Severity of Dyskinesia and D3R Signaling Changes Induced by L-DOPA Treatment of Hemiparkinsonian Rats Are Features Inherent to the Treated Subjects

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
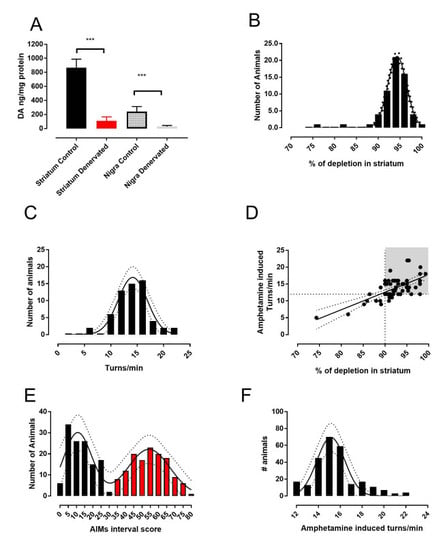
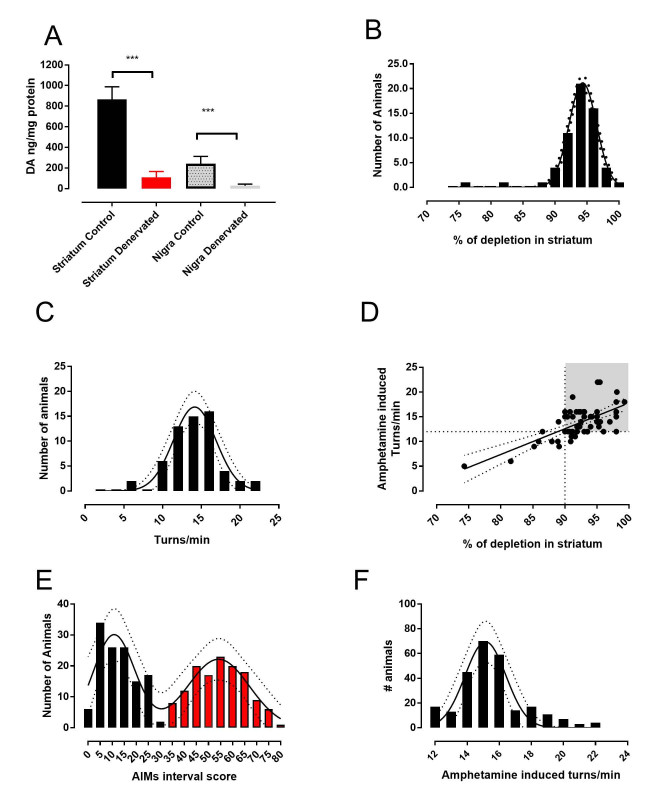
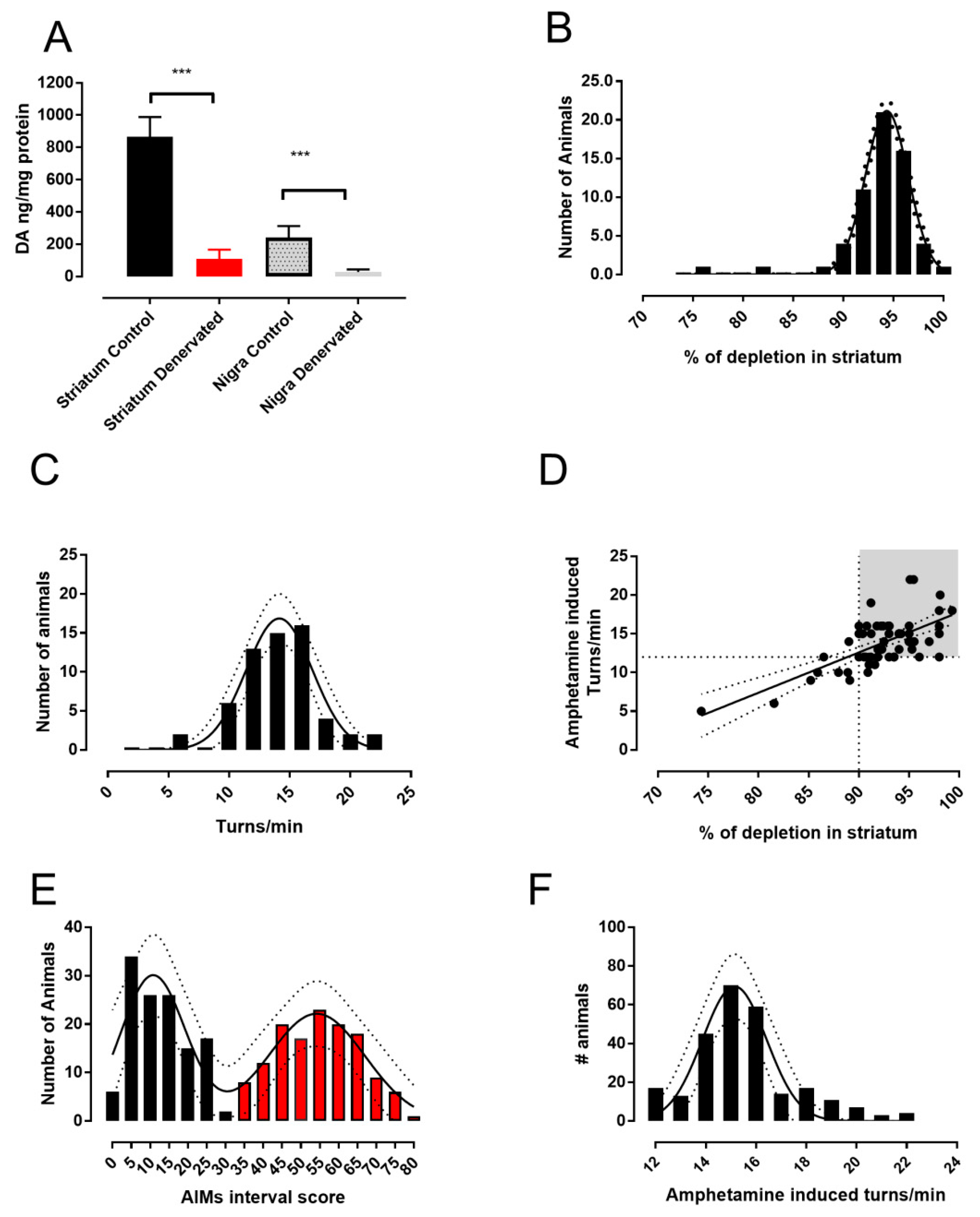
2.1. The 6-OHDA Lesion and Animal Selection
2.2. Determination of Dopamine Content
2.3. L-DOPA Treatment
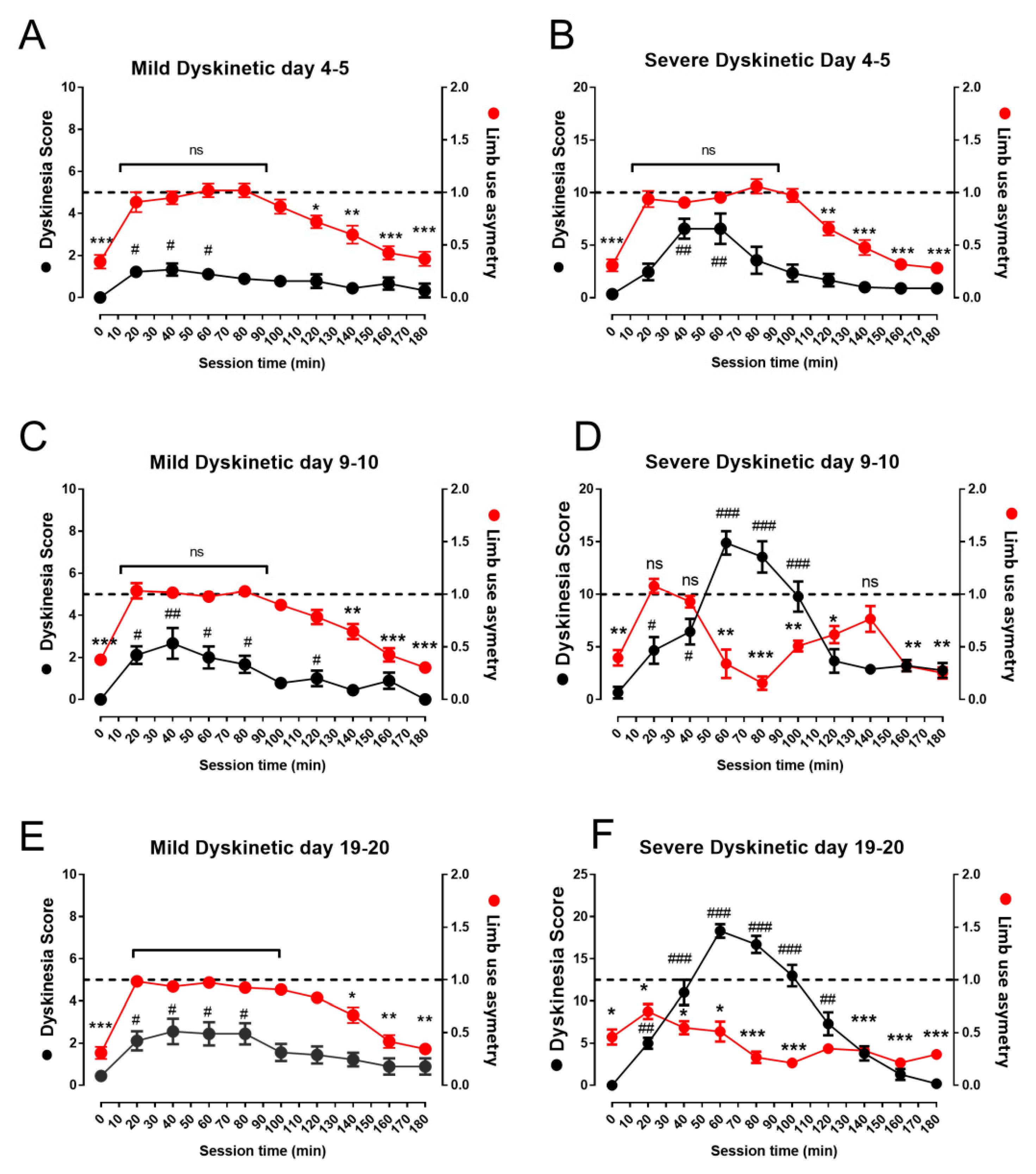
2.4. Behavioral Observations
2.5. cAMP Accumulation Assay
2.6. [3H]-GABA Release
2.7. Statistical Analysis
2.8. Drugs
3. Results
3.1. Analysis of L-DOPA- Induced AIMs Scores Yields Two Populations from a Single 6-OHDA-Lesioned Population
3.2. Therapeutic Response to L-DOPA is Different in Mild and Severe Dyskinetic Animals
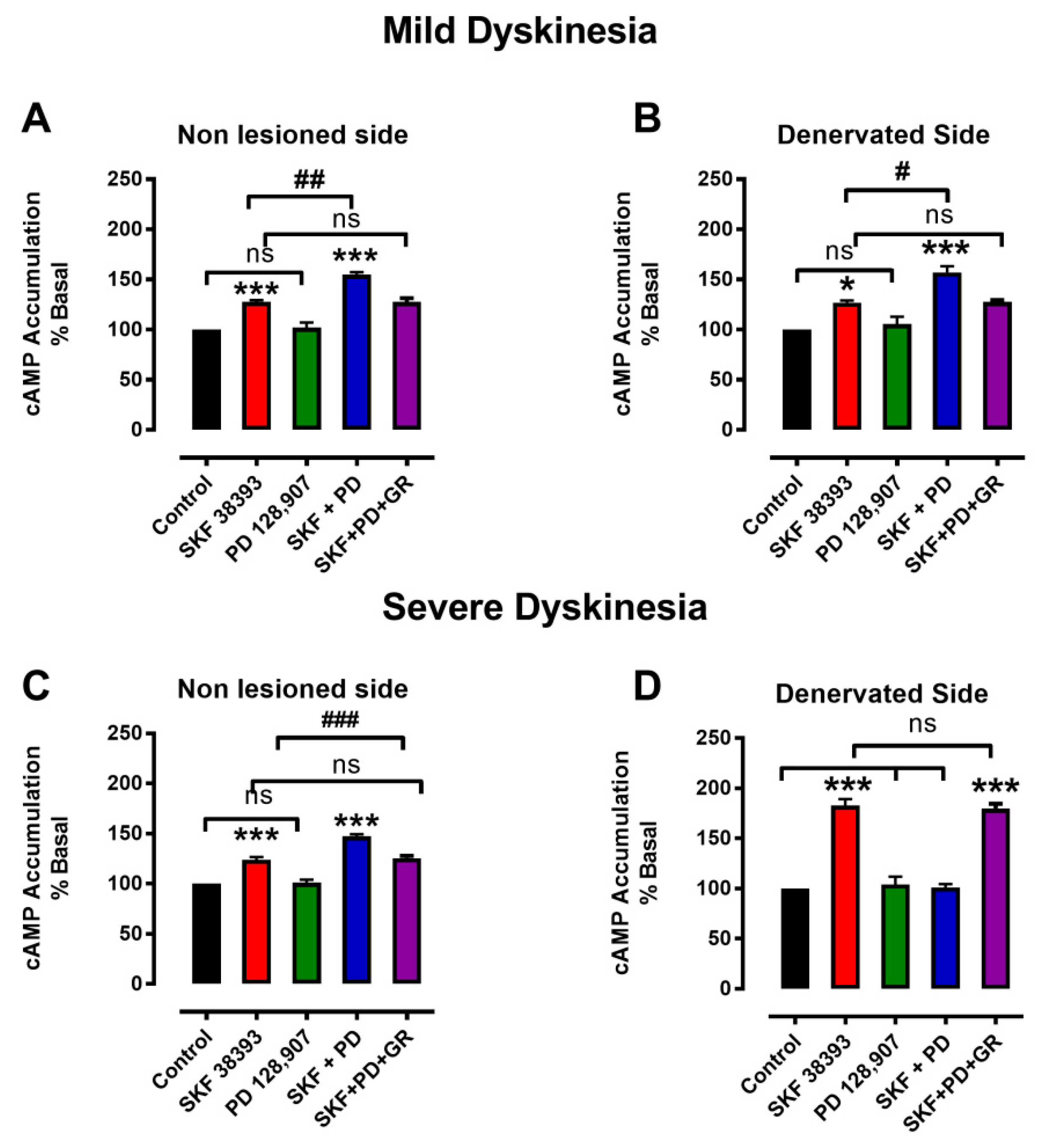
3.3. Denervation-Induced Changes in [3H]cAMP Accumulation by D3R Were Reverted in L-DOPA-Treated Animals with Mild Dyskinesia, but These Persist in Animals with Severe Dyskinesia
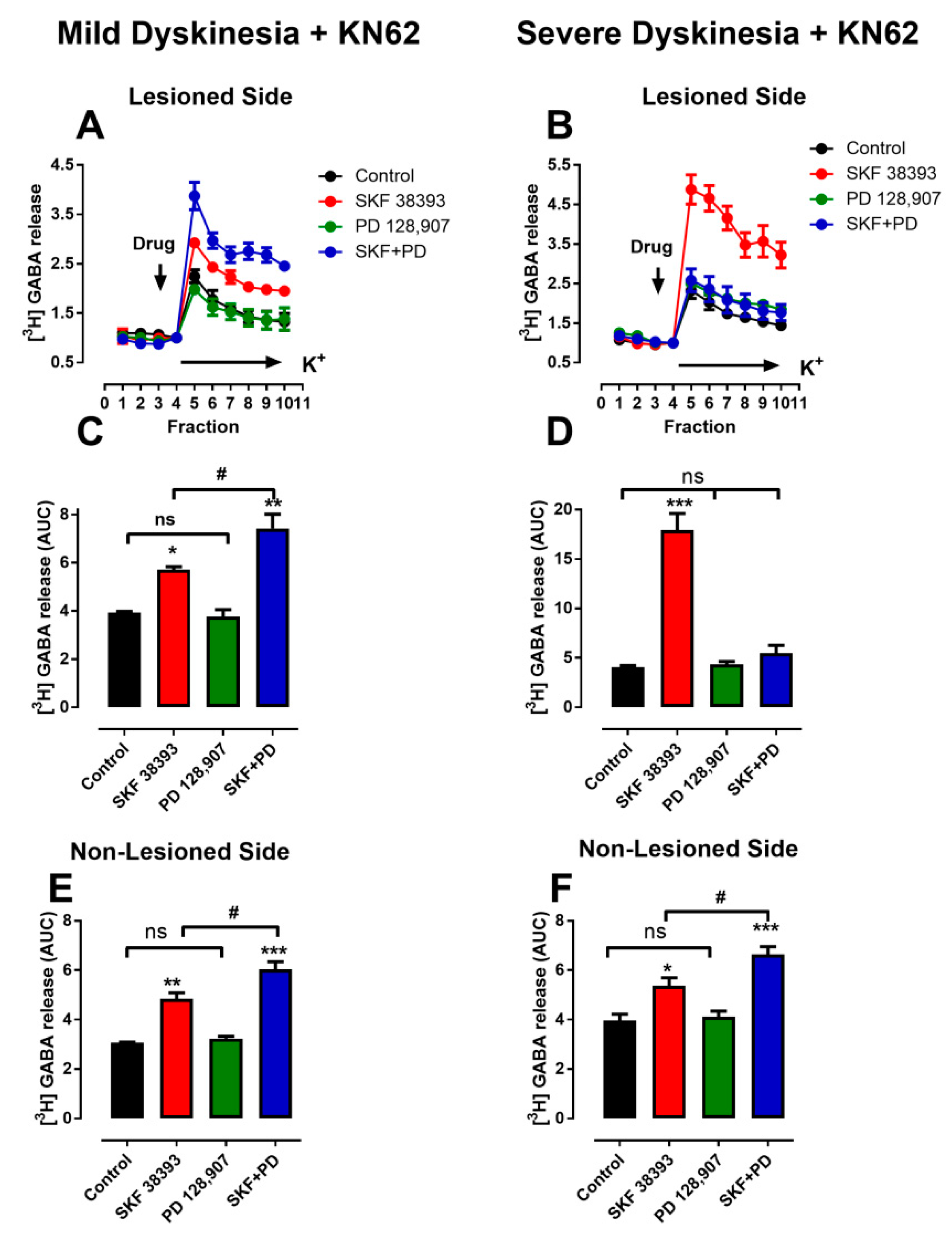
3.4. Denervation-Induced Changes in K+-Stimulated [3H]-GABA Release Were Reverted in L-DOPA- Treated Animals with Mild Dyskinesia, but These Persist in Animals with Severe Dyskinesia
4. Discussion
4.1. Separate LID Populations of Rats with the Same Degree of Lesion
4.2. LID and Therapeutic Response to L-DOPA
4.3. Changes in cAMP as Determinant of LID
4.4. D3R Changes and LIDs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Hirsch, E.C.; Farrer, M.; Schapira, A.H.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 16, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Alcacer, C.; Santini, E.; Valjent, E.; Gaven, F.; Girault, J.A.; Herve, D. Galpha(olf) mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J. Neurosci. 2012, 32, 5900–5910. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.F.; Dovero, S.; Charron, G.; Porras, G.; Gross, C.E.; Fernagut, P.O.; Bezard, E. Immediate-early gene expression in structures outside the basal ganglia is associated to l-DOPA-induced dyskinesia. Neurobiol. Dis. 2014, 62, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Carta, M.; Lindgren, H.S.; Lundblad, M.; Stancampiano, R.; Fadda, F.; Cenci, M.A. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J. Neurochem. 2006, 96, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Lee, C.S.; Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 1998, 10, 2694–2706. [Google Scholar] [CrossRef]
- Fiorentini, C.; Rizzetti, M.C.; Busi, C.; Bontempi, S.; Collo, G.; Spano, P.; Missale, C. Loss of synaptic D1 dopamine/N-methyl-d-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol. Pharmacol. 2006, 69, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Barbon, A.; Missale, C. Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Neurobiol. Dis. 2013, 54, 339–348. [Google Scholar] [CrossRef]
- Gardoni, F.; Picconi, B.; Ghiglieri, V.; Polli, F.; Bagetta, V.; Bernardi, G.; Cattabeni, F.; Di Luca, M.; Calabresi, P. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J. Neurosci. 2006, 26, 2914–2922. [Google Scholar] [CrossRef]
- Munoz, A.; Li, Q.; Gardoni, F.; Marcello, E.; Qin, C.; Carlsson, T.; Kirik, D.; Di Luca, M.; Bjorklund, A.; Bezard, E.; et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of L-DOPA-induced dyskinesia. Brain 2008, 131, 3380–3394. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Barajas, C.; Silva, I.; Lopez-Santiago, L.M.; Aceves, J.; Erlij, D.; Floran, B. L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol. Dis. 2011, 41, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Sancesario, G.; Morrone, L.A.; D’Angelo, V.; Castelli, V.; Ferrazzoli, D.; Sica, F.; Martorana, A.; Sorge, R.; Cavaliere, F.; Bernardi, G.; et al. Levodopa-induced dyskinesias are associated with transient down-regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: Reduced synthesis or increased catabolism? Neurochem. Int. 2014, 79, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Bono, F.; Busi, C.; Barbon, A.; Missale, C. Shp-2 knockdown prevents l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov. Disord. 2016, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.C.; Bachmann, C.G.; Linazasoro, G. Classifying risk factors for dyskinesia in Parkinson’s disease. Parkinsonism Relat. Disord. 2010, 16, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Putterman, D.B.; Munhall, A.C.; Kozell, L.B.; Belknap, J.K.; Johnson, S.W. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2007, 323, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Di Monte, D.A.; McCormack, A.; Petzinger, G.; Janson, A.M.; Quik, M.; Langston, W.J. Relationship among nigrostriatal denervation, parkinsonism, and dyskinesias in the MPTP primate model. Mov. Disord. 2000, 15, 459–466. [Google Scholar] [CrossRef]
- Schneider, J.S.; Gonczi, H.; Decamp, E. Development of levodopa-induced dyskinesias in parkinsonian monkeys may depend upon rate of symptom onset and/or duration of symptoms. Brain Res. 2003, 990, 38–44. [Google Scholar] [CrossRef]
- Smith, L.A.; Jackson, M.J.; Hansard, M.J.; Maratos, E.; Jenner, P. Effect of pulsatile administration of levodopa on dyskinesia induction in drug-naive MPTP-treated common marmosets: Effect of dose, frequency of administration, and brain exposure. Mov. Disord. 2003, 18, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.C.; Bonifati, V.; van Duijn, C.M. Parkinson’s disease: Piecing together a genetic jigsaw. Brain 2003, 126, 1722–1733. [Google Scholar] [CrossRef]
- Cai, G.; Wang, H.Y.; Friedman, E. Increased dopamine receptor signaling and dopamine receptor-G protein coupling in denervated striatum. J. Pharmacol. Exp. Ther. 2002, 302, 1105–1112. [Google Scholar] [CrossRef]
- Bordet, R.; Ridray, S.; Carboni, S.; Diaz, J.; Sokoloff, P.; Schwartz, J.C. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc. Natl. Acad. Sci. USA 1997, 94, 3363–3367. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Griffon, N.; Bezard, E.; Leriche, L.; Diaz, J.; Gross, C.; Sokoloff, P. Brain-derived neurotrophic factor controls dopamine D3 receptor expression: Therapeutic implications in Parkinson’s disease. Eur. J. Pharmacol. 2003, 480, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Cote, S.R.; Kuzhikandathil, E.V. Chronic levodopa treatment alters expression and function of dopamine D3 receptor in the MPTP/p mouse model of Parkinson’s disease. Neurosci. Lett. 2015, 585, 33–37. [Google Scholar] [CrossRef]
- Bezard, E.; Ferry, S.; Mach, U.; Stark, H.; Leriche, L.; Boraud, T.; Gross, C.; Sokoloff, P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat. Med. 2003, 9, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Azkona, G.; Sagarduy, A.; Aristieta, A.; Vazquez, N.; Zubillaga, V.; Ruiz-Ortega, J.A.; Perez-Navarro, E.; Ugedo, L.; Sanchez-Pernaute, R. Buspirone anti-dyskinetic effect is correlated with temporal normalization of dysregulated striatal DRD1 signalling in L-DOPA-treated rats. Neuropharmacology 2014, 79, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Cote, S.R.; Chitravanshi, V.C.; Bleickardt, C.; Sapru, H.N.; Kuzhikandathil, E.V. Overexpression of the dopamine D3 receptor in the rat dorsal striatum induces dyskinetic behaviors. Behav. Brain Res. 2014, 263, 46–50. [Google Scholar] [CrossRef]
- Marcellino, D.; Ferre, S.; Casado, V.; Cortes, A.; Le Foll, B.; Mazzola, C.; Drago, F.; Saur, O.; Stark, H.; Soriano, A.; et al. Identification of dopamine D1-D3 receptor heteromers. Indications for a role of synergistic D1-D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025. [Google Scholar] [CrossRef]
- Fiorentini, C.; Busi, C.; Gorruso, E.; Gotti, C.; Spano, P.; Missale, C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharmacol. 2008, 74, 59–69. [Google Scholar] [CrossRef]
- Cruz-Trujillo, R.; Avalos-Fuentes, A.; Rangel-Barajas, C.; Paz-Bermudez, F.; Sierra, A.; Escartin-Perez, E.; Aceves, J.; Erlij, D.; Floran, B. D3 dopamine receptors interact with dopamine D1 but not D4 receptors in the GABAergic terminals of the SNr of the rat. Neuropharmacology 2013, 67, 370–378. [Google Scholar] [CrossRef]
- Albarran, S.; Paz-Bermudez, F.; Erlij, D.; Aceves, J.; Florán, B. Dopamine D3 receptor prevents stimulation of [3H] GABA release in substantia nigra pars reticulata of hemiparkinsonian dyskinetic rats. Soc. Neurosc. Abstr. 2013. 240.06/M7. Available online: https://scholar.google.com/scholar?cluster=7837714118961510460&hl=es&as_sdt=2005&sciodt=0,5 (accessed on 1 September 2019).
- Avalos-Fuentes, A.; Albarran-Bravo, S.; Loya-Lopez, S.; Cortes, H.; Recillas-Morales, S.; Magana, J.J.; Paz-Bermudez, F.; Rangel-Barajas, C.; Aceves, J.; Erlij, D.; et al. Dopaminergic denervation switches dopamine D3 receptor signaling and disrupts its Ca(2+) dependent modulation by CaMKII and calmodulin in striatonigral projections of the rat. Neurobiol. Dis. 2015, 74, 336–346. [Google Scholar] [CrossRef]
- Hudson, J.L.; van Horne, C.G.; Stromberg, I.; Brock, S.; Clayton, J.; Masserano, J.; Hoffer, B.J.; Gerhardt, G.A. Correlation of apomorphine- and amphetamine-induced turning with nigrostriatal dopamine content in unilateral 6-hydroxydopamine lesioned rats. Brain Res. 1993, 626, 167–174. [Google Scholar] [CrossRef]
- Nash, J.E.; Johnston, T.H.; Collingridge, G.L.; Garner, C.C.; Brotchie, J.M. Subcellular redistribution of the synapse-associated proteins PSD-95 and SAP97 in animal models of Parkinson’s disease and L-DOPA-induced dyskinesia. FASEB J. 2005, 19, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Mela, F.; Marti, M.; Dekundy, A.; Danysz, W.; Morari, M.; Cenci, M.A. Antagonism of metabotropic glutamate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J. Neurochem. 2007, 101, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Hagell, P.; Widner, H. Clinical rating of dyskinesias in Parkinson’s disease: Use and reliability of a new rating scale. Mov. Disord. 1999, 14, 448–455. [Google Scholar] [CrossRef]
- Lundblad, M.; Andersson, M.; Winkler, C.; Kirik, D.; Wierup, N.; Cenci, M.A. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2002, 15, 120–132. [Google Scholar] [CrossRef]
- Schallert, T.; Fleming, S.M.; Leasure, J.L.; Tillerson, J.L.; Bland, S.T. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 2000, 39, 777–787. [Google Scholar] [CrossRef]
- Alexander, S.P. The measurement of cyclic AMP levels in biological preparations. Methods Mol. Biol. 1995, 41, 79–89. [Google Scholar]
- Nava-Asbell, C.; Paz-Bermudez, F.; Erlij, D.; Aceves, J.; Floran, B. GABA(B) receptor activation inhibits dopamine D1 receptor-mediated facilitation of [(3)H]GABA release in substantia nigra pars reticulata. Neuropharmacology 2007, 53, 631–637. [Google Scholar] [CrossRef]
- Ungerstedt, U.; Arbuthnott, G.W. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970, 24, 485–493. [Google Scholar] [CrossRef]
- Avalos-Fuentes, A.; Loya-Lopez, S.; Flores-Perez, A.; Recillas-Morales, S.; Cortes, H.; Paz-Bermudez, F.; Aceves, J.; Erlij, D.; Floran, B. Presynaptic CaMKIIalpha modulates dopamine D3 receptor activation in striatonigral terminals of the rat brain in a Ca(2)(+) dependent manner. Neuropharmacology 2013, 71, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.; Melamed, E.; Sahakian, B.J.; Wurtman, R.J. Circling behavior in rats with partial, unilateral nigro-striatal lesions: Effect of amphetamine, apomorphine, and DOPA. Pharmacol. Biochem. Behav. 1980, 12, 185–188. [Google Scholar] [CrossRef]
- Boix, J.; Padel, T.; Paul, G. A partial lesion model of Parkinson’s disease in mice—Characterization of a 6-OHDA-induced medial forebrain bundle lesion. Behav. Brain Res. 2015, 284, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Iancu, R.; Mohapel, P.; Brundin, P.; Paul, G. Behavioral characterization of a unilateral 6-OHDA-lesion model of Parkinson’s disease in mice. Behav. Brain Res. 2005, 162, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Levivier, M.; Jiang, H.; Ferreira, M.; Jackson-Lewis, V.; Donaldson, D.; Togasaki, D.M. Dose-dependent lesions of the dopaminergic nigrostriatal pathway induced by intrastriatal injection of 6-hydroxydopamine. Neuroscience 1995, 67, 631–647. [Google Scholar] [CrossRef]
- Ahlskog, J.E.; Muenter, M.D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 2001, 16, 448–458. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.S.; Williams, D.R.; Gallagher, D.A.; Massey, L.A.; Silveira-Moriyama, L.; Lees, A.J. Nonmotor symptoms as presenting complaints in Parkinson’s disease: A clinicopathological study. Mov. Disord. 2008, 23, 101–106. [Google Scholar] [CrossRef]
- Nadjar, A.; Gerfen, C.R.; Bezard, E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: A feature inherent to the treatment or the disease? Prog. Neurobiol. 2009, 87, 1–9. [Google Scholar] [CrossRef]
- dos Santos, E.U.D.; Duarte, E.B.C.; Miranda, L.M.R.; Asano, A.G.C.; Asano, N.M.J.; Maia, M.M.D.; de Souza, P.R.E. Influence of DRD1 and DRD3 Polymorphisms in the Occurrence of Motor Effects in Patients with Sporadic Parkinson’s Disease. NeuroMolecular Med. 2019, 21, 295–302. [Google Scholar] [CrossRef]
- Martín-Flores, N.; Fernández-Santiago, R.; Antonelli, F.; Cerquera, C.; Moreno, V.; Martí, M.J.; Ezquerra, M.; Malagelada, C. MTOR Pathway-Based Discovery of Genetic Susceptibility to L-DOPA-Induced Dyskinesia in Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2092–2100. [Google Scholar] [CrossRef]
- Cerasa, A.; Salsone, M.; Morelli, M.; Pugliese, P.; Arabia, G.; Gioia, C.M.; Novellino, F.; Quattrone, A. Age at onset influences neurodegenerative processes underlying PD with levodopa-induced dyskinesias. Parkinsonism Relat. Disord. 2013, 19, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Luquin, M.R.; Scipioni, O.; Vaamonde, J.; Gershanik, O.; Obeso, J.A. Levodopa-induced dyskinesias in Parkinson’s disease: Clinical and pharmacological classification. Mov. Disord. 1992, 7, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Lang, A.E. Levodopa-related motor complications--phenomenology. Mov. Disord. 2008, 23 (Suppl. 3), S509–S514. [Google Scholar] [CrossRef] [PubMed]
- Goubault, E.; Nguyen, H.P.; Bogard, S.; Blanchet, P.J.; Bezard, E.; Vincent, C.; Langlois, M.; Duval, C. Cardinal Motor Features of Parkinson’s Disease Coexist with Peak-Dose Choreic-Type Drug-Induced Dyskinesia. J. Parkinsons Dis. 2018, 8, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.M.; Engber, T.M.; Kask, A.M.; Chase, T.N. Motor fluctuations in levodopa treated parkinsonian rats: Relation to lesion extent and treatment duration. Brain Res. 1994, 662, 69–74. [Google Scholar] [CrossRef]
- Giorgi, M.; D’Angelo, V.; Esposito, Z.; Nuccetelli, V.; Sorge, R.; Martorana, A.; Stefani, A.; Bernardi, G.; Sancesario, G. Lowered cAMP and cGMP signalling in the brain during levodopa-induced dyskinesias in hemiparkinsonian rats: New aspects in the pathogenetic mechanisms. Eur. J. Neurosci. 2008, 28, 941–950. [Google Scholar] [CrossRef]
- Picconi, B.; Centonze, D.; Hakansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Feyder, M.; Bonito-Oliva, A.; Fisone, G. L-DOPA-Induced Dyskinesia and Abnormal Signaling in Striatal Medium Spiny Neurons: Focus on Dopamine D1 Receptor-Mediated Transmission. Front. Behav. Neurosci. 2011, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisone, G.; Bezard, E. Molecular mechanisms of l-DOPA-induced dyskinesia. Int. Rev. Neurobiol. 2011, 98, 95–122. [Google Scholar]
- Guitart, X.; Navarro, G.; Moreno, E.; Yano, H.; Cai, N.S.; Sanchez-Soto, M.; Kumar-Barodia, S.; Naidu, Y.T.; Mallol, J.; Cortes, A.; et al. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: The dopamine D1-D3 receptor heterotetramer. Mol. Pharmacol. 2014, 86, 417–429. [Google Scholar] [CrossRef]
- Solis, O.; Moratalla, R. Dopamine receptors: Homomeric and heteromeric complexes in L-DOPA-induced dyskinesia. J. Neural Transm. 2018, 125, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Griffon, N.; Pilon, C.; Sautel, F.; Schwartz, J.C.; Sokoloff, P. Two intracellular signaling pathways for the dopamine D3 receptor: Opposite and synergistic interactions with cyclic AMP. J. Neurochem. 1997, 68, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Diaz, J.; Bordet, R.; Griffon, N.; Perachon, S.; Pilon, C.; Ridray, S.; Sokoloff, P. Functional implications of multiple dopamine receptor subtypes: The D1/D3 receptor coexistence. Brain Res. Rev. 1998, 26, 236–242. [Google Scholar] [CrossRef]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- DeLong, M.R. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990, 13, 281–285. [Google Scholar] [CrossRef]
- Rangel-Barajas, C.; Silva, I.; Garcia-Ramirez, M.; Sanchez-Lemus, E.; Floran, L.; Aceves, J.; Erlij, D.; Floran, B. 6-OHDA-induced hemiparkinsonism and chronic L-DOPA treatment increase dopamine D1-stimulated [(3)H]-GABA release and [(3)H]-cAMP production in substantia nigra pars reticulata of the rat. Neuropharmacology 2008, 55, 704–711. [Google Scholar] [CrossRef]
- Mela, F.; Marti, M.; Bido, S.; Cenci, M.A.; Morari, M. In vivo evidence for a differential contribution of striatal and nigral D1 and D2 receptors to L-DOPA induced dyskinesia and the accompanying surge of nigral amino acid levels. Neurobiol. Dis. 2012, 45, 573–582. [Google Scholar] [CrossRef]
- Deniau, J.M.; Mailly, P.; Maurice, N.; Charpier, S. The pars reticulata of the substantia nigra: A window to basal ganglia output. Prog. Brain Res. 2007, 160, 151–172. [Google Scholar]
- Windels, F.; Kiyatkin, E.A. GABA, not glutamate, controls the activity of substantia nigra reticulata neurons in awake, unrestrained rats. J. Neurosci. 2004, 24, 6751–6754. [Google Scholar] [CrossRef]
- Kumar, R.; Riddle, L.R.; Griffin, S.A.; Chu, W.; Vangveravong, S.; Neisewander, J.; Mach, R.H.; Luedtke, R.R. Evaluation of D2 and D3 dopamine receptor selective compounds on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 2009, 56, 956–969. [Google Scholar] [CrossRef]
- Riddle, L.R.; Kumar, R.; Griffin, S.A.; Grundt, P.; Newman, A.H.; Luedtke, R.R. Evaluation of the D3 dopamine receptor selective agonist/partial agonist PG01042 on L-dopa dependent animal involuntary movements in rats. Neuropharmacology 2011, 60, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Van Gerpen, J.A.; Bower, J.H.; Ahlskog, J.E. Levodopa-dyskinesia incidence by age of Parkinson’s disease onset. Mov. Disord. 2005, 20, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Riddle, L.; Griffin, S.A.; Grundt, P.; Newman, A.H.; Luedtke, R.R. Evaluation of the D3 dopamine receptor selective antagonist PG01037 on L-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 2009, 56, 944–955. [Google Scholar] [CrossRef]
- Visanji, N.P.; Fox, S.H.; Johnston, T.; Reyes, G.; Millan, M.J.; Brotchie, J.M. Dopamine D3 receptor stimulation underlies the development of L-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol. Dis. 2009, 35, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Solis, O.; Garcia-Montes, J.R.; Gonzalez-Granillo, A.; Xu, M.; Moratalla, R. Dopamine D3 Receptor Modulates l-DOPA-Induced Dyskinesia by Targeting D1 Receptor-Mediated Striatal Signaling. Cereb. Cortex. 2015, 27, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Mela, F.; Millan, M.J.; Brocco, M.; Morari, M. The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect L-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology 2010, 58, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.; Liang, J.J.; Sierra, A.; Martinez-Fong, D.; Quirion, R.; Aceves, J.; Srivastava, L.K. Expression of dopamine receptors in the subthalamic nucleus of the rat: Characterization using reverse transcriptase-polymerase chain reaction and autoradiography. Neuroscience 1999, 91, 549–556. [Google Scholar] [CrossRef]
- Briones-Lizardi, L.J.; Cortes, H.; Avalos-Fuentes, J.A.; Paz-Bermudez, F.J.; Aceves, J.; Erlij, D.; Floran, B. Presynaptic control of [(3)H]-glutamate release by dopamine receptor subtypes in the rat substantia nigra. Central role of D1 and D3 receptors. Neuroscience 2019, 406, 563–579. [Google Scholar] [CrossRef]
- Ibanez-Sandoval, O.; Hernandez, A.; Floran, B.; Galarraga, E.; Tapia, D.; Valdiosera, R.; Erlij, D.; Aceves, J.; Bargas, J. Control of the subthalamic innervation of substantia nigra pars reticulata by D1 and D2 dopamine receptors. J. Neurophysiol. 2006, 95, 1800–1811. [Google Scholar] [CrossRef]
- Shen, K.Z.; Johnson, S.W. Regulation of polysynaptic subthalamonigral transmission by D2, D3 and D4 dopamine receptors in rat brain slices. J. Physiol. 2012, 590, 2273–2284. [Google Scholar] [CrossRef]
- Hubert, G.W.; Paquet, M.; Smith, Y. Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey Substantia nigra. J. Neurosci. 2001, 21, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Romano, C.; Sesma, M.A.; McDonald, C.T.; O’Malley, K.; Van den Pol, A.N.; Olney, J.W. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J. Comp. Neurol. 1995, 355, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Tallaksen-Greene, S.J.; Kaatz, K.W.; Romano, C.; Albin, R.L. Localization of mGluR1a-like immunoreactivity and mGluR5-like immunoreactivity in identified populations of striatal neurons. Brain Res. 1998, 780, 210–217. [Google Scholar] [CrossRef]
- van den Pol, A.N.; Romano, C.; Ghosh, P. Metabotropic glutamate receptor mGluR5 subcellular distribution and developmental expression in hypothalamus. J. Comp. Neurol. 1995, 362, 134–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Johnson, K.M. Regulation of striatal cyclic-3’,5’-adenosine monophosphate accumulation and GABA release by glutamate metabotropic and dopamine D1 receptors. J. Pharmacol. Exp. Ther. 1995, 275, 877–884. [Google Scholar] [PubMed]
- Garcia-Montes, J.R.; Solis, O.; Enriquez-Traba, J.; Ruiz-DeDiego, I.; Drucker-Colin, R.; Moratalla, R. Genetic Knockdown of mGluR5 in Striatal D1R-Containing Neurons Attenuates L-DOPA-Induced Dyskinesia in Aphakia Mice. Mol. Neurobiol. 2018, 56, 4037–7050. [Google Scholar] [CrossRef]
- Lanza, K.; Meadows, S.M.; Chambers, N.E.; Nuss, E.; Deak, M.M.; Ferre, S.; Bishop, C. Behavioral and cellular dopamine D1 and D3 receptor-mediated synergy: Implications for L-DOPA-induced dyskinesia. Neuropharmacology 2018, 138, 304–314. [Google Scholar] [CrossRef]
- Farre, D.; Munoz, A.; Moreno, E.; Reyes-Resina, I.; Canet-Pons, J.; Dopeso-Reyes, I.G.; Rico, A.J.; Lluis, C.; Mallol, J.; Navarro, G.; et al. Stronger Dopamine D1 Receptor-Mediated Neurotransmission in Dyskinesia. Mol. Neurobiol. 2015, 52, 1408–1420. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albarrán-Bravo, S.; Ávalos-Fuentes, J.A.; Cortés, H.; Rodriguez-Sánchez, M.; Leyva-García, N.; Rangel-Barajas, C.; Erlij, D.; Florán, B. Severity of Dyskinesia and D3R Signaling Changes Induced by L-DOPA Treatment of Hemiparkinsonian Rats Are Features Inherent to the Treated Subjects. Biomolecules 2019, 9, 431. https://doi.org/10.3390/biom9090431
Albarrán-Bravo S, Ávalos-Fuentes JA, Cortés H, Rodriguez-Sánchez M, Leyva-García N, Rangel-Barajas C, Erlij D, Florán B. Severity of Dyskinesia and D3R Signaling Changes Induced by L-DOPA Treatment of Hemiparkinsonian Rats Are Features Inherent to the Treated Subjects. Biomolecules. 2019; 9(9):431. https://doi.org/10.3390/biom9090431
Chicago/Turabian StyleAlbarrán-Bravo, Sacnité, José Arturo Ávalos-Fuentes, Hernán Cortés, Marina Rodriguez-Sánchez, Norberto Leyva-García, Claudia Rangel-Barajas, David Erlij, and Benjamín Florán. 2019. "Severity of Dyskinesia and D3R Signaling Changes Induced by L-DOPA Treatment of Hemiparkinsonian Rats Are Features Inherent to the Treated Subjects" Biomolecules 9, no. 9: 431. https://doi.org/10.3390/biom9090431