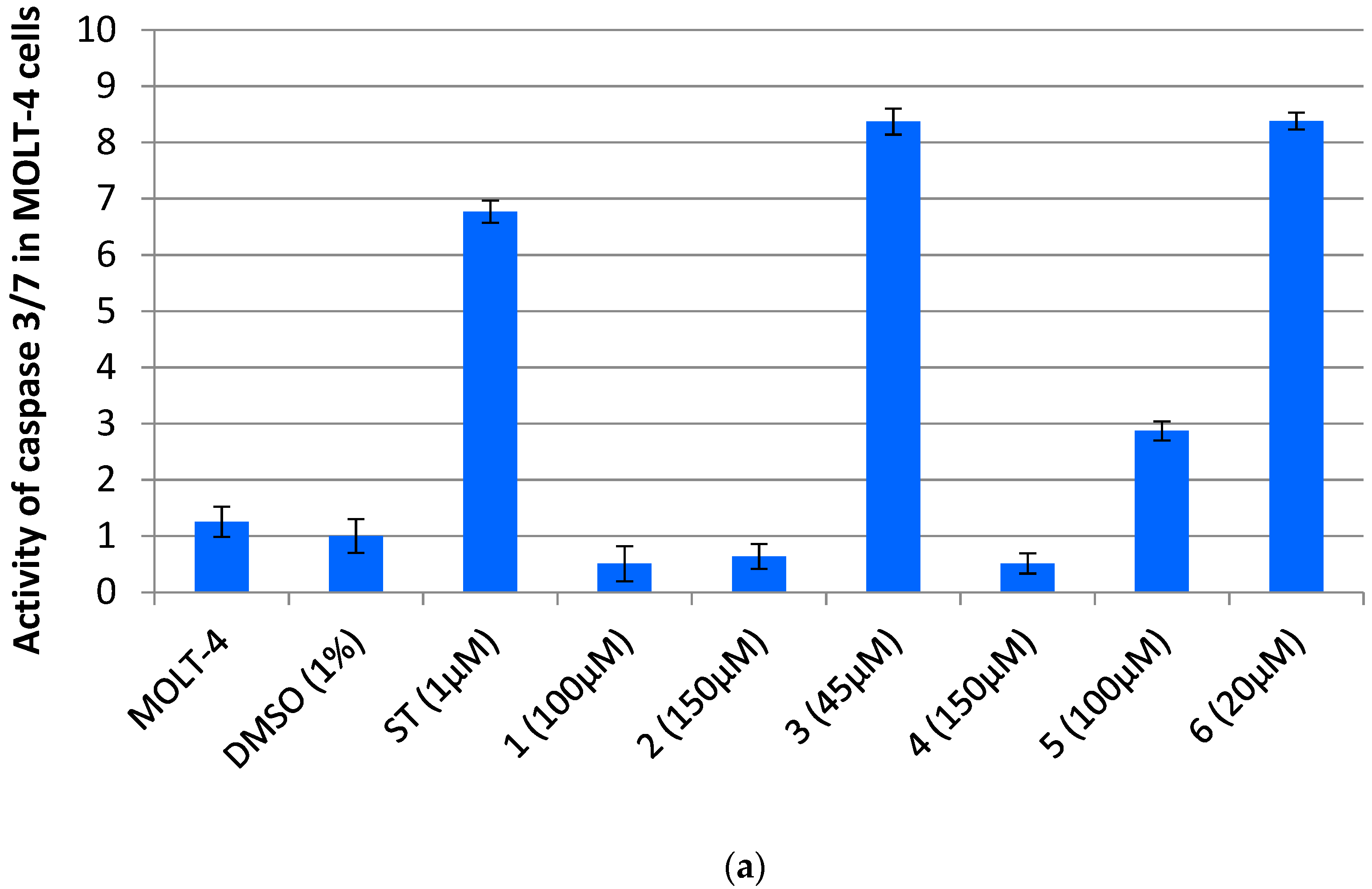
3.1. Dicarboximides Are Cytotoxic toward Leukemia Cells
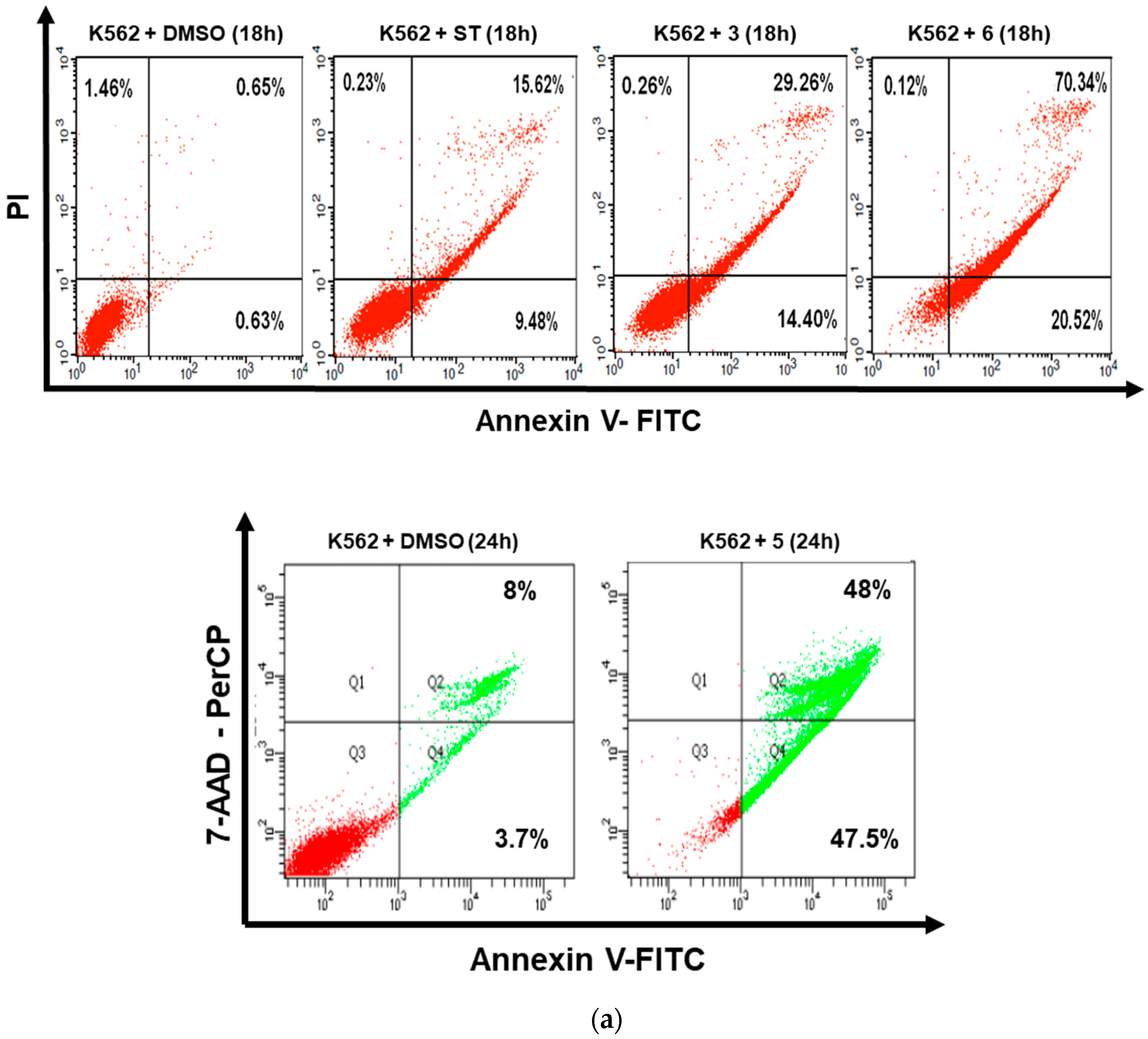
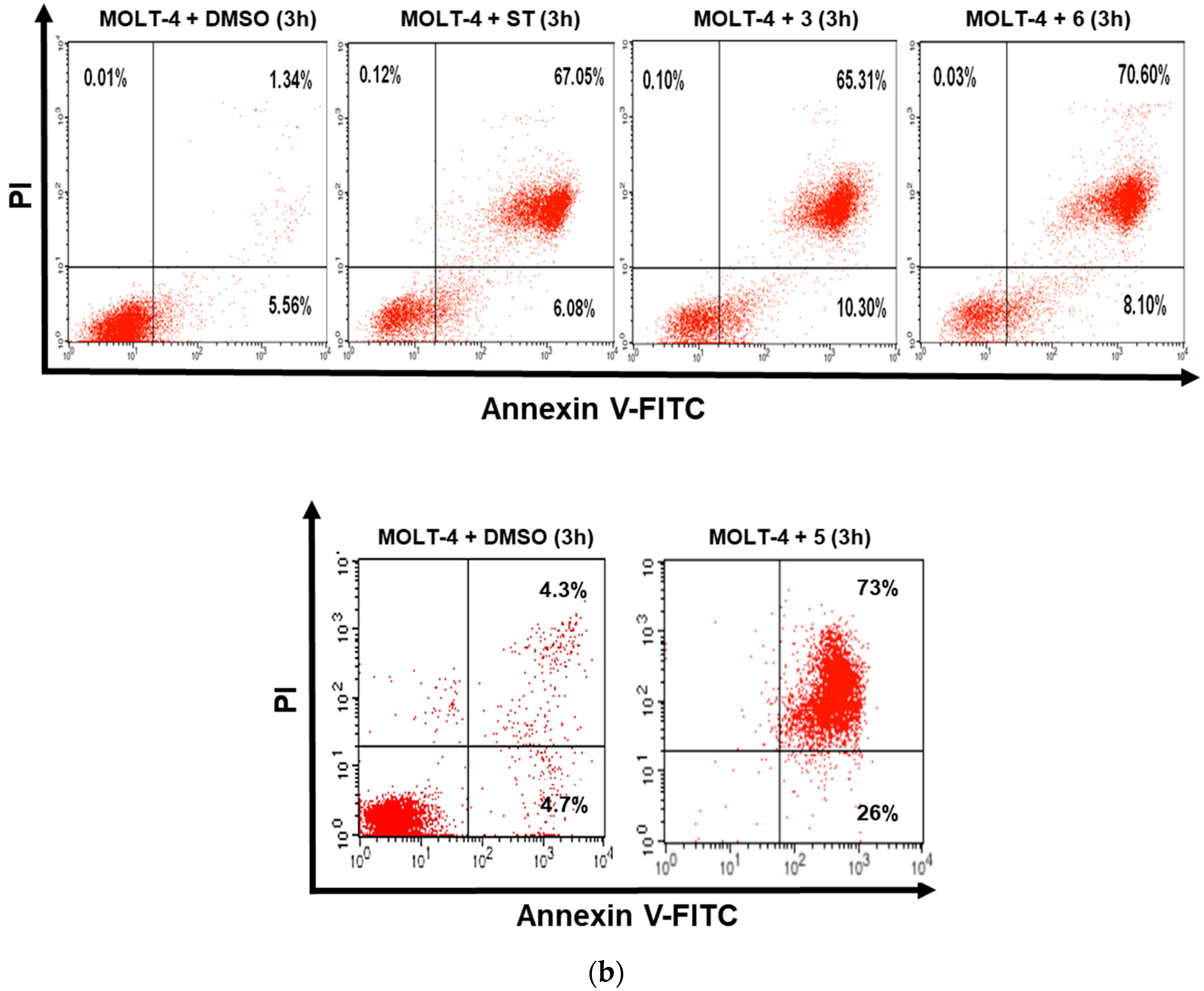
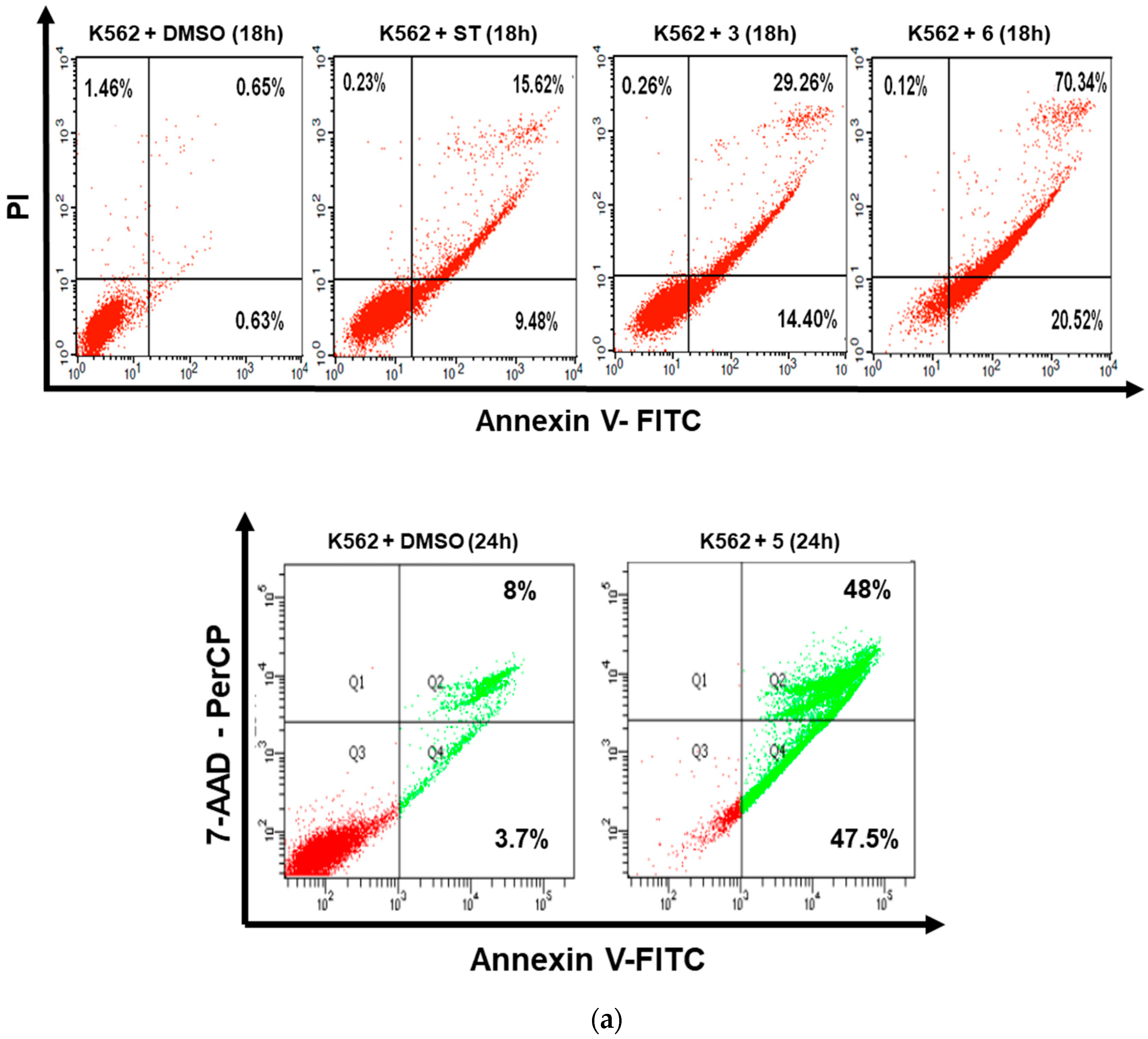
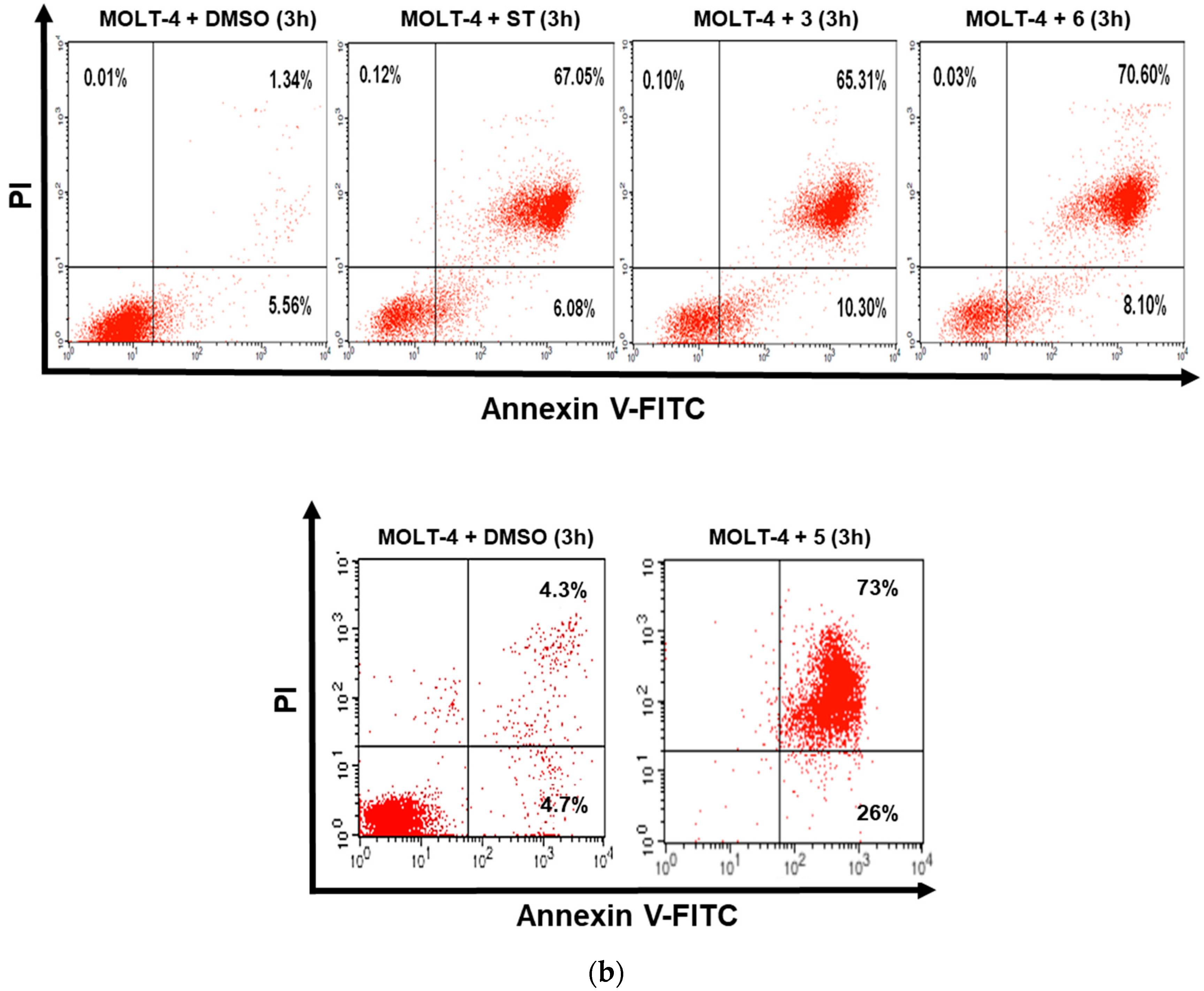
3.2. Dicarboximides Induce Apoptosis in Leukemia Cells
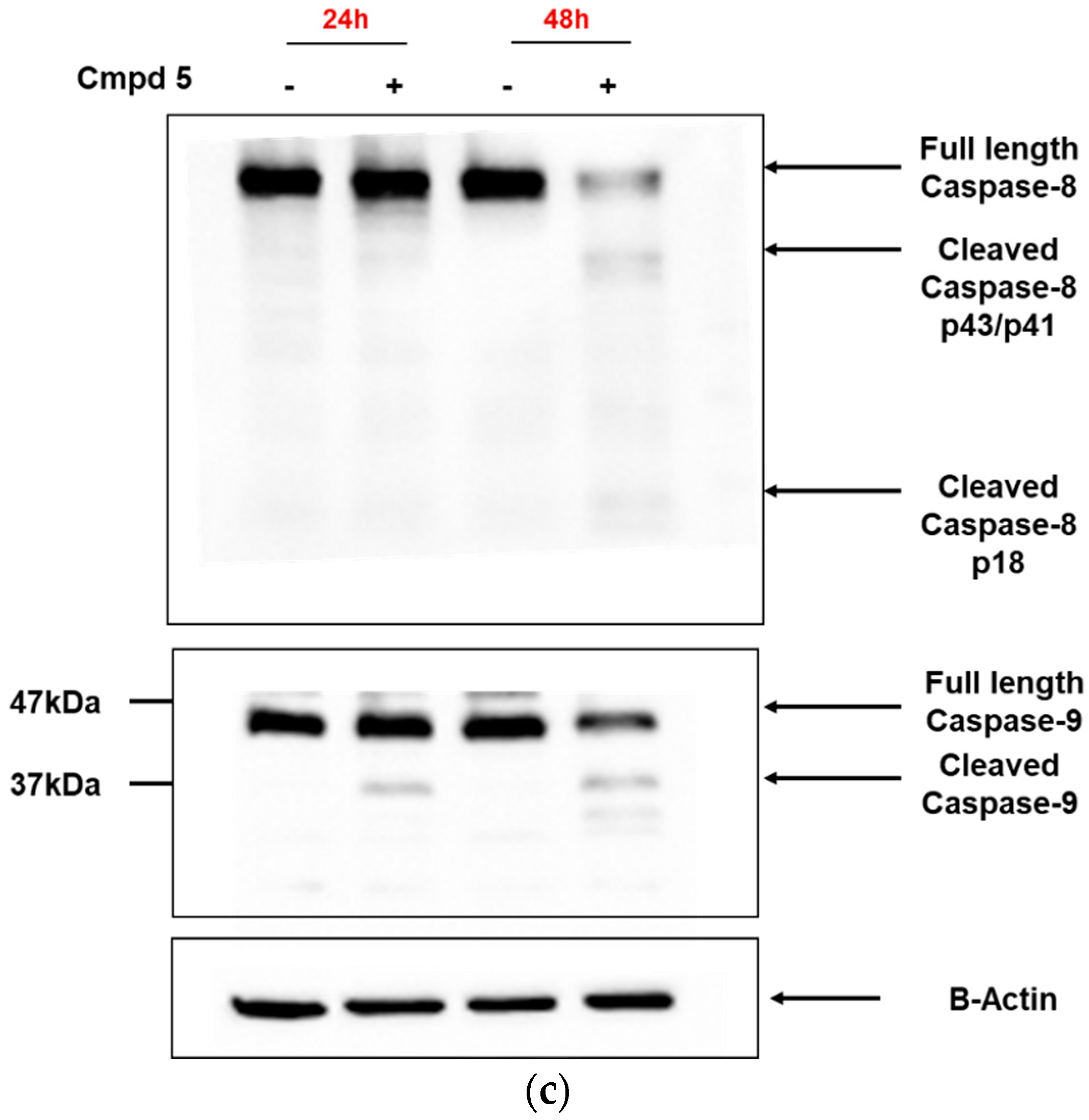
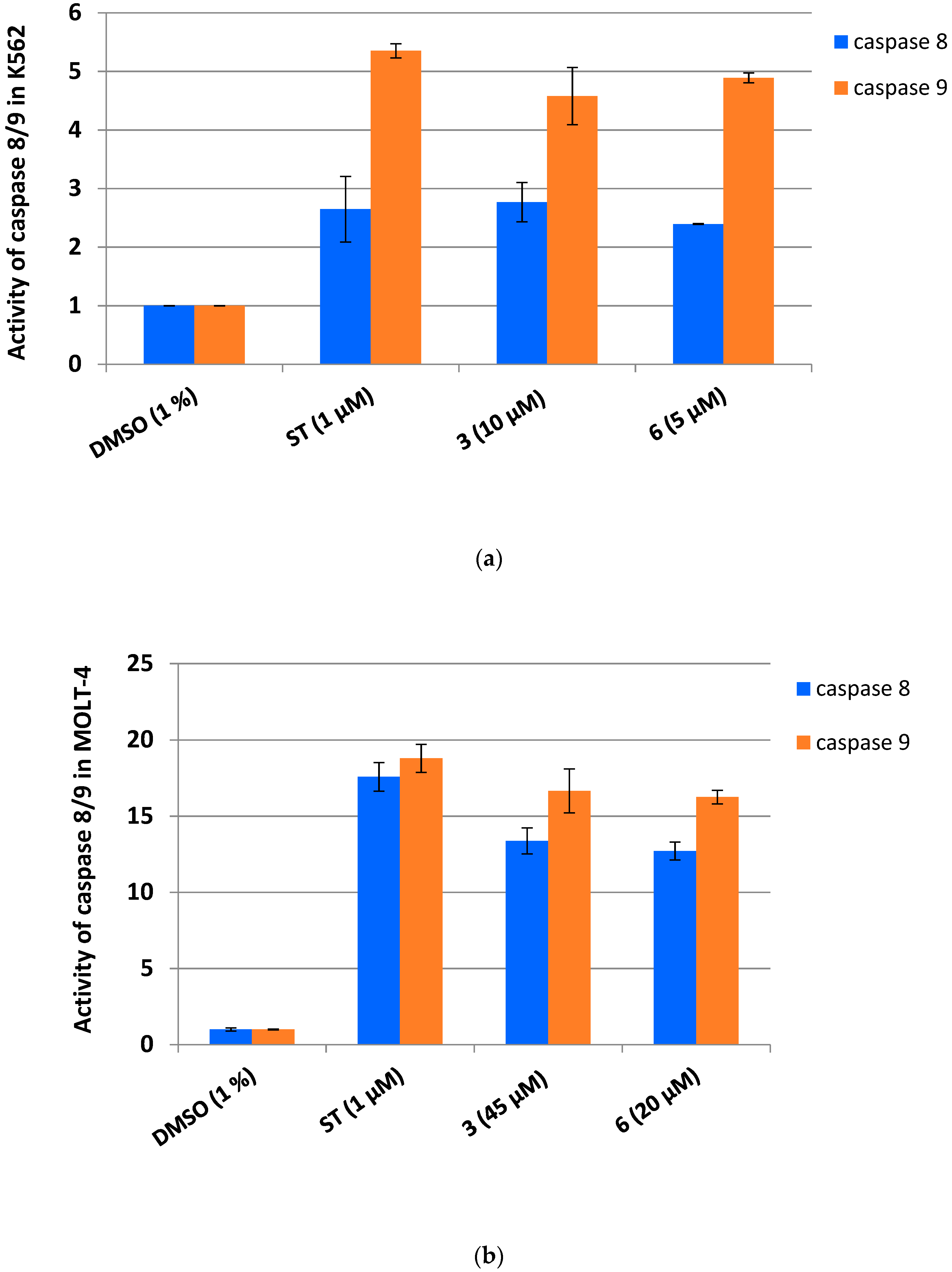
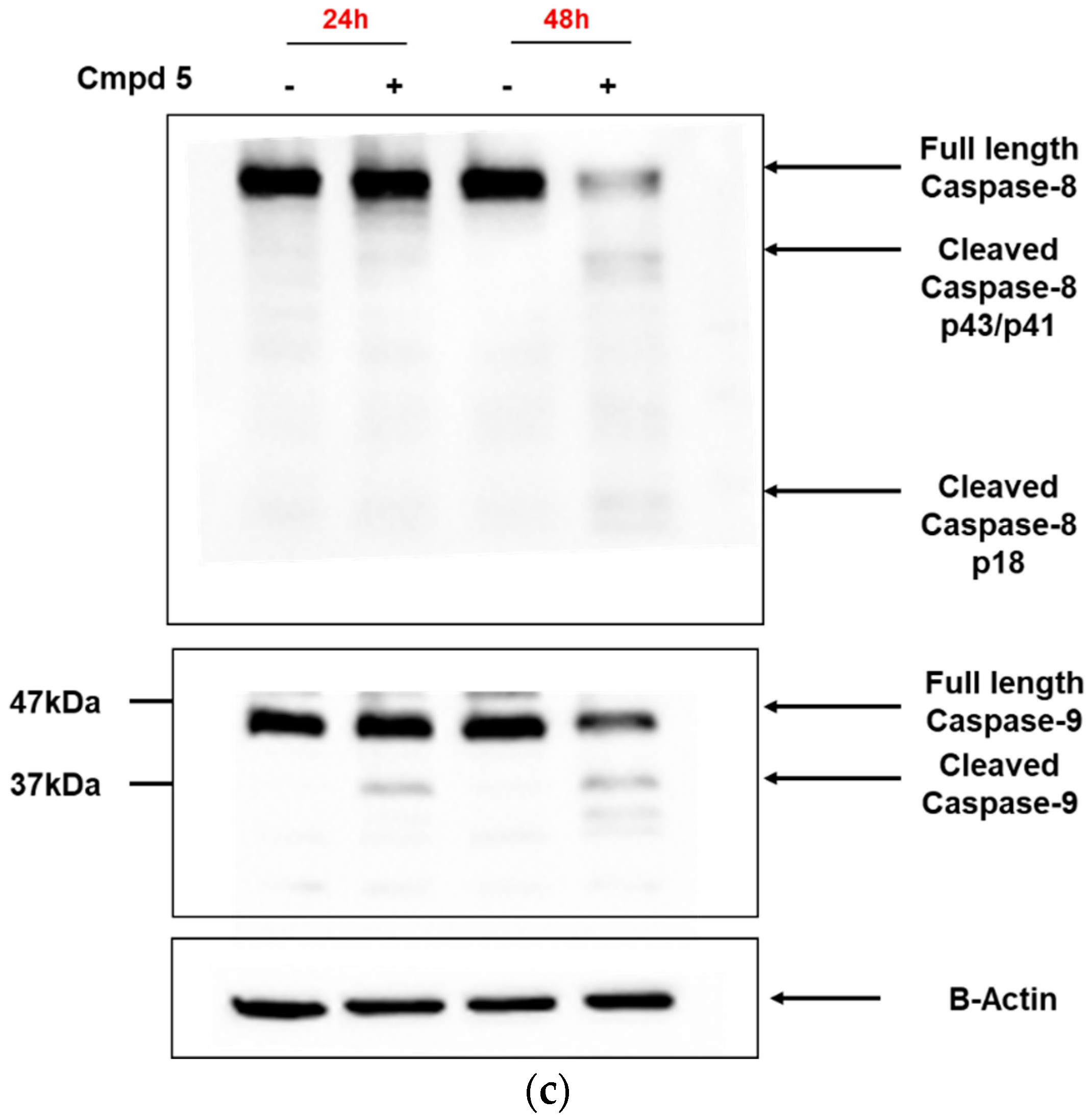
3.3. Dicarboximides Activate Apoptosis via Receptor and Mitochondrial Pathways
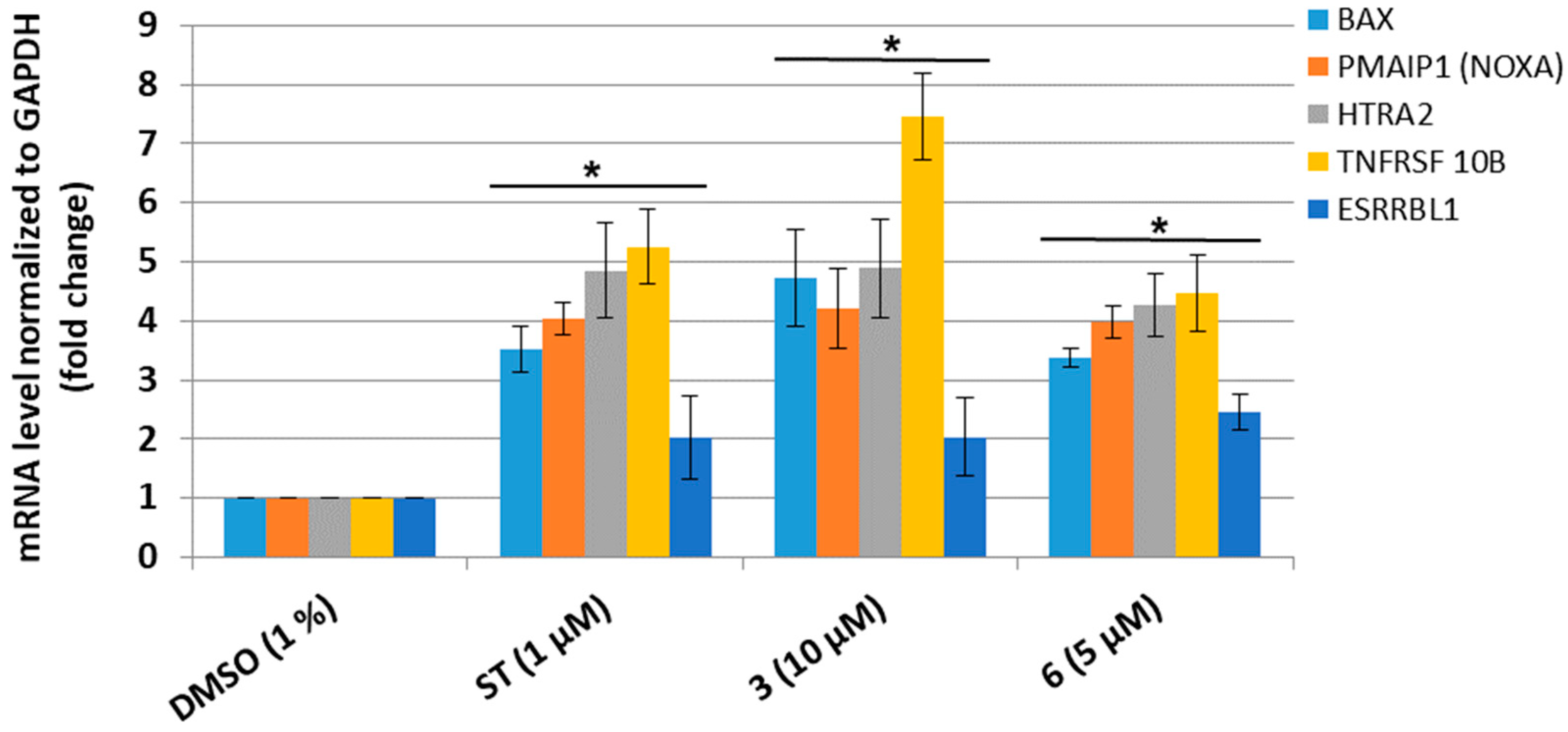
3.4. Dicarboximides Change the Expression Profile of Genes Involved in Apoptosis
3.5. Identification of Cellular Proteins Targeted by Dicarboximides—Pull-Down Assay
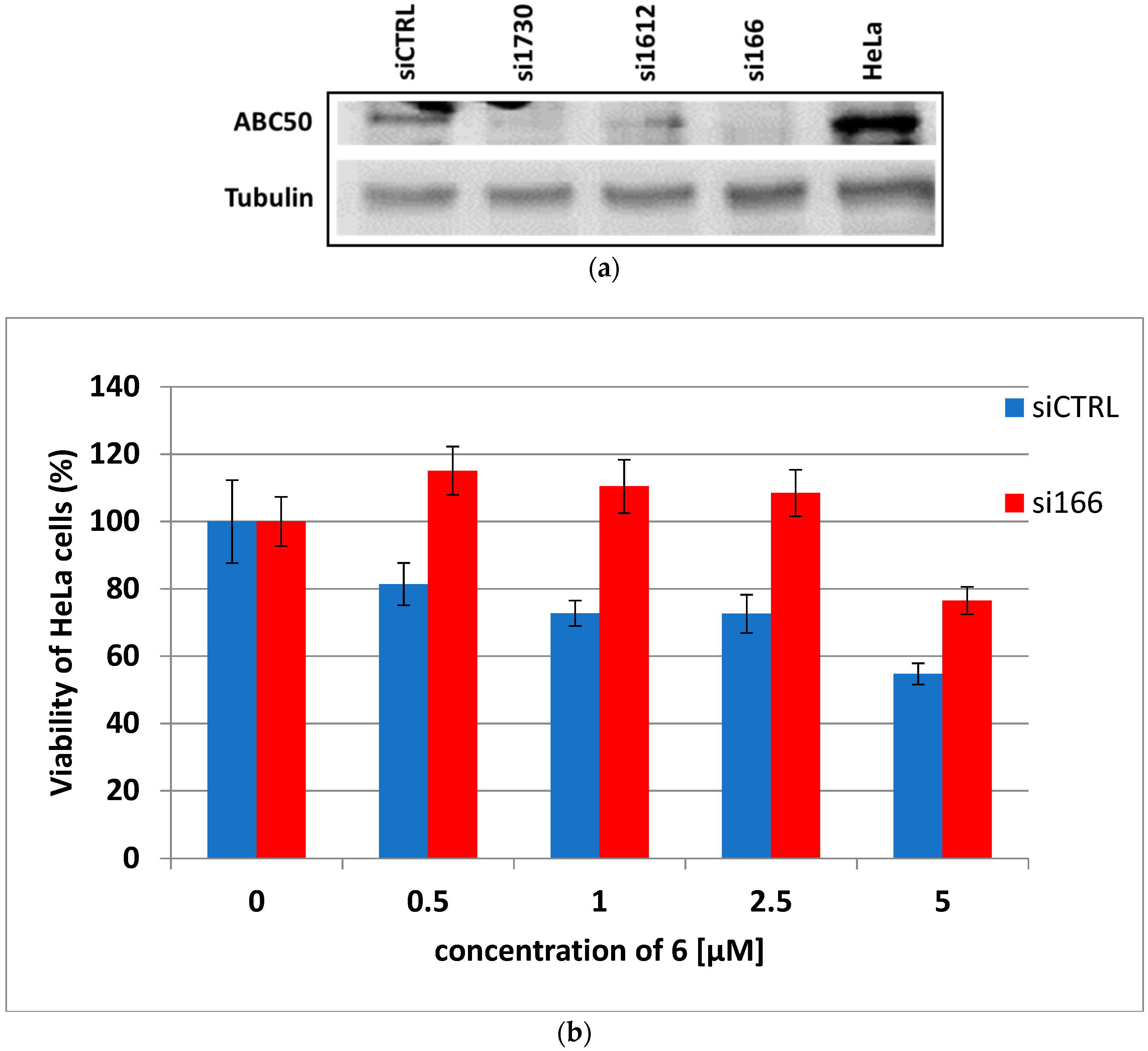
3.6. ABC50 Knockdown Abolishes HeLa Sensitivity to Dicarboximide 6
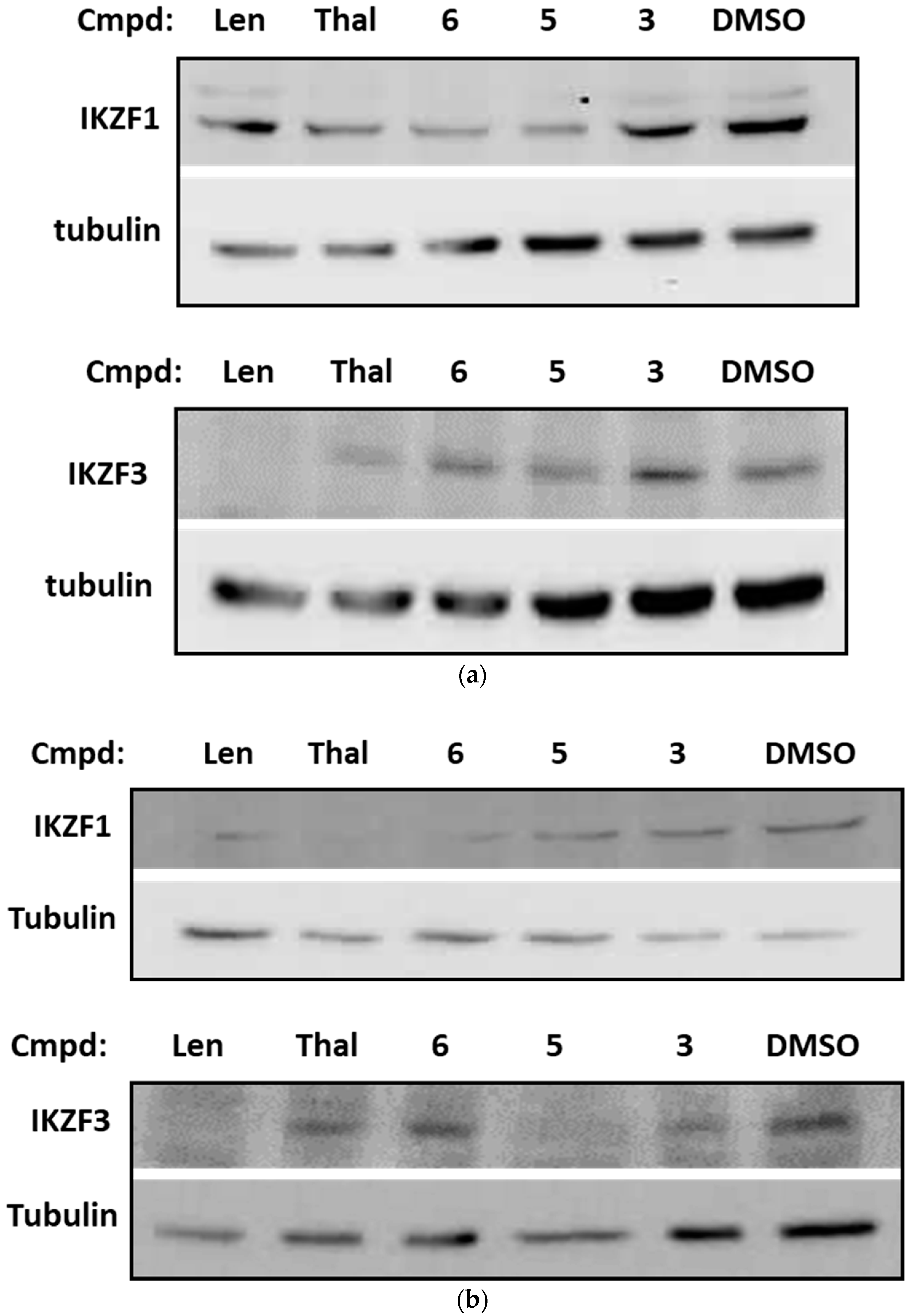
3.7. Dicarboximides Reduce the Level of IKZF1/3 Transcription Factors in Leukemia Cells
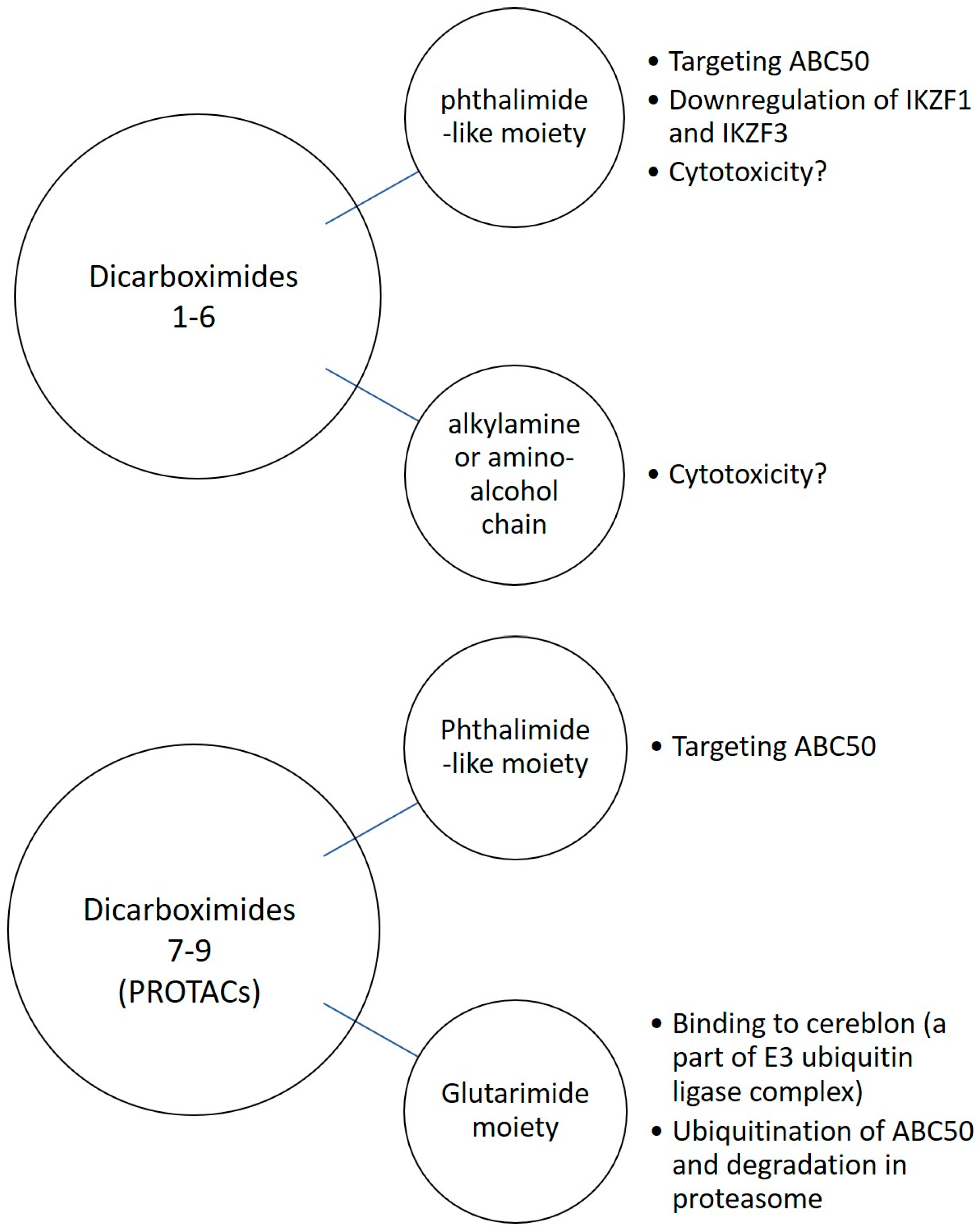
3.8. Glutarimide Derivatives of Dicarboximides as Modulators of ABC50 in Cancer Cells—Proteolysis-Targeting Chimeras (PROTACs) Approach
4. Discussion
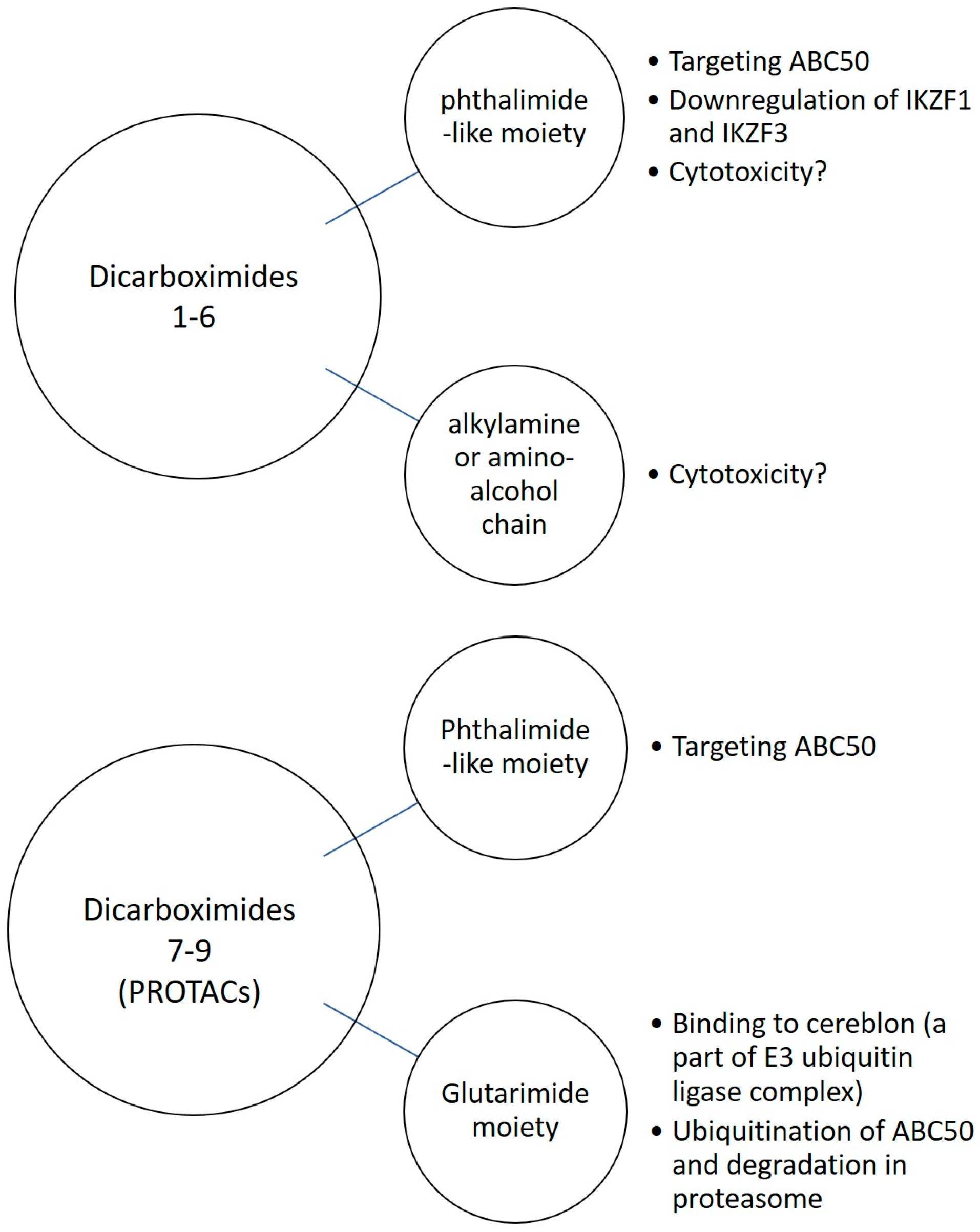
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2003, 5, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Bergmann, A. Apoptosis-induced compensatory proliferation. The cell is dead. Long live the cell. Trends Cell Biol. 2008, 18, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, P.; Rastogi, R.; Rajeshwar, P.S. Apoptosis: Molecular mechanism and pathogenicity. EXCLI J. 2009, 8, 155–181. [Google Scholar]
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nature Rev. Cancer. 2009, 9, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Broker, L.E.; Kruyt, F.A.; Giaccone, G. Cell death independent of caspases: A review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell. Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Saez, R.; Craig, J.; Kuhn, J. Phase I clinical investigation of amonafide. J. Clin. Oncol. 1989, 7, 1351–1358. [Google Scholar] [CrossRef]
- Llombart, M.; Poveda, A.; Forner, E. Phase I study of mitonafide in solid tumors. Investig. New Drugs. 1992, 10, 177–181. [Google Scholar] [CrossRef]
- Quintana-Espinoza, P.; García-Luis, J.; Amesty, A.; Martín-Rodríguez, P.; Lorenzo-Castrillejo, I.; Ravelo, A.G.; Fernández-Pérez, L.; Machín, F.; Estévez-Braun, A. Synthesis and study of antiproliferative, antitopoisomerase II, DNA-intercalating and DNA-damaging activities of arylnaphthalimides. Bioorg. Med. Chem. 2013, 1, 6484–6495. [Google Scholar] [CrossRef]
- Chen, Z.; Liang, X.; Zhang, H.; Xie, H.; Liu, J.; Xu, Y.; Zbu, W.; Wang, Y.; Wang, X.; Tan, S.; et al. A new class of naphthalimide-based antitumor agents that inhibit topoisomerase II and induce lysosomal membrane permeabilization and apoptosis. J. Med. Chem. 2010, 53, 2589–2600. [Google Scholar] [CrossRef]
- Xu, G.; He, Q.; Yang, B.; Hu, Y. Synthesis and Antitumor Activity of Novel 4-Chloro-3-Arylmaleimide Derivatives. Lett. Drug Des. Discovery 2009, 6, 51–55. [Google Scholar] [CrossRef]
- Sanchez-Martinez, C.; Shih, C.; Zhu, G.; Li, T.; Brooks, H.B.; Patel, B.; Schultz, R.M.; DeHahn, T.B.; Spencer, C.D.; Watkins, S.A.; et al. Studies on cyclin-dependent kinase inhibitors: Indolo-[2, 3-a] pyrrolo [3, 4-c] carbazoles versus bis-indolylmaleimides. Bioorg. Med. Chem. Lett. 2003, 13, 3841–3846. [Google Scholar] [CrossRef]
- Stewart, A.K. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef]
- Morgan, B.; Sun, L.; Avitahl, N.; Andrikopoulos, K.; Ikeda, T.; Gonzales, E.; Wu, P.; Neben, S.; Georgopoulos, K. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J. 1997, 16, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Kuran, B.; Krawiecka, M.; Kossakowski, J.; Pindel, Ł.; Młynarczyk, G.; Cieślak, M.; Kaźmierczak-Barańska, J.; Królewska, K. Synthesis and biological activity of a novel series of 6,7-dimethoxyquinazoline-2,4(1H,3H)-dione derivatives. Acta Pol. Pharm. 2012, 69, 145–148. [Google Scholar]
- Kuran, B.; Napiórkowska, M.; Kossakowski, J.; Cieślak, M.; Kaźmierczak-Barańska, J.; Królewska, K.; Nawrot, B. New, Substituted Derivatives of Dicarboximides and their Cytotoxic Properties. Anticancer Agents Med. Chem. 2016, 16, 852–864. [Google Scholar] [CrossRef]
- Kuran, B.; Krawiecka, M.; Kossakowski, J.; Cieslak, M.; Kazmierczak-Baranska, J.; Królewska, K.; Nawrot, B. Dicarboximides derivatives for use in the treatment of cancer. Patent application EP13176421.9, 2013. [Google Scholar]
- Alpegiani, M.; Mazzoni, A.; Vergani, D.; Cabri, W. Process for synthesis of thalidomide. United States Patent Application Publication Pub. No. US 2005/0272934A1, 8 December 2005. [Google Scholar]
- Królewska-Golińska, K.; Cieślak, M.J.; Sobczak, M.; Dolot, R.; Radzikowska-Cieciura, E.; Napiórkowska, M.; Wybrańska, I.; Nawrot, B. Novel benzo[b]furans with anti-microtubule activity upregulate expression of apoptotic genes and arrest leukemia cells in G2/M phase. Anticancer Agents Med. Chem. 2019, 19, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Dutta, S.; Shanmugavel, M.; Mondhe, D.M.; Sharma, P.R.; Singh, S.K.; Saxena, A.K.; Sanyal, U. 6-Nitro-2-(3-hydroxyoropyl)-1H-benz[de]isoquinoline-1,3-dione, a potent antitumor agent, induces cell cycle arrest and apoptosis. J. Exp.Clin. Cancer Res. 2010, 29, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Machado, K.E.; de Oliveira, K.N.; Andreossi, H.M.; Bubniak Ldos, S.; de Moraes, A.C.; Gaspar, P.C.; Andrade Eda, S.; Nunes, R.J.; Santos-Silva, M.C. Apoptotic events induced by maleimides on human acute leukemia cell lines. Chem. Res. Toxico. 2013, 26, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Lopez, Y.; Sarmiento-Silva, R.E.; Moran-Andrade, J.; Gomez-Garcıa, B. Staurosporine-induced apoptosis in P388D1 macrophages involves both extrinsic and intrinsic pathways. Cell Biol. 2009, 33, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Dong Zhang, X.; Gillespie, S.K.; Hersey, P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol. Cancer Ther. 2004, 3, 187–197. [Google Scholar]
- Tyzack, J.K.; Wang, X.; Belsham, G.J.; Proud, C.G. ABC50 interacts with eukaryotic initiation factor 2 and associates with the ribosome in an ATP-dependent manner. J Biol Chem. 2000, 275, 34131–34139. [Google Scholar] [CrossRef] [PubMed]
- Paytubi, S.; Wang, X.; Lam, Y.W.; Izquierdo, L.; Hunter, M.J.; Jan, E.; Hundal, H.S.; Proud, C.G. ABC50 promotes translation initiation in mammalian cells. J Biol Chem. 2009, 284, 24061–24073. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Vikström, I.; Chambers, J.M.; McArthur, K.; Ann Anderson, M.; Henley, K.J.; Happo, L.; Cluse, L.; Johnstone, R.W.; Roberts, A.W.; et al. Translation inhibitors induce cell death by multiple mechanisms and Mcl-1 reduction is only a minor contributor. Cell Death Dis. 2012, 3, e409. [Google Scholar] [CrossRef]
- Lee, M.N.; Roy, M.; Ong, S.E.; Mertins, P.; Villani, A.C.; Li, W.; Dotiwala, F.; Sen, J.; Doench, J.G.; Orzalli, M.H.; et al. Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nat. Immunol. 2013, 14, 179–185. [Google Scholar] [CrossRef]
- Richard, M.; Drouin, R.; Beaulieu, A.D. ABC50, a Novel Human ATP-Binding Cassette Protein Found in Tumor Necrosis Factor-a-Stimulated Synoviocytes. Genomics 1998, 53, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Li, Y.; Zhou, J.; Jiang, S.; Wang, Y.; Chen, Y.; Zhao, D.; Yang, C.; Qian, Q.; Ma, Y.; et al. Copy number variants of ABCF1, IL17REL, and FCGR3A are associated with the risk of gout. Protein Cell. 2017, 8, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, M.; Katsuyama, Y.; Hamano, H.; Umemura, T.; Kimura, A.; Yoshizawa, K.; Kiyosawa, K.; Fukushima, H.; Bahram, S.; Inoko, H.; et al. Two critical genes (HLA-DRB1 and ABCF1) in the HLA region are associated with the susceptibility to autoimmune pancreatitis. Immunogenetics 2007, 59, 45–52. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure * | Compound | Structure * |
|---|---|---|---|
| 1 | 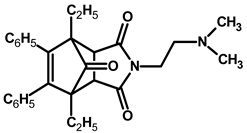 | 4 |  |
| 2 |  | 5 |  |
| 3 |  | 6 |  |
| 6a |  | 6b |  |
| 7 |  | 8 |  |
| 9 | 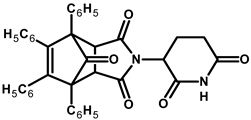 |
| Compound | HeLa | K562 | HL-60 | HUVEC | MOLT-4 | CFPAC | TI *** |
|---|---|---|---|---|---|---|---|
| cytarabine | >1000 * | 300 ± 19 * | 300 ± 18 * | >1000 * | nd | nd | 3.3 |
| bortezomib | 0.05 ± 0.01 * | 0.04 ± 0.01 * | 0.3 ± 0.1 * | <0.01 * | nd | nd | 0.25 |
| sorafenib | 20.0 ± 2.6 * | 3.0 ± 0.3 * | 15.0 ± 0.6 * | 20.0 ± 4.1 * | nd | nd | 6.7 |
| irinotecan | 200 ± 11 * | 10.0 ± 0.9 * | 70.0 ± 2.5 * | 30.0 ± 3.7 * | nd | nd | 3 |
| doxorubicin | 0.50 ± 0.01 * | 0.15 ± 0.01 * | 0.15 ± 0.03 * | 0.10 ± 0.03 * | nd | nd | 0.67 |
| 1 | >100 ** | 10 ± 0.59 ** | >100 ** | >100 ** | 20.0 ± 2.7 | >1000 | 10 |
| 2 | >100 ** | 4.5 ± 0.29 ** | >100 ** | >100 ** | 30.0 ± 2.3 | >1000 | 22 |
| 3 | >100 ** | 2.0 ± 0.31 ** | >100 ** | >100 ** | 9.0 ± 0.6 | >1000 | 50 |
| 4 | >100 ** | 8 ± 0.53 ** | >100 ** | >100 ** | 30.0 ± 2.0 | >1000 | 12.5 |
| 5 | >100 ** | 1.0 ± 0.05 ** | 2.0 ± 0.05 ** | >100 ** | 20.0 ± 2.3 | >1000 | 100 |
| 6 | 1.0 ± 0.2 ** | 1.0 ± 0.3 ** | 5.0 ± 0.24 ** | >100 ** | 4.0 ± 0.2 | 40.0 ± 3.1 | 100 |
| 6a | 20 ± 1.9 | 12 ± 1.5 | nd | >100 | 5 ± 0.59 | nd | 83 |
| 6b | 64 ± 10 | 7 ± 1.2 | nd | 3.8 ± 0.22 | 1.5 ± 0.06 | nd | 0.5 |
| 7 | 180 | 170 | nd | 200 | 150 | nd | 1.2 |
| 8 | >200 | 25 | nd | 65 | 18 | nd | 2.6 |
| 9 | >200 | >200 | nd | >200 | >200 | nd | nd |
| Band No. | Database | Entry | Score | Mass (Da) | No. of Significant Matches | No. of Significant Sequences | Exponentially Modified Protein Abundance Index (emPAI) | Description |
|---|---|---|---|---|---|---|---|---|
| 1 | Sprot | Q9Y490 | 2693 | 271,766 | 72 | 59 | 18,629 | Talin-1 |
| 2 | Sprot | Q8NE71 | 289 | 96,323 | 8 | 8 | 0.42 | ATP-binding cassette sub-family F member 1 (ABCF1 or ABC50) |
| 3 | Sprot | Q8NE71 | 565 | 96,323 | 15 | 14 | 0.85 | ABC50 |
| 4 | Sprot | Q8NE71 | 1923 | 96,323 | 43 | 29 | 32,905 | ABC50 |
| 5 | Sprot | Q8NE71 | 651 | 96,323 | 18 | 16 | 42,767 | ABC50 |
| 6 | Sprot | Q8NE71 | 495 | 96,323 | 12 | 11 | 0.62 | ABC50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cieślak, M.; Kaźmierczak-Barańska, J.; Królewska-Golińska, K.; Napiórkowska, M.; Stukan, I.; Wojda, U.; Nawrot, B. New Thalidomide-Resembling Dicarboximides Target ABC50 Protein and Show Antileukemic and Immunomodulatory Activities. Biomolecules 2019, 9, 446. https://doi.org/10.3390/biom9090446
Cieślak M, Kaźmierczak-Barańska J, Królewska-Golińska K, Napiórkowska M, Stukan I, Wojda U, Nawrot B. New Thalidomide-Resembling Dicarboximides Target ABC50 Protein and Show Antileukemic and Immunomodulatory Activities. Biomolecules. 2019; 9(9):446. https://doi.org/10.3390/biom9090446
Chicago/Turabian StyleCieślak, Marcin, Julia Kaźmierczak-Barańska, Karolina Królewska-Golińska, Mariola Napiórkowska, Iga Stukan, Urszula Wojda, and Barbara Nawrot. 2019. "New Thalidomide-Resembling Dicarboximides Target ABC50 Protein and Show Antileukemic and Immunomodulatory Activities" Biomolecules 9, no. 9: 446. https://doi.org/10.3390/biom9090446