For Better or Worse: The Potential for Dose Limiting the On-Target Toxicity of PI 3-Kinase Inhibitors


1
Department of Molecular Medicine and Pathology, Faculty of Medical and Health Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
2
Maurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
*
Author to whom correspondence should be addressed.
Biomolecules 2019, 9(9), 402; https://doi.org/10.3390/biom9090402
Submission received: 25 July 2019
/
Revised: 15 August 2019
/
Accepted: 21 August 2019
/
Published: 22 August 2019
(This article belongs to the Special Issue Phosphoinositide 3-kinase, a Field in Transition)
Abstract
:The hyper-activation of the phosphoinositide (PI) 3-kinase signaling pathway is a hallmark of many cancers and overgrowth syndromes, and as a result, there has been intense interest in the development of drugs that target the various isoforms of PI 3-kinase. Given the key role PI 3-kinases play in many normal cell functions, there is significant potential for the disruption of essential cellular functions by PI 3-kinase inhibitors in normal tissues; so-called on-target drug toxicity. It is, therefore, no surprise that progress within the clinical development of PI 3-kinase inhibitors as single-agent anti-cancer therapies has been slowed by the difficulty of identifying a therapeutic window. The aim of this review is to place the cellular, tissue and whole-body effects of PI 3-kinase inhibition in the context of understanding the potential for dose limiting on-target toxicities and to introduce possible strategies to overcome these.
1. Introduction
1.1. PI 3-Kinases are Essential to Life
Phosphoinositide 3-kinases (PI3Ks) play a critical role in pathways regulating cellular functions such as metabolism, growth and survival, cytoskeletal rearrangements and cell migration and are, therefore, essential to life [1]. They are also clearly implicated in cancer, immune dysfunction and overgrowth syndromes and, as such, PI3K inhibitors have been the focus of anti-cancer therapeutic developments [2]. The aim of this review is to discuss the essential nature of PI3K actions in cellular and whole-body function and place the effects of PI3K inhibition in context, highlighting the dose-limiting impacts of these therapeutics. Given their use in cancer and other pathologies, we also suggest how we can use our understanding of PI3K function in health and disease to tailor the use of PI3K inhibitors and utilize combination therapies.
1.2. The PI 3-Kinase Classes and their Signaling
There are three classes of PI3Ks grouped according to their primary lipid substrate specificity and structure (see Table 1). The Class I PI3Ks principally phosphorylate phosphatidylinositol 4,5-bisphosphate PI(4,5)P2 (aka PIP2) on the 3’OH of the inositol ring to produce PI(3,4,5)P3 (PIP3). The Class Ia PI3Ks are heterodimers consisting of a catalytic and regulatory subunit. The genes, PIK3CA, PIK3CB and PIK3CD code for three highly homologous 110 kDa catalytic subunits (p110α, β, or δ respectively; see Table 1). These are always found coupled to a regulatory adaptor subunit that has no catalytic activity. The genes PIK3R1, PIK3R2 and PIK3R3 code for the regulatory p85α, p85β, or p55γ proteins (see Figure 1). Two smaller forms of p85α derived from alternative promoter usage have also been identified and are termed p55α and p50α [3]. The Class Ib PI3K is also a dimer composed of a catalytic subunit, p110γ, coupled to a regulatory subunit (p101 or p84/p87PIKAP) [4,5,6]. Class II and III PI3K enzymes and a further group of kinases including mTOR and DNA-PK (sometimes referred to as Class IV PI3K) are related structurally to Class I PI3Ks [7,8,9], but are not the focus of this review. However, it should be borne in mind that structural similarities in the kinase domains of all three classes mean there is a strong potential for cross-reactivity with drugs designed to target Class I PI3Ks.
The Class I PI3Ks are acutely activated downstream of a range of growth factor receptors, cytokine receptors and by G-protein coupled receptors (GPCRs). All three Class Ia p110 catalytic subunits contain an N-terminal adaptor-binding domain (ABD; that binds to the inter-SH2 domain on p85), a Ras-binding domain (RBD), a C2 domain (putatively involved in membrane-binding), a helical domain with unknown function, and a catalytic kinase domain (see Figure 1). Growth factor receptors activate Class Ia PI3Ks via the regulatory adapter subunits, all of which contain two SH2 domains that bind directly to phosphotyrosine residues on the activated receptors and/or receptor substrates [10] (see Figure 1). This interaction localizes the p110 catalytic subunit to the plasma membrane and relieves the regulatory inhibition of p85, allowing p110 to phosphorylate PIP2 to PIP3 [11]. The Class Ib catalytic subunit p110γ together with p101, its main regulatory adapter, is mainly activated by GPCRs through interaction with Gβγ heterodimers [12]. GPCRs can also influence the activity of p110β via direct interactions with Gβγ [4]. Furthermore p110α, δ and γ can also be regulated by Ras whereas p110β interacts with the Rho subfamily of GTPases Rac and Ccd42 [13].
Once produced, PIP3 initiates a range of intracellular signaling events, largely by binding to PH domains contained in certain signaling proteins. The activation of Akt is a particularly important consequence of increases in PIP3 levels. Crosstalk also exists between the Ras/Raf/MEK/Erk pathway and PI3K (see Figure 2). The levels of cellular PIP3 are tightly controlled and PI3K-dependent signaling is terminated by the dephosphorylation of PIP3, carried out by the tumor suppressor phosphatase and tensin homologue (PTEN) and SH2-domain containing inositol-5-phosphatase (SHIP), generating PI(4,5)P2 and PI(3,4)P2, respectively [14]. In this way, many signaling systems converge to effect essential cellular processes through the PI3K pathway (see Figure 2).
2. Tissue Distribution and Biological Roles of Class I PI3-Kinases
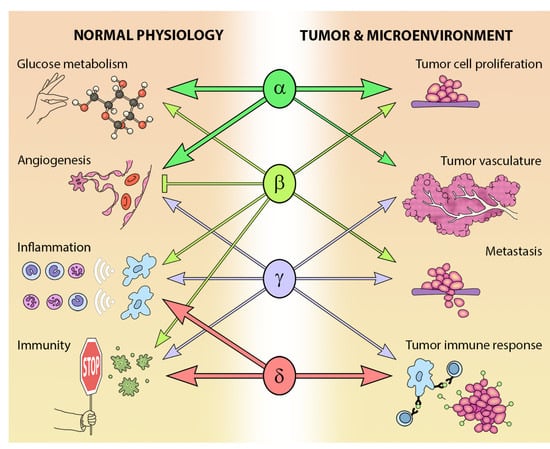
The different Class I PI3K isoforms vary in their tissue distribution and impact on normal physiology (see Figure 3). PI3Kα and β are widely distributed in mammalian tissues, whereas PI3Kδ and γ are mainly, but not exclusively, expressed in blood cells and their precursors [15] (see Table 1). Due to the ubiquitous expression and central role that PI3Kα and PI3Kβ play in physiology, it is not surprising that PIK3CA−/− mice are embryonic lethal [16] and PIK3CB−/− mice partially embryonic lethal [17,18]. Kinase dead homozygous p110αD933A knock-in mutants are also embryonic lethal by E12.5, as a result of dysfunctional vascular development [19]. Crossing PIK3CA+/− and PIK3CB+/− mice results in the expected Mendelian ratio of offspring, indicating that the expression of one allele of each isoform is sufficient for normal development [20].
While PI3Kδ is mainly found in leukocytes [15], this isoform has been reported in neurons [21] and cancer cells of non-leukocyte origin, such as breast cancer cells and melanoma [15]. PI3Kδ kinase-dead knock-in mice are viable and their phenotype is restricted to defects in immune signaling and response [22,23,24].
In addition to a wide distribution in hematopoietic tissues, PIK3CG is also expressed in a limited number of other tissues including the heart and endothelium [25,26], as well as tumors including cancer of the pancreas and breast [27,28,29]. As might be expected from its expression profile, germ-line deletion of PIK3CG results in defects in innate immune and inflammatory responses [26,30,31,32,33]. These defects are relatively well tolerated and PIK3CG−/− mice exhibit normal viability, fertility and longevity [30,31,32].
3. PI3-Kinase in Cancer
The PI3K pathway is commonly dysregulated in cancer and different isoforms have varied involvement in tumors and the tumor microenvironment (see Figure 3). PIK3CA is reported to be one of the most commonly mutated oncogenes in human cancer [34]. Increased copy number and/or overexpression in the Class I PI3Ks also contributes to hyper-activation of the PI3K pathway in many cancers [35,36,37,38,39] (see Figure 4). The hotspot mutations E545K and H1047R in the helical and kinase domains of p110α (PIK3CA exons 9 and 20) were first described by Samuels et al. in 2004 [40,41] and these remain a focus of oncogenic research. More recently, a number of next-generation sequencing programs have identified a range of rarer mutations in the other Class I isoforms (data available at www.cbioportal.org) [42,43], with the most frequent being the hotspot mutation D1067A/V/Y occurring at the C-terminus of p110β [44], while another C-terminal mutation in p110β (E1051G/K) has also been described in several cancers. Multiple mutations have also been recently described in PIK3CD (N334K, E525K, R821C/H, E1021K) [45,46] and in the accessory domains of PIK3CG (R544*/P/Q) and PIK3CB (R604G/P). While numerous mutations causing amino acid changes have been found in p110β, δ and γ, these are far less common than mutations in p110α [39,41,42] and the impact of these mutations on enzyme activity is poorly understood.
The importance of the activating mutations in PIK3CA is further highlighted by the identification of gain-of-function PIK3CA mutations in a range of human tissue overgrowth syndromes [47,48,49,50]. Common activating mutations include those in the catalytic and helical domains of p110α that have been previously associated with oncogenic transformation (H1047L, H1047R, E542K, E543K and C420R), as well as a novel syndrome-specific mutation p110α R115P [49]. The PI3Kα E545K mutation has also been repeatedly implicated in overgrowth of the brain [51] along with other mutations in the PI3K pathway (AKT3 c.49C>T p. E17K and MTOR c.4448C>T p.C1483Y).
Mutations also occur in the genes for the regulatory subunits PIK3R1, PIK3R2, PIK3R3 and PIK3R5 (see Figure 4) [52,53,54]. These result in elevation of the lipid kinase activity and oncogenic transformation—primarily through activation of p110α [55,56]. This is consistent with the theory that these mutations in p85 weaken the inhibitory interaction between p85α and p110α while preserving the stabilizing/activating interaction between p85α, SH2 and adapter-binding domain of p110α [57]. Inherited mutations have also been identified in p85 that reduce its ability to transduce signals from growth factor receptors to p110, causing reductions in insulin signaling and an insulin-resistant phenotype [58].
The observation that the PI3K pathway is activated in many different cancers through mutation and/or overexpression (see Figure 4) has driven the development of a range of PI3K pathway inhibitors as potential cancer therapies [59] (see Figure 2). However, the clinical efficacy of single agents targeting PI3K, AKT and mTOR has been limited [60]; indeed, phase 1b clinical trials BEZ 235 were halted due to “significant toxicity with no objective responses” [61].
The availability of PI3K crystal structures [62,63,64,65] has resulted in the supersession of early, pan-specific PI3K inhibitors by potent compounds targeting specific Class I isoforms in an effort to limit side effect profiles. There are signs this approach is more successful, because while one pan-PI3K inhibitor (copanlisib/Aliqopa®) has been approved for relapsed follicular lymphoma [66], several isoform-specific inhibitors are now on the market; the PI3Kδ inhibitor idelalisib (Zydelig®) has been approved to target chronic lymphocytic leukemia (CLL) [67,68], the dual PI3Kδ/γ inhibitor duvelisib (Copiktra®) for relapsed or refractory CLL or small lymphatic lymphoma [69] and the PI3Kα inhibitor alpelisib (Piqray®) for HR+/HER2− advanced breast cancers with PIK3CA mutations [70].
While non-specific off-target actions of drugs always have the potential to negatively affect the therapeutic window, the pleiotropic importance of PI3K in cellular processes means that on-target effects of PI3K inhibitors also significantly limit their therapeutic use (see Table 2). This review will present the cellular, tissue and whole-body effects of PI 3-kinase inhibition in the context of understanding the potential for dose limiting on-target toxicities and introduce possible strategies to overcome these.
4. Targets of PI3-Kinase Inhibitors
4.1. Effects of PI3K Attenuation on Whole Body Glucose Metabolism
In general, the inhibition of PI3K has been observed to counteract the effects of nutritional excess and results in normoglycemia, reduced fatty liver, and reduced adiposity [71]. The actions of insulin on glucose metabolism are mediated by PI3K [14] so it is not surprising that in clinical trials, hyperglycemia was a common effect of many early pan PI3K inhibitors [72,73,74,75,76,77], as well as dual-specificity PI3K/mTOR inhibitors [78] and PI3Kα-specific inhibitors [79,80]. Mild hyperglycemia has also been observed after administration of the PI3Kβ inhibitor GSK2636771 [81]. Indeed, the metabolic impact of inhibitors targeting the PI3K/Akt/mTOR (PAM) pathway has led to management guidance being provided by the PAM Task Force convened by the National Cancer Institute Investigational Drug Steering Committee [82].
The metabolic effects of short- and long-term PI3K attenuation have been studied preclinically in mice either by the genetic knock-down of PI3K activity [83,84], or pharmacologic inhibition [85,86]. Interestingly, both groups observed that the short-term effect of PI3Kα knock-down results in greater metabolic disturbance than chronic PI3K reduction (summarized in Table 3) [83,84,85,86]. This strongly suggests that some degree of feedback compensation is occurring over time, a finding that has been supported by a recent study that actively targeted this feedback loop using dietary and pharmacological strategies to improve the efficacy of PI3K inhibition in the treatment of tumor-bearing mice [87]. It is of note that the pharmacologic inhibition of PI3Kβ, δ and γ had much less of an effect on glucose metabolism, indicating that metabolic disturbance will be of most relevance with drugs targeting PI3Kα [85,86]; however, the genetic knock-out of PI3Kγ has been shown to ameliorate the development of diet-induced insulin resistance and Type 2 diabetes [88]. The many sites at which PI3K inhibition could impact on glucose homeostasis are discussed below.
4.1.1. The Role of PI3K in Insulin Secretion
The regulated secretion of insulin from β-cells is a key process for regulating glucose homeostasis; however, there are contradictory findings regarding the role of PI3K in insulin secretion. Acute studies (30–90 minutes of inhibitor exposure) using β-cell lines or isolated islets indicate that PI3K inhibition increases insulin secretion [89,90,91,92,93], with the majority of this effect being due to PI3Kα-mediated effects [94]. These in vitro studies align with the increased insulin secretion and islet hyperplasia observed in kinase-dead p110βK805R/K805R mice [17], however, PI3Kβ inhibition by shRNA in cultured β-cells has been shown to result in a decrease in insulin secretion [94]. It is further proposed that PI3Kγ signaling via GPCRs controls constitutive insulin secretion by coordinating intracellular processes of trafficking and secretion [95,96] and mediating GIP-induced insulin secretion [97]. This picture is further complicated by feedback systems in responsive metabolic systems; hyperglycemia resulting from inhibition of PI3K is corrected through increased insulin secretion which, in turn, activates PI3K/mTOR signaling, reducing the impact of the primary PI3K inhibition [87]. This feedback response would be blunted in obese patients exhibiting underlying insulin resistance and those with diabetes where the insulin secretory capacity is compromised.
4.1.2. Central Metabolic Effects on Appetite
Appetite loss is already a serious issue in the clinical management of cancer patients [98], so it is important to understand how anti-cancer drugs will affect appetite. Decreased appetite and/or dysgeusia (taste distortion) has been observed after the clinical administration of pan PI3K inhibitors [73,75,77,99], a dual PI3K/mTOR inhibitor (BEZ235) [78], as well as PI3Kα-isoform-specific inhibitors [79,80] and the PI3Kβ inhibitor GSK2636771 [81]. This negative effect on appetite would not necessarily have been anticipated, as several preclinical studies indicated that pan-PI3K inhibitors block leptin and insulin signaling in the hypothalamus, slightly increasing food uptake [100,101,102,103,104]. Furthermore, the tissue-specific knock-out of PI3Kβ in hypothalamic pro-opiomelanocortin (POMC)-expressing neurons resulted in leptin resistance and a diet-induced increase in adiposity [105]. One acute inhibitor dosing study that does align with the clinical trial results showed that dual PI3K/mTOR inhibitor BEZ235, PI3Kα inhibitor PIK75 and DNA-PK/PI3Kα/mTOR inhibitor PI-103 all significantly decreased food intake, while inhibitors of PI3Kβ, γ and δ had no effect [86]. Reassuringly, these acute effects on food intake were not sustained with long-term dosing, again implying that some degree of compensation is occurring [85].
4.1.3. Metabolic Effects in Muscle and Adipose Tissue
Inhibition with PI3Kα-specific inhibitors (PIK-90, PI-103, or PIK-75) blocked insulin-stimulated glucose uptake in vivo [106], resulting in insulin resistance. This is supported by findings that PI3Kα is the dominant isoform required to mediate insulin and IGF-I signal transduction in muscle and adipocytes, which are key organs in the regulation of glucose disposal [84,106,107]. Increased fat mass has been demonstrated in young PI3Kα mutant mice [84]. However, this is compensated for in chronic administration and older PI3Kα mutant mice, which show a lean phenotype with reduced adiposity [83,85]. Inhibition of PI3Kα using PIK-75 also completely abolished adipocyte differentiation as assessed by morphology, transcript and protein levels of adipocyte markers [108]. Therefore, pan-inhibitors (LY294002 and wortmannin), which attenuate insulin-stimulated glucose uptake in muscle and adipocytes in a dose-dependent manner [109,110], most likely exert these actions through their inhibition of PI3Kα.
Evidence has emerged that indicates a role for PI3Kβ in skeletal muscle differentiation and myogenesis [111]. This observation correlates with the finding that PI3Kβ levels are significantly reduced in the muscle and adipose of men born with low birthweight [112,113], a group who have been identified as being susceptible to an increased risk of developing diabetes [114]. However, the inhibition of PI3Kβ has not been shown to have any effect on insulin-induced Akt phosphorylation in muscle or adipose [106] or adipocyte differentiation [108].
It has also been shown that PI3Kγ acts within adipose tissue to promote fat mass gain. PI3Kγ KO mice are leaner than their control littermates; they have increased energy expenditure and, therefore, show reduced fat gain, despite normal caloric intake. Furthermore, PIK3CGKD/KD mice fed a high-fat diet exhibit less weight gain and are apparently protected from insulin resistance and diet-induced steatosis [115].
4.1.4. Metabolic Effects in the Liver
Insulin action in the liver is critical for maintaining normoglycemia, as glucose storage (glycogenesis), breakdown (glycolysis) and production (glycogenolysis and gluconeogenesis) in the liver are all regulated by insulin [116]. A liver-specific KO of PI3Kα results in a diabetic syndrome with decreased insulin sensitivity, impaired glucose tolerance, increased gluconeogenesis and leptinemia and decreased lipidemia [117]. In support of these genetic findings, the acute treatment of mice with PI3Kα or pan-PI3K inhibitors results in the increased gluconeogenic production of glucose from pyruvate [86]. Once again, these acute effects differ from those in a chronic model, with the long-term low-dose use of pan-PI3K inhibitors resulting in reduced liver-steatosis in mice and monkeys [118].
While the role of PI3Kα in liver metabolism appears definitive, the role of PI3Kβ in the liver remains unclear. Both genetic and pharmacologic studies exist which indicate that PI3Kβ has little effect on liver metabolism [86,117]. However, other studies have shown that mouse models lacking functional PI3Kβ either globally, or specifically in the liver, have defects in glucose metabolism [17,119,120]. Furthermore, it appears many of the cellular effects of PIK3CB liver-specific KO are mediated through a lipid kinase-independent function, since the impaired insulin sensitivity and glucose homeostasis were not mediated via Akt phosphorylation [119] and could be due to the alternative signaling of PI3Kβ through G-protein coupled receptors [121].
PI3Kγ has also been implicated in liver metabolism. Both PI3Kγ kinase-dead mice [115] and PI3Kγ-KO mice [88] fed a high-fat diet demonstrate a reduction in hepatic steatosis. However, a recent study has reported large changes in liver structure and function in a pancreatic neoplasia-bearing mouse model with the partial or complete knock down of PI3Kγ [122].
Taken together, these studies all indicate that disruption of the PAM pathway results in an inability for the liver to sense satiety and ultimately impacts whole-body metabolic homeostasis.
4.2. Effects of PI3K Attenuation in the Gut
Some of the most common side effects of the clinical use of PI3K/mTOR inhibitors are colitis, diarrhea, nausea and vomiting (see Table 2). While these are common effects for many types of drugs, the mechanism for idelalisib-induced colitis is thought to be mediated at least in part through the enhanced inflammatory response occurring in response to gut pathogens [123], and there is some evidence that points to this dose-limiting toxicity being a PI3K class effect, since PI3Ks play roles in gut immunity, motility and neuro-transmission [124].
The role of PI3K signaling in immune cells of the intestinal mucosa has been extensively reviewed [125]. In the gut, PI3K signals downstream of Toll-like receptors and T-cell receptors to mediate immune homeostasis in the face of commensal and pathogenic bacteria. Evidence suggests that PI3Kγ [125] and PI3Kδ [126,127] are important isoforms in intestinal inflammation and that deregulation of the PI3K pathway can result in inflammatory bowel disease and its associated cancers.
Interstitial cells of Cajal (ICC) are required for normal gut motility and in turn, the normal development of ICC is largely dependent upon c-Kit signaling [128]. Pan-PI3K inhibitors wortmannin and LY294002 cause the loss of ICC and the suppression of slow wave in mouse jejunal muscle strips [129]; however, the deletion of c-Kit-induced PI3K signaling (via disruption of PI3K binding to Y719F on c-Kit) was found to have no effect on the function or development of ICC in mice [128]. This latter finding indicates that c-Kit and PI3K are exerting their effects in an independent manner.
With regards to neurotransmission, PI3Kα negatively regulates the secretion of the intestinal peptide neurotensin, which stimulates GI secretion, motility and the growth of the small intestine and pancreas [130]. Furthermore, decreased PI3K activity is proposed to cause apoptosis of enteric neurons in the intestine, resulting in delayed gastric emptying and more rapid intestinal transit [131].
Therefore, mechanisms involved in PI3K-mediated effects on gut motility indicate contrasting roles. On the one hand, PI3K inhibitors have been shown to slow gut motility via the loss of ICC [129], while on the other, gut motility would be expected to increase as a result of PI3K-mediated down regulation of neurotensin [130] and the associated loss of intestinal innervation [132].
4.3. Effects of PI3K Attenuation in the Brain
In addition to the central effects on appetite that have already been covered in this review, pan PI3K inhibitors have also been linked to mood alterations. Wider class inhibition also commonly results in fatigue—a symptom that could have a psychological component. Certain PI3K inhibitors are known to cross the blood–brain barrier including BKM120, XL147, XL765, GDC0084, PQR309 and BEZ235 [133,134,135]. BKM120 has a depressive effect in some humans [76,135] and long-term use of PI3Kα inhibitors (BEZ235, PIK75 and PI-103) and wortmannin in mice was found to produce signs of depression [85,136], while rats treated with wortmannin and LY294002 also display learning and memory defects [137] and a reduced fear response [138]. Taken together, these data highlight the need for the psychological monitoring of patients in PI3K drug trials, particularly if they are taking drugs that are known to cross the blood–brain barrier.
4.4. Effects of PI3K Attenuation in Airways
Pneumonitis, or inflammation of lung tissue and alveoli, has been reported as a side effect associated with the PI3K inhibitor, idelalisib (Zydelig®), across multiple clinical trials, even causing death in a small number of patients (3/760; 0.4%) in an early clinical trial [139]. While the actual mechanism of idelalisib-induced pneumonitis remains unclear, it is now recognized that there is an increased risk of infection due to the immunomodulatory effects of this PI3Kδ inhibitor—likely mediated, at least in part, through the enhanced inflammatory response occurring in response to pathogens present in the airways [123,140,141]. It is also known that the PI3K pathway plays an important role in airway smooth muscle (ASM) development, contractility and inflammation, with many studies focusing on regulation of the PI3K pathway as a way to control asthma and chronic obstructive pulmonary disease (COPD) [142]. Therefore, there are potential benefits and risks of targeting PI3K in this tissue.
ASM hyperplasia within the bronchial wall of asthmatics is thought to be due to increased muscle proliferation, which is under the control of both the ERK and PI3K pathways. A number of mitogens acting through RTKs and GPCRs, and to a lesser-known extent, cytokine receptors, activate the PI3K and ERK signaling pathways to stimulate the proliferation of ASM [143,144]. In asthmatics, there is some evidence that it is upregulation of the PI3K (rather than the ERK) pathway that results in muscle hyperplasia [145]. PI3Kα, δ and γ all interact with RAS via their RAS-binding domain (RBD), whereas the RBD of PI3Kβ does not interact with RAS, but rather with RAC1 and CDC42 from the RHO family of GTPases. Even so, PI3Kβ is still implicated in lung pathology since mice with RBD mutant PI3Kβ are resistant to experimental lung fibrosis (a pathology linked with lysophosphatidic acid signaling through GPCRs) [119,146].
In addition to stimulating ASM growth and differentiation, the PI3K pathway is involved in ASM contractility, which in turn, is implicated in airway hyper responsiveness (AHR) and as a pro-inflammatory signaling pathway in the airway [147]. The role of eosinophils in asthma pathology has been well documented since the original observations of Huber and Koessler in 1922 [148] and it is known that PI3Kγ is essential for triggering eosinophil influx [149,150]. Furthermore, PI3Kδ has a role in regulating eosinophil trafficking and recruitment during allergic airway inflammation [151] and AHR [152].
While the role of PI3K inhibition has been thoroughly explored in relation to asthma and COPD, the severe hypersensitivity pneumonitis experienced by a small number of patients receiving idelalisib rings a note of caution [139]. Furthermore, since the idelalisib-induced pneumonitis is consistent with mTOR inhibitor-induced pneumonitis [153], this biological side-effect appears to be a rare but severe class effect. An expert panel commenting on the management of cancer patients undergoing mTOR inhibitor treatment has recommended clinical trial exclusion or close monitoring of patients with pre-existing lung disease, severe pulmonary compromise or active lung infection [154] and provides a framework for the management of patients on idelalisib, which may be adopted for broader PI3K inhibitor use [155].
4.5. Effects of PI3K Attenuation on Inflammation, Immunity and the Hematopoietic System
PI3K isoforms play multiple roles in the immune system and these could potentially be exploited to directly target certain leukemias (as has been done with idelalisib and duvelisib) or to modulate the actions of the highly successful immunotherapies that have been developed recently. Conversely, there is the possibility that PI3K inhibitors could negatively impact the patient’s immune system. Readers are referred to an excellent review on the roles of PI3K signaling in inflammatory and autoimmune diseases and hematological malignancies [156]; however, for completeness, critical findings will be described here.
Given the largely localized expression of PI3Kδ and γ in leukocytes [15], it is to be expected that these isoforms are the most important in immune regulation and the hematopoietic system. As mentioned previously, knocking out PI3Kδ or γ does not affect viability, fertility or longevity in mice [22,30,31,32]. However, under conditions of immune challenge, these mice exhibit deregulation of B and T cells, NK cells, dendritic and mast cells, macrophages, basophils, eosinophils and neutrophils [156].
PI3Kδ plays a key role in agonist-induced B-cell receptor (BCR) signaling [22,23,157,158,159], but agonist-independent or ‘tonic’ BCR signaling is not affected by PI3Kδ KO [160]. This is due to redundancy in the Class I PI3K family, as it has been confirmed that in the absence of PI3Kδ or PI3Kα (but not PI3Kβ), compensation occurs to promote early B-cell development in marrow and B-cell survival in the spleen [160,161]. In the absence of both PI3Kα and PI3Kδ, pre-BCR signaling failed to promote the developmental progression of B-cell progenitors [160].
PI3Kγ transduces a central pro-inflammatory signal involved in leukocyte chemotaxis, mast cell degranulation and endothelial cell activation [115] and can suppress inflammation in a variety of mouse models of disease, including atherosclerosis [33,162], rheumatoid arthritis [163], glomerulonephritis [164], anaphylaxis [165] and multiple sclerosis [166]. Although neutrophils are enriched in PI3Kδ and γ KO models, these cells also express abundant amounts of PI3Kα and β and it is thought that all Class I PI3K isoforms may contribute to GM-CSF-mediated neutrophil survival [167]. Furthermore, the global suppression of Class I PI3K activity below a certain threshold is required to abrogate this survival effect since it is not until PI3Kα, β and δ were inhibited that effects were seen [167].
Due to their hematopoietic tissue and cancer-specific expression patterns, PI3Kδ and γ inhibitors have been used to target relapsed or refractory lymphoma including, but not limited to, CLL, mantle cell lymphoma and non-Hodgkin lymphoma [67,168,169,170,171,172,173]. Unsurprisingly, these PI3Kδ and γ isoform-specific inhibitors are also noted for hematologic toxicities including anemia, thrombocytopenia, leukocytosis, hemolysis and neutropenia [168,169,173,174]. These hematopoietic toxicities are often noted in trial outcomes as common laboratory abnormalities [173,175], although none, other than neutropenia, are serious enough to be noted as a warning in the US prescribing information for idelalisib [139,176]. Furthermore, respiratory infection was observed in 20% of patients receiving idelalisib on a trial for relapsed or refractory mantle cell lymphoma [172] and PI3Kδ inhibition with AMG 319 resulted in elevated T-reg cells (>10% of CD4+); however, the T-reg cells of most patients normalized with continued treatment, indicating immune restoration [169].
Genetic and pharmacological blockade studies show that PI3K regulates the development, activation and differentiation of B- and T-cells [177]. This can have both positive and negative effects. On the positive side, PI3K inhibition can help attenuate immune response, but on the negative side, it can enhance inflammation, disrupt peripheral tolerance and promote autoimmunity [177,178]. This enhanced inflammatory response occurs in the parts of the body most exposed to pathogens (skin, airways and gut) and can exhibit strong side effects upon PI3Kδ inhibition, resulting in therapy-limiting rashes, pneumonitis and colitis [123].
Interestingly, PI3Kδ inhibition has been found to have a positive anti-inflammatory effect in ischemic brains and it has been proposed that PI3Kδ inhibition could help treat ischemic strokes [179] via a mechanism involving tumor necrosis factor-α (TNF-α). It is also suggested as a therapy for people suffering from activated PI3K-delta syndrome (APDS), who have activating mutations in PIK3CD [180].
4.6. Effects of PI3K Attenuation in the Skin
Our skin is the largest organ in our body and provides protection from pathogens, promotes thermoregulation and prevents dehydration. Therefore, while a mild skin rash might not be serious enough to be dose limiting, severe grades of rash can impact on daily living [181]. While rashes have been reported as a side effect of other targeted therapies, chemotherapy, immunotherapy, radiation therapy and stem cell transplants [181], maculopapular rash is one of the common dose-limiting toxicities reported for pan PI3K and dual PI3K/mTOR inhibitors [74,75,76,99,182] and for the PI3Kδ inhibitor idelalisib [172], but not for other isoform-specific inhibitors. Since rashes are commonly associated with multiple different therapies, the rash linked with pan PI3K inhibitor drug use is unlikely to be a class effect. Further reassurance can be derived from the fact that isoform-specific inhibitors (other than PI3Kδ) have not been reported to result in a rash. As mentioned previously, the mechanism for idelalisib-induced rash is thought to be mediated at least in part through the enhanced inflammatory response occurring in response to skin pathogens. On a positive note, where rashes are encountered as a side effect, it has been suggested that clinicians could use this effect as a pharmacodynamic biomarker for drug titration [183] in the same way rash outbreak is used to titrate the dose of EGFR inhibitors [184].
4.7. Effects of PI3K Chronic Attenuation in Bone
Whilst no adverse clinical effects have been noted in bone, preclinical studies show that PI3K plays a role in both osteoblasts and osteoclasts and is involved in bone formation and resorption [185], with PI3Kα being the dominant isoform in skeletal bone [186]. In osteoclasts, PI3Ks are activated by cytokines and growth factors (e.g., CSF-1, RANKL and alphavB3 integrin), resulting in osteoclast survival, development and motility [185]. PI3Kα inhibitors (BEZ-235, PIK75 and PI-103) reduce bone resorption and promote differentiation and survival of osteoblasts [185,186,187].
The effect of the long-term administration of PI3K inhibitors on various parameters of bone function in mice suggests that pan-PI3K inhibitors might be detrimental to skeletal health [86]. Smith et al. showed that the pan-PI3K inhibitors (ZSTK474, PI-103, and BEZ235) decreased bone density and either decreased or tended to decrease bone strength [86]. Furthermore, two PI3Kα inhibitors (PIK75 and A66) also reduced bone density and strength, while PI3Kβ and δ inhibitors did not show any consistent effects on bone [86]. This indicates that PI3Kα is the isoform that is most important in regulating bone mass and strength, which is consistent with findings which demonstrate that PI3Kα is by far the most prevalent Class Ia PI3K isoform expressed in bone [86,186]. These findings are supported by other studies which demonstrated that the genetic activation of skeletal PI3K signaling is associated with increased bone formation [188], while those that abrogate PI3K signaling by knockdown of Akt are accompanied by decreased bone formation and bone density [189,190,191].
While no increase in bone mass was observed using the PI3Kγ inhibitor AS252424 [86], mice lacking PI3Kγ exhibit increased bone mass and density, through the modulation of osteoclastogenesis [192].
Studies to date have not fully elucidated the mechanism(s) of the skeletal effects of PI3Kα inhibitors, however, these preclinical findings suggest that the evaluation of skeletal health (bone turnover markers and bone density) be undertaken in ongoing and planned clinical trials of pan PI3K inhibitors and PI3Kα selective inhibitors.
4.8. Effects of Chronic PI3K Attenuation in the Heart
PI3Ks are widely distributed throughout the cardiovascular system, with cardiac cells expressing PI3Kα, β and γ [193]. PI3Kα is essential for cardiomyocyte viability and growth mediated via PAM signaling plays an important role in cardiac hypertrophy [25,194,195,196,197]. The knockout of PI3Kα or β in cardiac myocytes (either during development or in adults) results in changes in heart structure, leading to heart failure and death [198]. PI3Kα protects against myocardial infarction [199] and regulates the expression of genes essential to maintaining cardiac structure and Z-disc alignment and signaling [200], while PI3Kα and β are essential for maintaining the organized network of T-tubules by regulating junctophilin-2 localization, which is vital for efficient Ca2+ induced Ca2+ release and ventricular contraction [198]. Preclinical studies have also shown that pharmacological PI3Kγ inhibition impacts the heart; AS605240 suppressed Akt phosphorylation in an in vivo model of myocardial infarction—decreasing inflammation and increasing cardiomyocyte apoptosis [201].
In general, it is thought that PI3Kα is the most important isoform for maintaining cardiomyocyte size, while PI3Kγ is involved in cardiac function and contractility [202,203]. PI3Kα is positively associated with heart health [204] and the loss of PI3Kα accelerates pathological ventricular remodeling and heart failure in rodent models of chronic adrenergic stimulation, primary cardiomyopathy and pressure overload [205]. It has been proposed that the use of PI3Kα inhibitors, while likely to be safe in patients with normal cardiac function, may cause cardiac dysfunction and possibly heart failure in patients with pre-existing cardiac disease [206,207]. Conversely, PI3Kγ inhibition has been associated with improved cardiac function [208,209].
5. Conclusions and Future Directions
The activation of the PI3K pathway in cancer has led to a huge investment in developing inhibitors targeting this pathway. Despite these intensive efforts, very few PI3K inhibitors have been approved for clinical use. As discussed above, a major factor affecting the clinical utility of these drugs has been the potential for on-target toxicities that become dose-limiting due to the fact that the PI3K pathway is so important in such a wide range of physiological and metabolic responses. Some of these on-target toxicities may eventually be clinically manageable through dietary and pharmacological strategies to control the metabolic effects [87]. Patients who are already metabolically, or immunologically challenged should be excluded from treatment [82,123,139] and biomarkers could be developed to identify further groups at risk of adverse side effects. Conversely, there may be a subset of patients with diseases that are particularly sensitive to PI3K inhibitors; in the case of cancer therapy, this is a concept known as oncogene addiction [210]. There is emerging evidence that patients with H1047R mutations in p110α respond better to lower doses of PI3K inhibitors than other tumors do [211]. Furthermore, there are promising signs that on-target toxicities can indeed be ameliorated; it has recently been shown that the PI3Kα inhibitor BYL719 (alpelasib/Piqray®) can be administered in a way that minimizes side effects while delivering clinical benefit in patients suffering from overgrowth syndromes driven by somatic PIK3CA mutations [212]. Alternative dosing strategies may also be successful. In this regard, it is notable that the dosing regimen for copanlisib (Aliqopa®)—the only pan-PI3K inhibitor to receive FDA approval so far—is administered intravenously once per week, rather than by oral daily dosing (routinely used to achieve the maximum tolerated dose). However, the narrow therapeutic window for PI3K inhibitors and the fact that they largely induce cytostasis rather than cell death, has seen a current trend in clinic-to-trial progression of using PI3K inhibitors at lower tolerable doses in combination with other specific inhibitors [60,213]. Combination therapies may also offer the possibility of dosing PI3K inhibitors metronomically [214] to achieve short-term synergistic effects with other agents. Another strategy for improving the tolerability of PI3K-targeted drugs could be to make PI3K inhibitor pro-drugs that are only activated in the tumor tissue. This has been achieved chemotherapeutically by adding chemical triggers to the inhibitor which make the intact pro-drug inactive; however, as these pro-drugs break apart, they release their chemotherapeutic agent to achieve high concentrations of the active drug in the tumor relative to the periphery [215]. Pro-drug strategies that take advantage of the hypoxic environment present in many tumors are a particularly attractive approach in this regard [216]. Finally, it may be possible to take advantage of the small structural differences caused by oncogenic mutations in PI3K to develop drugs that selectively target the oncogenic forms and thus spare normal signaling via PI3K.
In conclusion, PI3K inhibition is an effective strategy for targeting cancer cells and overgrowth syndromes at multiple levels, but the reality of on-target toxicity in non-tumor tissues means much work remains to be done to develop new treatment strategies if this class of drugs is ever to be used as an effective part of a chronic treatment regime for cancer or overgrowth therapies.
Author Contributions
Conceptualization, P.R.S. and C.M.B.; writing—original draft preparation, C.M.B. and P.R.S.; writing—review and editing, C.M.B., K.L.L. and P.R.S.; funding acquisition, P.R.S.
Funding
The authors have received salary funding from the Health Research Council of New Zealand and the Maurice Wilkins Centre for Molecular Biodiscovery.
Acknowledgments
The authors would like to thank Mia Jüllig for preparing figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Flanagan, J.U.; Shepherd, P.R. Structure, function and inhibition of the phosphoinositide 3-kinase p110alpha enzyme. Biochem. Soc. Trans. 2014, 42, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Abell, K.; Bilancio, A.; Clarkson, R.W.; Tiffen, P.G.; Altaparmakov, A.I.; Burdon, T.G.; Asano, T.; Vanhaesebroeck, B.; Watson, C.J. Stat3-induced apoptosis requires a molecular switch in PI(3)K subunit composition. Nat. Cell Biol. 2005, 7, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef] [PubMed]
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr. Biol. 2005, 15, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Dorner, M.B.; Schaefer, M. Characterization of p87PIKAP, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006, 281, 9977–9986. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [Green Version]
- Munson, M.J.; Ganley, I.G. MTOR, PIK3C3, and autophagy: Signaling the beginning from the end. Autophagy 2015, 11, 2375–2376. [Google Scholar] [CrossRef]
- Gulluni, F.; De Santis, M.C.; Margaria, J.P.; Martini, M.; Hirsch, E. Class II PI3K Functions in Cell Biology and Disease. Trends Cell Biol. 2019, 29, 339–359. [Google Scholar] [CrossRef]
- Carpenter, C.L.; Auger, K.R.; Chanudhuri, M.; Yoakim, M.; Schaffhausen, B.; Shoelson, S.; Cantley, L.C. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J. Biol. Chem. 1993, 268, 9478–9483. [Google Scholar]
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85/p110 phosphatidylinositol 3’-kinase: Stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol. 1998, 18, 1379–1387. [Google Scholar] [CrossRef]
- Suire, S.; Condliffe, A.M.; Ferguson, G.J.; Ellson, C.D.; Guillou, H.; Davidson, K.; Welch, H.; Coadwell, J.; Turner, M.; Chilvers, E.R.; et al. Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat. Cell Biol. 2006, 8, 1303–1309. [Google Scholar] [CrossRef]
- Soler, A.; Angulo-Urarte, A.; Graupera, M. PI3K at the crossroads of tumor angiogenesis signaling pathways. Mol. Cell. Oncol. 2015, 2, e975624. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, P.R. Mechanisms regulating phosphoinositide 3-kinase signalling in insulin-sensitive tissues. Acta Physiol. Scand. 2005, 183, 3–12. [Google Scholar] [CrossRef]
- Sawyer, C.; Sturge, J.; Bennett, D.C.; O’Hare, M.J.; Allen, W.E.; Bain, J.; Jones, G.E.; Vanhaesebroeck, B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110delta. Cancer Res. 2003, 63, 1667–1675. [Google Scholar]
- Bi, L.; Okabe, I.; Bernard, D.J.; Wynshaw-Boris, A.; Nussbaum, R.L. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 1999, 274, 10963–10968. [Google Scholar] [CrossRef] [PubMed]
- Ciraolo, E.; Iezzi, M.; Marone, R.; Marengo, S.; Curcio, C.; Costa, C.; Azzolino, O.; Gonella, C.; Rubinetto, C.; Wu, H.; et al. Phosphoinositide 3-kinase p110beta activity: Key role in metabolism and mammary gland cancer but not development. Sci. Signal. 2008, 1, ra3. [Google Scholar] [CrossRef]
- Guillermet-Guibert, J.; Smith, L.B.; Halet, G.; Whitehead, M.A.; Pearce, W.; Rebourcet, D.; Leon, K.; Crepieux, P.; Nock, G.; Stromstedt, M.; et al. Novel Role for p110beta PI 3-Kinase in Male Fertility through Regulation of Androgen Receptor Activity in Sertoli Cells. PLoS Genet. 2015, 11, e1005304. [Google Scholar] [CrossRef]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Okabe, I.; Bernard, D.J.; Nussbaum, R.L. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm. Genome 2002, 13, 169–172. [Google Scholar] [PubMed]
- Veerasingham, S.J.; Yamazato, M.; Berecek, K.H.; Wyss, J.M.; Raizada, M.K. Increased PI3-kinase in presympathetic brain areas of the spontaneously hypertensive rat. Circ. Res. 2005, 96, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.A.; Rawlings, D.; Reynolds, H.; Vigorito, E.; et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Nashed, B.F.; Zhang, T.; Al-Alwan, M.; Srinivasan, G.; Halayko, A.J.; Okkenhaug, K.; Vanhaesebroeck, B.; Hayglass, K.T.; Marshall, A.J. Role of the phosphoinositide 3-kinase p110delta in generation of type 2 cytokine responses and allergic airway inflammation. Eur. J. Immunol. 2007, 37, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Oudit, G.Y.; Kozieradzki, I.; Sarao, R.; Sun, H.; Sasaki, T.; Hirsch, E.; Suzuki, A.; Shioi, T.; Irie-Sasaki, J.; et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 2002, 110, 737–749. [Google Scholar] [CrossRef]
- Patrucco, E.; Notte, A.; Barberis, L.; Selvetella, G.; Maffei, A.; Brancaccio, M.; Marengo, S.; Russo, G.; Azzolino, O.; Rybalkin, S.D.; et al. PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 2004, 118, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Edling, C.E.; Selvaggi, F.; Buus, R.; Maffucci, T.; Di Sebastiano, P.; Friess, H.; Innocenti, P.; Kocher, H.M.; Falasca, M. Key role of phosphoinositide 3-kinase class IB in pancreatic cancer. Clin. Cancer Res. 2010, 16, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Brazzatti, J.A.; Klingler-Hoffmann, M.; Haylock-Jacobs, S.; Harata-Lee, Y.; Niu, M.; Higgins, M.D.; Kochetkova, M.; Hoffmann, P.; McColl, S.R. Differential roles for the p101 and p84 regulatory subunits of PI3Kgamma in tumor growth and metastasis. Oncogene 2012, 31, 2350–2361. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mazzocca, A.; Lupo, L.; Edling, C.E.; Azzariti, A.; Antonaci, S.; Falasca, M.; Giannelli, G. PI3K class IB controls the cell cycle checkpoint promoting cell proliferation in hepatocellular carcinoma. Int. J. Cancer 2012, 130, 2505–2513. [Google Scholar] [CrossRef]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, H.; Xie, W.; Zhang, Z.; Smrcka, A.V.; Wu, D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 2000, 287, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.D.; Sukhova, G.K.; Libby, P.; Schvartz, E.; Lichtenstein, A.H.; Field, S.J.; Kennedy, C.; Madhavarapu, S.; Luo, J.; Wu, D.; et al. Deletion of the phosphoinositide 3-kinase p110gamma gene attenuates murine atherosclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8077–8082. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 2010, 347, 21–41. [Google Scholar] [PubMed]
- Vogt, P.K.; Hart, J.R.; Gymnopoulos, M.; Jiang, H.; Kang, S.; Bader, A.G.; Zhao, L.; Denley, A. Phosphatidylinositol 3-kinase: The oncoprotein. Curr. Top. Microbiol. Immunol. 2010, 347, 79–104. [Google Scholar] [PubMed]
- Vogt, P.K.; Gymnopoulos, M.; Hart, J.R. PI 3-kinase and cancer: Changing accents. Curr. Opin. Genet. Dev. 2009, 19, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.A.; St Clair, F.; Munday, A.D.; Thomas, R.J.; Mitchell, C.A. Increased levels of phosphatidylinositol 3-kinase activity in colorectal tumors. Cancer 1998, 83, 41–47. [Google Scholar] [CrossRef]
- Kang, S.; Denley, A.; Vanhaesebroeck, B.; Vogt, P.K. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2006, 103, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Velculescu, V.E. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004, 3, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.L.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kdelta and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Heurtier, L.; Lamrini, H.; Chentout, L.; Deau, M.C.; Bouafia, A.; Rosain, J.; Plaza, J.M.; Parisot, M.; Dumont, B.; Turpin, D.; et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause Activated PI3K-delta Syndrome 1 (APDS1). Haematologica 2017, 102, e278–e281. [Google Scholar] [CrossRef] [PubMed]
- Lindhurst, M.J.; Parker, V.E.; Payne, F.; Sapp, J.C.; Rudge, S.; Harris, J.; Witkowski, A.M.; Zhang, Q.; Groeneveld, M.P.; Scott, C.E.; et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat. Genet. 2012, 44, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Kurek, K.C.; Luks, V.L.; Ayturk, U.M.; Alomari, A.I.; Fishman, S.J.; Spencer, S.A.; Mulliken, J.B.; Bowen, M.E.; Yamamoto, G.L.; Kozakewich, H.P.; et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am. J. Hum. Genet. 2012, 90, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.J.; Paria, N.; Burns, D.K.; Israel, B.A.; Cornelia, R.; Wise, C.A.; Ezaki, M. Somatic gain-of-function mutations in PIK3CA in patients with macrodactyly. Hum. Mol. Genet. 2013, 22, 444–451. [Google Scholar] [CrossRef]
- Maclellan, R.A.; Luks, V.L.; Vivero, M.P.; Mulliken, J.B.; Zurakowski, D.; Padwa, B.L.; Warman, M.L.; Greene, A.K.; Kurek, K.C. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast. Reconstr. Surg. 2014, 133, 12e–19e. [Google Scholar] [CrossRef]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urick, M.E.; Rudd, M.L.; Godwin, A.K.; Sgroi, D.; Merino, M.; Bell, D.W. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011, 71, 4061–4067. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Janakiraman, V.; Kljavin, N.M.; Chaudhuri, S.; Stern, H.M.; Wang, W.; Kan, Z.; Dbouk, H.A.; Peters, B.A.; Waring, P.; et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell 2009, 16, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.; Hennessy, B.T.; Li, J.; Yu, S.; Myers, A.P.; Djordjevic, B.; Lu, Y.; Stemke-Hale, K.; Dyer, M.D.; Zhang, F.; et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011, 1, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Chaussade, C.; Cho, K.; Mawson, C.; Rewcastle, G.W.; Shepherd, P.R. Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem. Biophys. Res. Commun. 2009, 381, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Kang, S.; Vogt, P.K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar] [CrossRef] [PubMed]
- Baynes, K.C.; Beeton, C.A.; Panayotou, G.; Stein, R.; Soos, M.; Hansen, T.; Simpson, H.; O’Rahilly, S.; Shepherd, P.R.; Whitehead, J.P. Natural variants of human p85 alpha phosphoinositide 3-kinase in severe insulin resistance: A novel variant with impaired insulin-stimulated lipid kinase activity. Diabetologia 2000, 43, 321–331. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hu, J.; Zhong, H.A. Advances in the Development of Class I Phosphoinositide 3-Kinase (PI3K) Inhibitors. Curr. Top. Med. Chem. 2016, 16, 1413–1426. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Molina, A.M.; Lakhman, Y.; Patil, S.; Woo, K.; DeLuca, J.; Lee, C.H.; Hsieh, J.J.; Feldman, D.R.; Motzer, R.J.; et al. A Phase Ib Study of BEZ235, a Dual Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in Patients With Advanced Renal Cell Carcinoma. Oncologist 2016, 21, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Williams, R.; Berndt, A.; Miller, S.; Hon, W.C.; Zhang, X. Form and flexibility in phosphoinositide 3-kinases. Biochem. Soc. Trans. 2009, 37, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, A.; Miller, S.; Williams, O.; Le, D.D.; Houseman, B.T.; Pacold, J.I.; Gorrec, F.; Hon, W.C.; Liu, Y.; Rommel, C.; et al. The p110delta structure: Mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- Hon, W.C.; Berndt, A.; Williams, R.L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31, 3655–3666. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. FDA Approves New Treatment for Adults with Relapsed Follicular Lymphoma. 2018. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-relapsed-follicular-lymphoma (accessed on 14 August 2019).
- Brown, J.R.; Byrd, J.C.; Coutre, S.E.; Benson, D.M.; Flinn, I.W.; Wagner-Johnston, N.D.; Spurgeon, S.E.; Kahl, B.S.; Bello, C.; Webb, H.K.; et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood 2014, 123, 3390–3397. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.W.; Przepiorka, D.; de Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Duvelisib (Copiktra, Verastem, Inc.) for Adult Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL). 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/duvelisib-copiktra-verastem-inc-adult-patients-relapsed-or-refractory-chronic-lymphocytic-leukemia (accessed on 14 August 2019).
- U.S. Food & Drug Administration. FDA Approves Alpelisib for Metastatic Breast Cancer. 2019. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-alpelisib-metastatic-breast-cancer (accessed on 14 August 2019).
- Ortega-Molina, A.; Efeyan, A.; Lopez-Guadamillas, E.; Munoz-Martin, M.; Gomez-Lopez, G.; Canamero, M.; Mulero, F.; Pastor, J.; Martinez, S.; Romanos, E.; et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012, 15, 382–394. [Google Scholar] [CrossRef]
- Kim, R.D.; Alberts, S.R.; Renshaw, F.G.; Genvresse, I.; Reif, S.; Kaplan, J.; Grilley-Olson, J.E. Phase 1 dose escalation study of copanlisib (BAY 80-6946) in combination with gemcitabine or gemcitabine-cisplatin in advanced cancer patients. J. Clin. Oncol. 2014, 32, 2610. [Google Scholar] [CrossRef]
- Garcia, V.M.; Baird, R.D.; Shah, K.J.; Basu, B.; Tunariu, N.; Blanco, M.; Cassier, P.A.; Pedersen, J.V.; Puglisi, M.; Sarker, D.; et al. A phase I study evaluating GDC-0941, an oral phosphoinositide-3 kinase (PI3K) inhibitor, in patients with advanced solid tumors or multiple myeloma. J. Clin. Oncol. 2011, 29, 3021. [Google Scholar] [CrossRef]
- Shapiro, G.; Kwak, E.; Baselga, J.; Rodon, J.; Scheffold, C.; Laird, A.D.; Bedell, C.; Edelman, G. Phase I dose-escalation study of XL147, a PI3K inhibitor administered orally to patients with solid tumors. J. Clin. Oncol. 2009, 27. Suppl. Abstract 3500. [Google Scholar]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Inada-Inoue, M.; Mitsuma, A.; Yoshino, T.; Ohtsu, A.; Suenaga, N.; Sato, M.; Kakizume, T.; Robson, M.; Quadt, C.; et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014, 105, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Wagner, A.J.; LoRusso, P.M.; Tibes, R.; Jin, J.; Ware, J.A.; Yan, Y.; Derynck, M.K.; Dolezal, M.V.; Demetri, G.D. A first-in-human Phase I study to evaluate the pan-PI3K inhibitor GDC-0941 administered QD or BID in patients with advanced solid tumours. EJC Suppl. 2009, 7, 122. [Google Scholar] [CrossRef]
- Wunderle, L.; Badura, S.; Lang, F.; Wolf, A.; Schleyer, E.; Serve, H.; Goekbuget, N.; Pfeifer, H.; Bug, G.; Ottmann, O.G. Safety and Efficacy Of BEZ235, a Dual PI3-Kinase /mTOR Inhibitor, In Adult Patients With Relapsed Or Refractory Acute Leukemia: Results Of a Phase I Study. Blood 2013, 122, 2675. [Google Scholar]
- Gonzalez-Angulo, A.M.; Juric, D.; Argiles, G.; Schellens, J.H.M.; Burris, H.A.; Berlin, J.; Middleton, M.R.; Schuler, M.H.; Van Geel, R.; Helgason, T.; et al. Safety, pharmacokinetics, and preliminary activity of the alpha-specific PI3K inhibitor BYL719: Results from the first-in-human study. J. Clin. Oncol. 2013, 31, 2531. [Google Scholar]
- Juric, D.; Gonzalez-Angulo, A.M.; Burris, H.A.; Schuler, M.; Schellens, J.; Berlin, J.; Gupta, A.; Seggewiss-Bernhardt, R.; Adamo, B.; Gil-Martin, M.; et al. Preliminary safety, pharmacokinetics and anti-tumor activity of BYL719, an alpha-specific P13K inhibitor in combination with fulvestrant: Results from a phase I study. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Arkenau, H.T.; Mateo, J.; Lemech, C.R.; Infante, J.R.; Burris, H.A.; Bang, Y.J.; Eder, J.P.; Herbst, R.S.; Sharma, S.; Chung, H.C.; et al. A phase I/II, first-in-human dose-escalation study of GSK2636771 in patients (pts) with PTEN-deficient advanced tumors. J. Clin. Oncol. 2014, 32, 2514. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J. Clin. Oncol. 2012, 30, 2919–2928. [Google Scholar] [CrossRef]
- Foukas, L.C.; Bilanges, B.; Bettedi, L.; Pearce, W.; Ali, K.; Sancho, S.; Withers, D.J.; Vanhaesebroeck, B. Long-term p110alpha PI3K inactivation exerts a beneficial effect on metabolism. EMBO Mol. Med. 2013, 5, 563–571. [Google Scholar] [CrossRef]
- Foukas, L.C.; Claret, M.; Pearce, W.; Okkenhaug, K.; Meek, S.; Peskett, E.; Sancho, S.; Smith, A.J.; Withers, D.J.; Vanhaesebroeck, B. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 2006, 441, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Ong, W.K.; Costa, J.L.; Watson, M.; Cornish, J.; Grey, A.; Gamble, G.D.; Dickinson, M.; Leung, S.; Rewcastle, G.W.; et al. Extended treatment with selective phosphatidylinositol 3-kinase and mTOR inhibitors has effects on metabolism, growth, behaviour and bone strength. FEBS J. 2013, 280, 5337–5349. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.C.; Ong, W.K.; Rewcastle, G.W.; Kendall, J.D.; Han, W.; Shepherd, P.R. Effects of acutely inhibiting PI3K isoforms and mTOR on regulation of glucose metabolism in vivo. Biochem. J. 2012, 442, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Ueki, K.; Okazaki, Y.; Iwane, A.; Kubota, N.; Ohsugi, M.; Awazawa, M.; Kobayashi, M.; Sasako, T.; Kaneko, K.; et al. Blockade of class IB phosphoinositide-3 kinase ameliorates obesity-induced inflammation and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 5753–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, K.; Yamashita, T.; Tsubamoto, Y.; Terauchi, Y.; Hirose, K.; Kubota, N.; Yamashita, S.; Taka, J.; Satoh, S.; Sekihara, H.; et al. Phosphatidylinositol 3-kinase suppresses glucose-stimulated insulin secretion by affecting post-cytosolic [Ca(2+)] elevation signals. Diabetes 2002, 51, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Zawalich, W.S.; Zawalich, K.C. A link between insulin resistance and hyperinsulinemia: Inhibitors of phosphatidylinositol 3-kinase augment glucose-induced insulin secretion from islets of lean, but not obese, rats. Endocrinology 2000, 141, 3287–3295. [Google Scholar] [CrossRef]
- Hagiwara, S.; Sakurai, T.; Tashiro, F.; Hashimoto, Y.; Matsuda, Y.; Nonomura, Y.; Miyazaki, J. An inhibitory role for phosphatidylinositol 3-kinase in insulin secretion from pancreatic B cell line MIN6. Biochem. Biophys. Res. Commun. 1995, 214, 51–59. [Google Scholar] [CrossRef]
- Collier, J.J.; White, S.M.; Dick, G.M.; Scott, D.K. Phosphatidylinositol 3-kinase inhibitors reveal a unique mechanism of enhancing insulin secretion in 832/13 rat insulinoma cells. Biochem. Biophys. Res. Commun. 2004, 324, 1018–1023. [Google Scholar] [CrossRef]
- Nunoi, K.; Yasuda, K.; Tanaka, H.; Kubota, A.; Okamoto, Y.; Adachi, T.; Shihara, N.; Uno, M.; Xu, L.M.; Kagimoto, S.; et al. Wortmannin, a PI3-kinase inhibitor: Promoting effect on insulin secretion from pancreatic beta cells through a cAMP-dependent pathway. Biochem. Biophys. Res. Commun. 2000, 270, 798–805. [Google Scholar] [CrossRef]
- Kolic, J.; Spigelman, A.F.; Plummer, G.; Leung, E.; Hajmrle, C.; Kin, T.; Shapiro, A.M.; Manning Fox, J.E.; MacDonald, P.E. Distinct and opposing roles for the phosphatidylinositol 3-OH kinase catalytic subunits p110alpha and p110beta in the regulation of insulin secretion from rodent and human beta cells. Diabetologia 2013, 56, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Pigeau, G.M.; Kolic, J.; Ball, B.J.; Hoppa, M.B.; Wang, Y.W.; Ruckle, T.; Woo, M.; Manning Fox, J.E.; MacDonald, P.E. Insulin granule recruitment and exocytosis is dependent on p110gamma in insulinoma and human beta-cells. Diabetes 2009, 58, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E. Control of secretory granule access to the plasma membrane by PI3 kinase-gamma. Islets 2009, 1, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Kolic, J.; Spigelman, A.F.; Smith, A.M.; Manning Fox, J.E.; MacDonald, P.E. Insulin secretion induced by glucose-dependent insulinotropic polypeptide requires phosphatidylinositol 3-kinase gamma in rodent and human beta-cells. J. Biol. Chem. 2014, 289, 32109–32120. [Google Scholar] [CrossRef] [PubMed]
- Inui, A. Cancer anorexia-cachexia syndrome: Are neuropeptides the key? Cancer Res. 1999, 59, 4493–4501. [Google Scholar]
- Shapiro, G.I.; Rodon, J.; Bedell, C.; Kwak, E.L.; Baselga, J.; Brana, I.; Pandya, S.S.; Scheffold, C.; Laird, A.D.; Nguyen, L.T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G., Jr.; Seeley, R.J.; Schwartz, M.W. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef]
- Niswender, K.D.; Morton, G.J.; Stearns, W.H.; Rhodes, C.J.; Myers, M.G., Jr.; Schwartz, M.W. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef]
- Xu, A.W.; Kaelin, C.B.; Takeda, K.; Akira, S.; Schwartz, M.W.; Barsh, G.S. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J. Clin. Investig. 2005, 115, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Wang, D.Q.; Tso, P.; Jandacek, R.J.; Woods, S.C.; Liu, M. Apolipoprotein E reduces food intake via PI3K/Akt signaling pathway in the hypothalamus. Physiol. Behav. 2011, 105, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.W.; Williams, K.W.; Ye, C.; Luo, J.; Balthasar, N.; Coppari, R.; Cowley, M.A.; Cantley, L.C.; Lowell, B.B.; Elmquist, J.K. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J. Clin. Investig. 2008, 118, 1796–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Qassab, H.; Smith, M.A.; Irvine, E.E.; Guillermet-Guibert, J.; Claret, M.; Choudhury, A.I.; Selman, C.; Piipari, K.; Clements, M.; Lingard, S.; et al. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 2009, 10, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Matheny, R.W., Jr.; Adamo, M.L. PI3K p110 alpha and p110 beta have differential effects on Akt activation and protection against oxidative stress-induced apoptosis in myoblasts. Cell Death Differ. 2010, 17, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Shepherd, P.R.; Chaussade, C. Investigating the role of class-IA PI 3-kinase isoforms in adipocyte differentiation. Biochem. Biophys. Res. Commun. 2009, 379, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, P.R.; Nave, B.T.; Siddle, K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3-L1 adipocytes: Evidence for the involvement of phosphoinositide 3-kinase and p70 ribosomal protein-S6 kinase. Biochem. J. 1995, 305, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.D.; Hansen, P.A.; Holloszy, J.O. Wortmannin inhibits insulin-stimulated but not contraction-stimulated glucose transport activity in skeletal muscle. FEBS Lett. 1995, 361, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Matheny, R.W., Jr.; Riddle-Kottke, M.A.; Leandry, L.A.; Lynch, C.M.; Abdalla, M.N.; Geddis, A.V.; Piper, D.R.; Zhao, J.J. Role of phosphoinositide 3-OH kinase p110beta in skeletal myogenesis. Mol. Cell. Biol. 2015, 35, 1182–1196. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Jensen, C.B.; Tingey, K.J.; Martin-Gronert, M.S.; Grunnet, L.; Brons, C.; Storgaard, H.; Vaag, A.A. Decreased protein levels of key insulin signalling molecules in adipose tissue from young men with a low birthweight: Potential link to increased risk of diabetes? Diabetologia 2006, 49, 2993–2999. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Jensen, C.B.; Tingey, K.J.; Storgaard, H.; Madsbad, S.; Vaag, A.A. Low birthweight is associated with specific changes in muscle insulin-signalling protein expression. Diabetologia 2005, 48, 547–552. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becattini, B.; Marone, R.; Zani, F.; Arsenijevic, D.; Seydoux, J.; Montani, J.P.; Dulloo, A.G.; Thorens, B.; Preitner, F.; Wymann, M.P.; et al. PI3Kgamma within a nonhematopoietic cell type negatively regulates diet-induced thermogenesis and promotes obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Sopasakis, V.R.; Liu, P.; Suzuki, R.; Kondo, T.; Winnay, J.; Tran, T.T.; Asano, T.; Smyth, G.; Sajan, M.P.; Farese, R.V.; et al. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Lopez-Guadamillas, E.; Mattison, J.A.; Mitchell, S.J.; Munoz-Martin, M.; Iglesias, G.; Gutierrez, V.M.; Vaughan, K.L.; Szarowicz, M.D.; Gonzalez-Garcia, I.; et al. Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell Metab. 2015, 21, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Selinger, E.S.; Ballou, L.M.; Lin, R.Z. Ablation of PI3K p110-alpha prevents high-fat diet-induced liver steatosis. Diabetes 2011, 60, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.; Okkenhaug, K.; Vanhaesebroeck, B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Mancinelli, G.; Cordoba-Chacon, J.; Viswakarma, N.; Castellanos, K.; Grimaldo, S.; Kumar, S.; Principe, D.; Dorman, M.J.; McKinney, R.; et al. p110gamma deficiency protects against pancreatic carcinogenesis yet predisposes to diet-induced hepatotoxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 14724–14733. [Google Scholar] [CrossRef]
- Reinwald, M.; Silva, J.T.; Mueller, N.J.; Fortun, J.; Garzoni, C.; de Fijter, J.W.; Fernandez-Ruiz, M.; Grossi, P.; Aguado, J.M. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: An infectious diseases perspective (Intracellular signaling pathways: Tyrosine kinase and mTOR inhibitors). Clin. Microbiol. Infect. 2018, 24, S53–S70. [Google Scholar] [CrossRef]
- Shi, X.Z.; Sarna, S.K. G protein-mediated dysfunction of excitation-contraction coupling in ileal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G899–G905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, C.M.; Rogers, J.T.; Walker, W.A. The role of phosphoinositide 3-kinase signaling in intestinal inflammation. J. Signal Transduct. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, E.C.; Kobayashi, T.; Russo, S.M.; Sheikh, S.Z.; Gipson, G.R.; Kennedy, S.T.; Uno, J.K.; Mishima, Y.; Borst, L.B.; Liu, B.; et al. Innate PI3K p110delta regulates Th1/Th17 development and microbiota-dependent colitis. J. Immunol. 2014, 192, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Uno, J.K.; Rao, K.N.; Matsuoka, K.; Sheikh, S.Z.; Kobayashi, T.; Li, F.; Steinbach, E.C.; Sepulveda, A.R.; Vanhaesebroeck, B.; Sartor, R.B.; et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology 2010, 139, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, S.J.; Rich, A.; Distad, M.A.; Miller, S.M.; Schmalz, P.F.; Szurszewski, J.H.; Sha, L.; Blume-Jensen, P.; Farrugia, G. Kit/stem cell factor receptor-induced phosphatidylinositol 3’-kinase signalling is not required for normal development and function of interstitial cells of Cajal in mouse gastrointestinal tract. Neurogastroenterol. Motil. 2003, 15, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.M.; Brennan, M.F.; Jackson, V.M.; Sanders, K.M. Role of PI3-kinase in the development of interstitial cells and pacemaking in murine gastrointestinal smooth muscle. J. Physiol. 1999, 516, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, J.; Cassidy, M.G.; Rychahou, P.; Starr, M.E.; Liu, J.; Li, X.; Epperly, G.; Weiss, H.L.; Townsend, C.M., Jr.; et al. PI3K p110alpha/Akt signaling negatively regulates secretion of the intestinal peptide neurotensin through interference of granule transport. Mol. Endocrinol. 2012, 26, 1380–1393. [Google Scholar] [CrossRef] [PubMed]
- Rayner, C.K.; Horowitz, M. Gastrointestinal motility and glycemic control in diabetes: The chicken and the egg revisited? J. Clin. Investig. 2006, 116, 299–302. [Google Scholar] [CrossRef]
- Anitha, M.; Gondha, C.; Sutliff, R.; Parsadanian, A.; Mwangi, S.; Sitaraman, S.V.; Srinivasan, S. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J. Clin. Investig. 2006, 116, 344–356. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Gil del Alcazar, C.R.; Hardebeck, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA double-strand break repair by the dual PI3K/mTOR inhibitor NVP-BEZ235 as a strategy for radiosensitization of glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, S.S.; Lin, K.; Roming, S.L.; Vellipuram, R.; Harney, J.P. Effects of PI3K inhibition and low docosahexaenoic acid on cognition and behavior. Physiol. Behav. 2010, 100, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xing, T.; Wang, M.; Miao, Y.; Tang, M.; Chen, J.; Li, G.; Ruan, D.Y. Local infusion of ghrelin enhanced hippocampal synaptic plasticity and spatial memory through activation of phosphoinositide 3-kinase in the dentate gyrus of adult rats. Eur. J. Neurosci. 2011, 33, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Kritman, M.; Maroun, M. Inhibition of the PI3 kinase cascade in corticolimbic circuit: Temporal and differential effects on contextual fear and extinction. Int. J. Neuropsychopharmacol. 2013, 16, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Coutre, S.E.; Barrientos, J.C.; Brown, J.R.; de Vos, S.; Furman, R.R.; Keating, M.J.; Li, D.; O’Brien, S.M.; Pagel, J.M.; Poleski, M.H.; et al. Management of adverse events associated with idelalisib treatment: Expert panel opinion. Leuk. Lymphoma 2015, 56, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Gilead Sciences International Pty Ltd. Product Information v4.0. ZYDELIG (100 mg and 150 mg Idelalisib) Tablets. 2017. Available online: https://www.tga.gov.au/sites/default/files/auspar-idelalisib-171019-pi.pdf (accessed on 25 July 2019).
- Maschmeyer, G.; De Greef, J.; Mellinghoff, S.C.; Nosari, A.; Thiebaut-Bertrand, A.; Bergeron, A.; Franquet, T.; Blijlevens, N.M.A.; Maertens, J.A.; European Conference on Infections in Leukemia (ECIL). Infections associated with immunotherapeutic and molecular targeted agents in hematology and oncology. A position paper by the European Conference on Infections in Leukemia (ECIL). Leukemia 2019, 33, 844–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Kinases as Novel Therapeutic Targets in Asthma and Chronic Obstructive Pulmonary Disease. Pharmacol. Rev. 2016, 68, 788–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, S.J.; Martin, J.G.; Bonacci, J.V.; Chan, V.; Fixman, E.D.; Hamid, Q.A.; Herszberg, B.; Lavoie, J.P.; McVicker, C.G.; Moir, L.M.; et al. Proliferative aspects of airway smooth muscle. J. Allergy Clin. Immunol. 2004, 114, S2–S17. [Google Scholar] [CrossRef] [PubMed]
- Tagaya, E.; Tamaoki, J. Mechanisms of airway remodeling in asthma. Allergol. Int. 2007, 56, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.K.; Lee, J.H.; Ge, Q.; Ramsay, E.E.; Poniris, M.H.; Parmentier, J.; Roth, M.; Johnson, P.R.; Hunt, N.H.; Black, J.L.; et al. Dual ERK and phosphatidylinositol 3-kinase pathways control airway smooth muscle proliferation: Differences in asthma. J. Cell. Physiol. 2008, 216, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, R.; de Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO Families of GTPases Directly Regulate Distinct Phosphoinositide 3-Kinase Isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.T.; Yang, C.M. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.L.; Koessler, K.K. The pathology of bronchial asthma. Arch. Intern. Med. 1922, 30, 689–760. [Google Scholar] [CrossRef]
- Lim, D.H.; Cho, J.Y.; Song, D.J.; Lee, S.Y.; Miller, M.; Broide, D.H. PI3K gamma-deficient mice have reduced levels of allergen-induced eosinophilic inflammation and airway remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L210–L219. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Edwards, M.J.; Sawicka, E.; Duggan, N.; Hirsch, E.; Wymann, M.P.; Owen, C.; Trifilieff, A.; Walker, C.; Westwick, J.; et al. Essential role of phosphoinositide 3-kinase gamma in eosinophil chemotaxis within acute pulmonary inflammation. Immunology 2009, 126, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.N.; Ha, S.G.; Ge, X.N.; Reza Hosseinkhani, M.; Bahaie, N.S.; Greenberg, Y.; Blumenthal, M.N.; Puri, K.D.; Rao, S.P.; Sriramarao, P. The p110delta subunit of PI3K regulates bone marrow-derived eosinophil trafficking and airway eosinophilia in allergen-challenged mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1179–L1191. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lee, H.K.; Hayflick, J.S.; Lee, Y.C.; Puri, K.D. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006, 20, 455–465. [Google Scholar] [CrossRef]
- Zhang, X.; Ran, Y.G.; Wang, K.J. Risk of mTOR inhibitors induced severe pneumonitis in cancer patients: A meta-analysis of randomized controlled trials. Future Oncol. 2016, 12, 1529–1539. [Google Scholar] [CrossRef]
- Duran, I.; Goebell, P.J.; Papazisis, K.; Ravaud, A.; Weichhart, T.; Rodriguez-Portal, J.A.; Budde, K. Drug-induced pneumonitis in cancer patients treated with mTOR inhibitors: Management and insights into possible mechanisms. Expert Opin. Drug Saf. 2014, 13, 361–372. [Google Scholar] [CrossRef]
- Cuneo, A.; Barosi, G.; Danesi, R.; Fagiuoli, S.; Ghia, P.; Marzano, A.; Montillo, M.; Poletti, V.; Viale, P.; Zinzani, P.L. Management of adverse events associated with idelalisib treatment in chronic lymphocytic leukemia and follicular lymphoma: A multidisciplinary position paper. Hematol. Oncol. 2019, 37, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.G.; Blunt, M.D.; Carter, E.; Ward, S.G. Inhibition of PI3K signaling spurs new therapeutic opportunities in inflammatory/autoimmune diseases and hematological malignancies. Pharmacol. Rev. 2012, 64, 1027–1054. [Google Scholar] [CrossRef] [PubMed]
- Jou, S.T.; Carpino, N.; Takahashi, Y.; Piekorz, R.; Chao, J.R.; Carpino, N.; Wang, D.; Ihle, J.N. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol. Cell. Biol. 2002, 22, 8580–8591. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Ali, K.; Vanhaesebroeck, B. Antigen receptor signalling: A distinctive role for the p110delta isoform of PI3K. Trends Immunol. 2007, 28, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pauls, S.D.; Lafarge, S.T.; Landego, I.; Zhang, T.; Marshall, A.J. The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: Activation mechanisms, regulation and impact on cellular functions. Front. Immunol. 2012, 3, 224. [Google Scholar] [CrossRef]
- Ramadani, F.; Bolland, D.J.; Garcon, F.; Emery, J.L.; Vanhaesebroeck, B.; Corcoran, A.E.; Okkenhaug, K. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci. Signal. 2010, 3, ra60. [Google Scholar] [CrossRef]
- Limon, J.J.; Fruman, D.A. B cell receptor signaling: Picky about PI3Ks. Sci. Signal. 2010, 3, pe25. [Google Scholar] [CrossRef]
- Fougerat, A.; Gayral, S.; Gourdy, P.; Schambourg, A.; Ruckle, T.; Schwarz, M.K.; Rommel, C.; Hirsch, E.; Arnal, J.F.; Salles, J.P.; et al. Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 2008, 117, 1310–1317. [Google Scholar] [CrossRef]
- Camps, M.; Ruckle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Francon, B.; et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef]
- Barber, D.F.; Bartolome, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Ruckle, T.; Schwarz, M.K.; Rodriguez, S.; et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef]
- Laffargue, M.; Calvez, R.; Finan, P.; Trifilieff, A.; Barbier, M.; Altruda, F.; Hirsch, E.; Wymann, M.P. Phosphoinositide 3-kinase gamma is an essential amplifier of mast cell function. Immunity 2002, 16, 441–451. [Google Scholar] [CrossRef]
- Comerford, I.; Litchfield, W.; Kara, E.; McColl, S.R. PI3Kgamma drives priming and survival of autoreactive CD4(+) T cells during experimental autoimmune encephalomyelitis. PLoS ONE 2012, 7, e45095. [Google Scholar] [CrossRef] [PubMed]
- Juss, J.K.; Hayhoe, R.P.; Owen, C.E.; Bruce, I.; Walmsley, S.R.; Cowburn, A.S.; Kulkarni, S.; Boyle, K.B.; Stephens, L.; Hawkins, P.T.; et al. Functional redundancy of class I phosphoinositide 3-kinase (PI3K) isoforms in signaling growth factor-mediated human neutrophil survival. PLoS ONE 2012, 7, e45933. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Flinn, I.; Patel, M.R.; Younes, A.; Foss, F.M.; Oki, Y.; Sweeney, J.; Allen, K.; Dunbar, J.; Kelly, P.F.; et al. Preliminary safety and efficacy of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-delta, gamma in patients with relapsed/refractory lymphoma. J. Clin. Oncol. 2013, 31. Suppl. Abstract 8518. [Google Scholar]
- Lanasa, M.C.; Glenn, M.; Mato, A.R.; Allgood, S.D.; Wong, S.; Amore, B.; Means, G.; Stevens, E.; Yan, C.; Friberg, G.; et al. First-In-Human Study Of AMG 319, a Highly Selective, Small Molecule Inhibitor Of PI3K delta, In Adult Patients With Relapsed Or Refractory Lymphoid Malignancies. Blood 2013, 122, 678. [Google Scholar]
- Flinn, I.; Kimby, E.; Cotter, F.E.; Giles, F.J.; Janssens, A.; Pulczynski, E.J.; Ysebeart, L.; Pluta, A.; Marco, J.A.G.; Taylor, K.; et al. A phase III, randomized, controlled study evaluating the efficacy and safety of idelalisib (GS-1101) in combination with ofatumumab for previously treated chronic lymphocytic leukemia (CLL). J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N. Duvelisib, a novel oral dual inhibitor of PI3K-delta, γ, is clinically active in advanced hematologic malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fukuhara, N.; Nagai, H.; Izutsu, K.; Kobayashi, Y.; Higuchi, Y.; Harigae, H.; Tokunaga, T.; Adewoye, H.; Robeson, M.; et al. Phase 1b and Pharmacokinetic Study of Idelalisib in Japanese Patients with Relapsed or Refractory (R/R) Indolent B-Cell Non-Hodgkin Lymphoma (iNHL) or Chronic Lymphocytic Leukemia (CLL). Blood 2015, 126, 5089. [Google Scholar]
- Gilead Sciences Inc. ZYDELIG- idelalisib tablet, film coated. Drug label information. Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=efbdafa9-d18c-4e85-b4a2-1e620fc74e50#S5.8 (accessed on March 2017).
- So, L.; Fruman, D.A. PI3K signalling in B- and T-lymphocytes: New developments and therapeutic advances. Biochem. J. 2012, 442, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, P.C.; Manzanero, S.; Mohannak, N.; Narayana, V.K.; Nguyen, T.H.; Kvaskoff, D.; Brennan, F.H.; Ruitenberg, M.J.; Gelderblom, M.; Magnus, T.; et al. PI3Kdelta inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nat. Commun. 2014, 5, 3450. [Google Scholar] [CrossRef] [PubMed]
- Michalovich, D.; Nejentsev, S. Activated PI3 Kinase Delta Syndrome: From Genetics to Therapy. Front. Immunol. 2018, 9, 369. [Google Scholar] [CrossRef]
- Wujcik, C. Targeted therapy. In Cancer Nursing: Principles and Practice, 8th ed.; Yarbro, C.H., Wujcik, D., Gobel, B., Eds.; Jones and Bartlett Learning: Burlington, MA, USA, 2016; pp. 667–671. [Google Scholar]
- Britten, C.D.; Adjei, A.A.; Millham, R.; Houk, B.E.; Borzillo, G.; Pierce, K.; Wainberg, Z.A.; LoRusso, P.M. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Investig. New Drugs 2014, 32, 510–517. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tejpar, S.; Vanbeckevoort, D.; Peeters, M.; Humblet, Y.; Gelderblom, H.; Vermorken, J.B.; Viret, F.; Glimelius, B.; Gallerani, E.; et al. Intrapatient cetuximab dose escalation in metastatic colorectal cancer according to the grade of early skin reactions: The randomized EVEREST study. J. Clin. Oncol. 2012, 30, 2861–2868. [Google Scholar] [CrossRef]
- Golden, L.H.; Insogna, K.L. The expanding role of PI3-kinase in bone. Bone 2004, 34, 3–12. [Google Scholar] [CrossRef]
- Grey, A.; Chaussade, C.; Empson, V.; Lin, J.M.; Watson, M.; O’Sullivan, S.; Rewcastle, G.; Naot, D.; Cornish, J.; Shepherd, P. Evidence for a role for the p110-alpha isoform of PI3K in skeletal function. Biochem. Biophys. Res. Commun. 2010, 391, 564–569. [Google Scholar] [CrossRef]
- Martin, S.K.; Fitter, S.; Bong, L.F.; Drew, J.J.; Gronthos, S.; Shepherd, P.R.; Zannettino, A.C. NVP-BEZ235, a dual pan class I PI3 kinase and mTOR inhibitor, promotes osteogenic differentiation in human mesenchymal stromal cells. J. Bone Miner. Res. 2010, 25, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bruxvoort, K.J.; Zylstra, C.R.; Liu, J.; Cichowski, R.; Faugere, M.C.; Bouxsein, M.L.; Wan, C.; Williams, B.O.; Clemens, T.L. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc. Natl. Acad. Sci. USA 2007, 104, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulici, V.; Hoenselaar, K.D.; Agoston, H.; McErlain, D.D.; Umoh, J.; Chakrabarti, S.; Holdsworth, D.W.; Beier, F. The role of Akt1 in terminal stages of endochondral bone formation: Angiogenesis and ossification. Bone 2009, 45, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, N.; Kugimiya, F.; Oshima, Y.; Ohba, S.; Ikeda, T.; Saito, T.; Shinoda, Y.; Kawasaki, Y.; Ogata, N.; Hoshi, K.; et al. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2007, 2, e1058. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Chang, W.; Hurley, M.; Vignery, A.; Wu, D. Important roles of PI3Kgamma in osteoclastogenesis and bone homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 12901–12906. [Google Scholar] [CrossRef] [PubMed]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Zhang, L.; Tarnavski, O.; Sherwood, M.C.; Kang, P.M.; Izumo, S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 12355–12360. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Shioi, T.; Huang, W.Y.; Zhang, L.; Tarnavski, O.; Bisping, E.; Schinke, M.; Kong, S.; Sherwood, M.C.; Brown, J.; et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J. Biol. Chem. 2004, 279, 4782–4793. [Google Scholar] [CrossRef]
- O’Neill, B.T.; Kim, J.; Wende, A.R.; Theobald, H.A.; Tuinei, J.; Buchanan, J.; Guo, A.; Zaha, V.G.; Davis, D.K.; Schell, J.C.; et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007, 6, 294–306. [Google Scholar] [CrossRef]
- Zhang, D.; Contu, R.; Latronico, M.V.; Zhang, J.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; Gu, Y.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Jia, Z.; Wang, W.; Ballou, L.M.; Jiang, Y.P.; Chen, B.; Mathias, R.T.; Cohen, I.S.; Song, L.S.; Entcheva, E.; et al. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS ONE 2011, 6, e24404. [Google Scholar] [CrossRef] [PubMed]
- Holgado, B.L.; Martinez-Munoz, L.; Sanchez-Alcaniz, J.A.; Lucas, P.; Perez-Garcia, V.; Perez, G.; Rodriguez-Frade, J.M.; Nieto, M.; Marin, O.; Carrasco, Y.R.; et al. CXCL12-Mediated Murine Neural Progenitor Cell Movement Requires PI3Kbeta Activation. Mol. Neurobiol. 2013, 48, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Waardenberg, A.J.; Bernardo, B.C.; Ng, D.C.; Shepherd, P.R.; Cemerlang, N.; Sbroggio, M.; Wells, C.A.; Dalrymple, B.P.; Brancaccio, M.; Lin, R.C.; et al. Phosphoinositide 3-kinase (PI3K(p110alpha)) directly regulates key components of the Z-disc and cardiac structure. J. Biol. Chem. 2011, 286, 30837–30846. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Katare, R.; Meloni, M.; Damilano, F.; Hirsch, E.; Emanueli, C.; Madeddu, P. Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ. Res. 2010, 106, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Penninger, J.M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 2009, 82, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T.; Kang, P.M.; Douglas, P.S.; Hampe, J.; Yballe, C.M.; Lawitts, J.; Cantley, L.C.; Izumo, S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000, 19, 2537–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Weeks, K.L.; Gao, X.M.; Williams, R.B.; Bernardo, B.C.; Kiriazis, H.; Matthews, V.B.; Woodcock, E.A.; Bouwman, R.D.; Mollica, J.P.; et al. PI3K(p110 alpha) protects against myocardial infarction-induced heart failure: Identification of PI3K-regulated miRNA and mRNA. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 724–732. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Jay, P.Y. PI3K(p110alpha) inhibitors as anti-cancer agents: Minding the heart. Cell Cycle 2007, 6, 910–913. [Google Scholar] [CrossRef]
- Ciraolo, E.; Morello, F.; Hirsch, E. Present and future of PI3K pathway inhibition in cancer: Perspectives and limitations. Curr. Med. Chem. 2011, 18, 2674–2685. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects From Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, B. Relevance of the concept of oncogene addiction to hormonal carcinogenesis and molecular targeting in cancer prevention and therapy. Adv. Exp. Med. Biol. 2008, 617, 3–13. [Google Scholar]
- Rodon, J.; Perez, J.; Kurzrock, R. Combining targeted therapies: Practical issues to consider at the bench and bedside. Oncologist 2010, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 558, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Bjerke, L.; Clarke, P.A.; Workman, P. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr. Opin. Pharmacol. 2015, 23, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hosford, S.R.; Dillon, L.M.; Shee, K.; Liu, S.C.; Bean, J.R.; Salphati, L.; Pang, J.; Zhang, X.; Nannini, M.A.; et al. Strategically Timing Inhibition of Phosphatidylinositol 3-Kinase to Maximize Therapeutic Index in Estrogen Receptor Alpha-Positive, PIK3CA-Mutant Breast Cancer. Clin. Cancer Res. 2016, 22, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.V.; Ferry, D.M.; Edmunds, S.J.; Gu, Y.; Singleton, R.S.; Patel, K.; Pullen, S.M.; Hicks, K.O.; Syddall, S.P.; Atwell, G.J.; et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin. Cancer Res. 2007, 13, 3922–3932. [Google Scholar] [CrossRef]
- Spiegelberg, L.; Houben, R.; Niemans, R.; de Ruysscher, D.; Yaromina, A.; Theys, J.; Guise, C.P.; Smaill, J.B.; Patterson, A.V.; Lambin, P.; et al. Hypoxia-activated prodrugs and (lack of) clinical progress: The need for hypoxia-based biomarker patient selection in phase III clinical trials. Clin. Transl. Radiat. Oncol. 2019, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of Class I PI3K catalytic and regulatory subunits. Abbreviations: ABD, adaptor binding domain; BH, breakpoint cluster region homology domain (Rho-Gap-like domain); C2, C2 domain; Gβγ BD, Gβγ binding domain; HD, helical domain; KD, kinase domain; iSH2: inter-SH2 domain (p110 binding domain); PR, proline-rich domain; RBD, RAS binding domain.
Figure 1.
Schematic representation of Class I PI3K catalytic and regulatory subunits. Abbreviations: ABD, adaptor binding domain; BH, breakpoint cluster region homology domain (Rho-Gap-like domain); C2, C2 domain; Gβγ BD, Gβγ binding domain; HD, helical domain; KD, kinase domain; iSH2: inter-SH2 domain (p110 binding domain); PR, proline-rich domain; RBD, RAS binding domain.
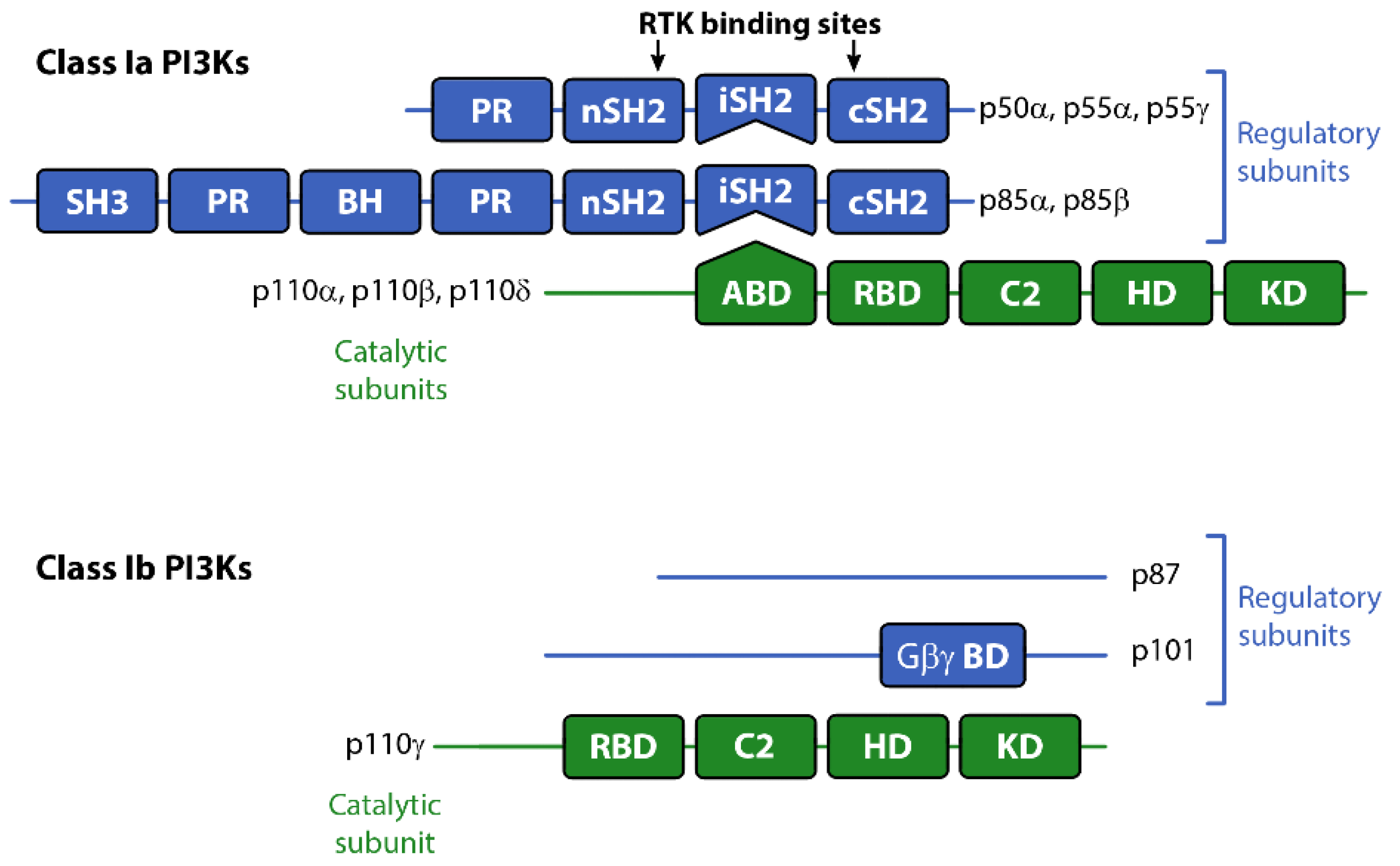
Figure 2.
Signaling pathways activated by different isoforms of Class I PI3K and PI3K inhibitors that target specific components of these pathways. Class 1a isoforms of PI3K are attracted to the membrane by the activation of RTKs (including IR/IGF1R), while PI3Kγ (and to a lesser extent PI3Kβ) is recruited by GPCR activation. The membrane proximity of PI3K results in the phosphorylation of membrane-bound PIP2 to PIP3, which mediates the activation of downstream protein kinases involved in a wide range of cellular processes. The various pan PI3K and isoform-specific inhibitors listed are ≥Phase 2 clinical trial according to www.clinicaltrials.gov (accessed August 2019). Abbreviations: AKT, Protein kinase B; Erk, extracellular signal-regulated kinase; GPCR, G-protein coupled receptor; IGF1R, insulin-like growth factor 1 receptor; IRS1, insulin receptor; MEK, MAPK/ERK kinase; mTOR, mammalian target of rapamycin; mTorc1, mammalian target of rapamycin complex 1; mTorc2, mammalian target of rapamycin complex 2; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-triphosphate; PTEN, phosphatase and tensin homologue; RTK, receptor tyrosine kinase.
Figure 2.
Signaling pathways activated by different isoforms of Class I PI3K and PI3K inhibitors that target specific components of these pathways. Class 1a isoforms of PI3K are attracted to the membrane by the activation of RTKs (including IR/IGF1R), while PI3Kγ (and to a lesser extent PI3Kβ) is recruited by GPCR activation. The membrane proximity of PI3K results in the phosphorylation of membrane-bound PIP2 to PIP3, which mediates the activation of downstream protein kinases involved in a wide range of cellular processes. The various pan PI3K and isoform-specific inhibitors listed are ≥Phase 2 clinical trial according to www.clinicaltrials.gov (accessed August 2019). Abbreviations: AKT, Protein kinase B; Erk, extracellular signal-regulated kinase; GPCR, G-protein coupled receptor; IGF1R, insulin-like growth factor 1 receptor; IRS1, insulin receptor; MEK, MAPK/ERK kinase; mTOR, mammalian target of rapamycin; mTorc1, mammalian target of rapamycin complex 1; mTorc2, mammalian target of rapamycin complex 2; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-triphosphate; PTEN, phosphatase and tensin homologue; RTK, receptor tyrosine kinase.
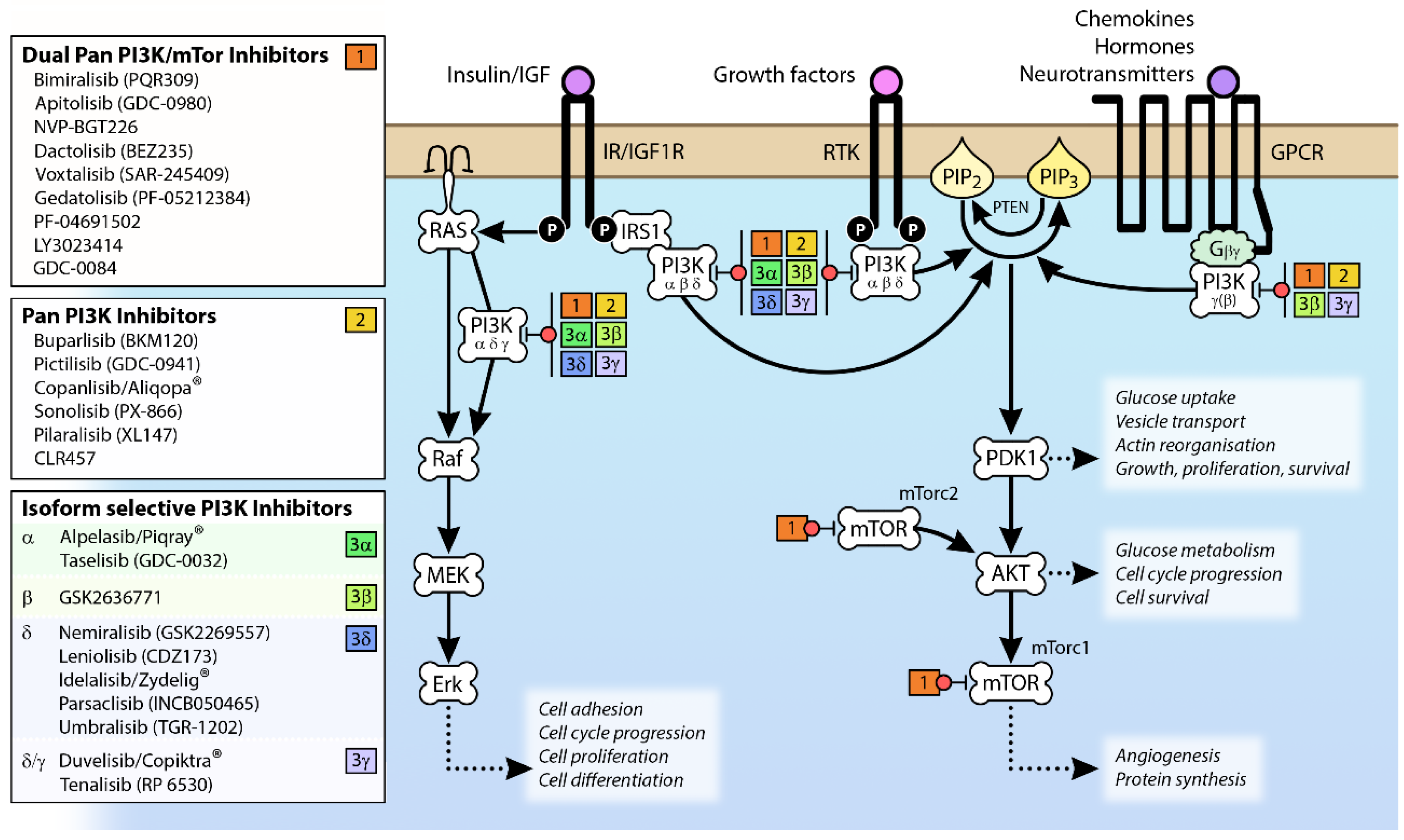
Figure 3.
Isoform-specific roles of Class I PI3Ks in normal and cancer physiology.
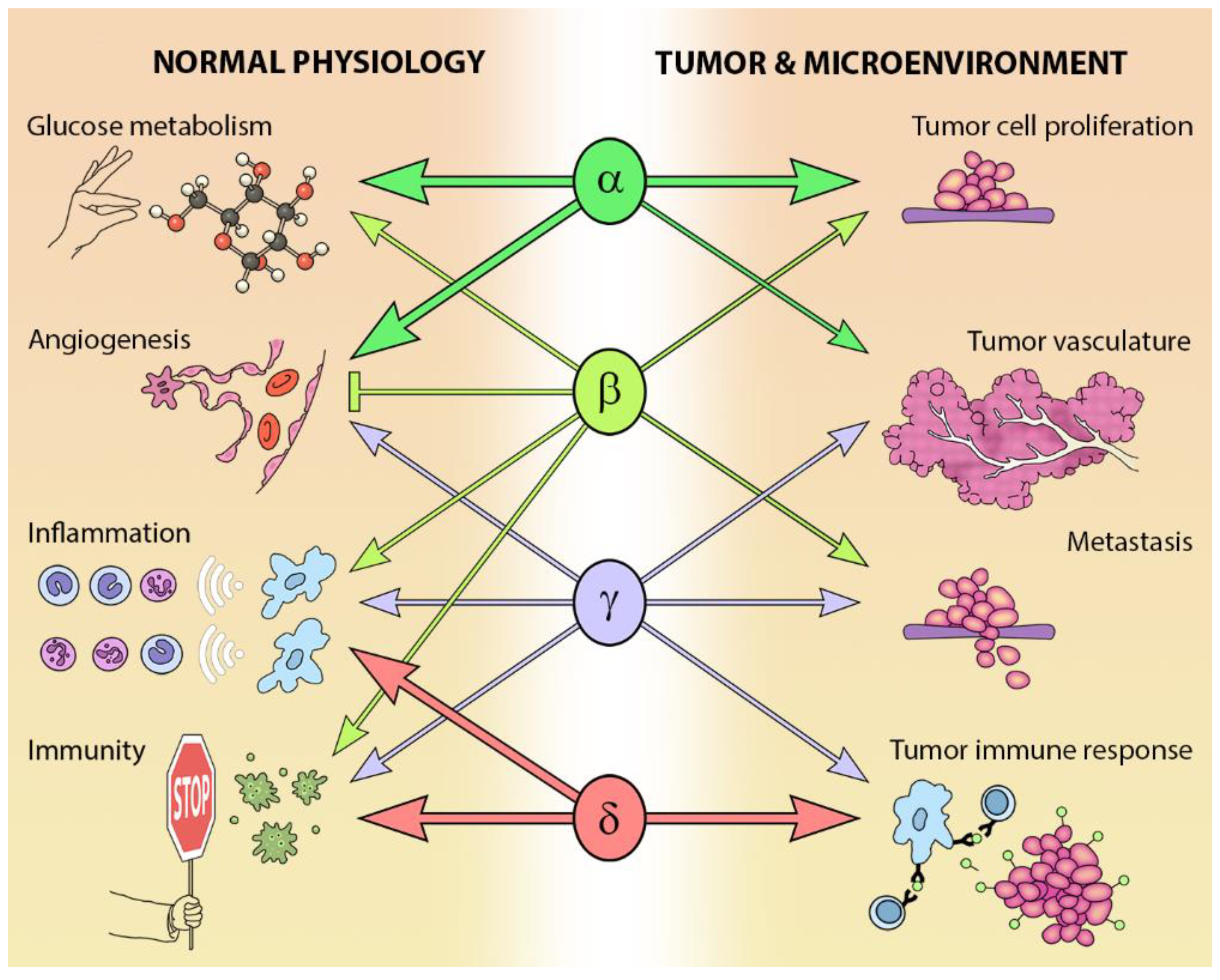
Figure 4.
Alterations of Class I PI3K in cancer. Alterations in expression, copy number and protein sequence in (A) catalytic subunits and (B) regulatory subunits reported in the named cancers (≥5% frequency). Information summarized from www.cbioportal.org (accessed July 2019). Abbreviations: CRC, colorectal cancer; GBM, glioblastoma; NM, non-melanoma; UT, urinary tract.
Figure 4.
Alterations of Class I PI3K in cancer. Alterations in expression, copy number and protein sequence in (A) catalytic subunits and (B) regulatory subunits reported in the named cancers (≥5% frequency). Information summarized from www.cbioportal.org (accessed July 2019). Abbreviations: CRC, colorectal cancer; GBM, glioblastoma; NM, non-melanoma; UT, urinary tract.
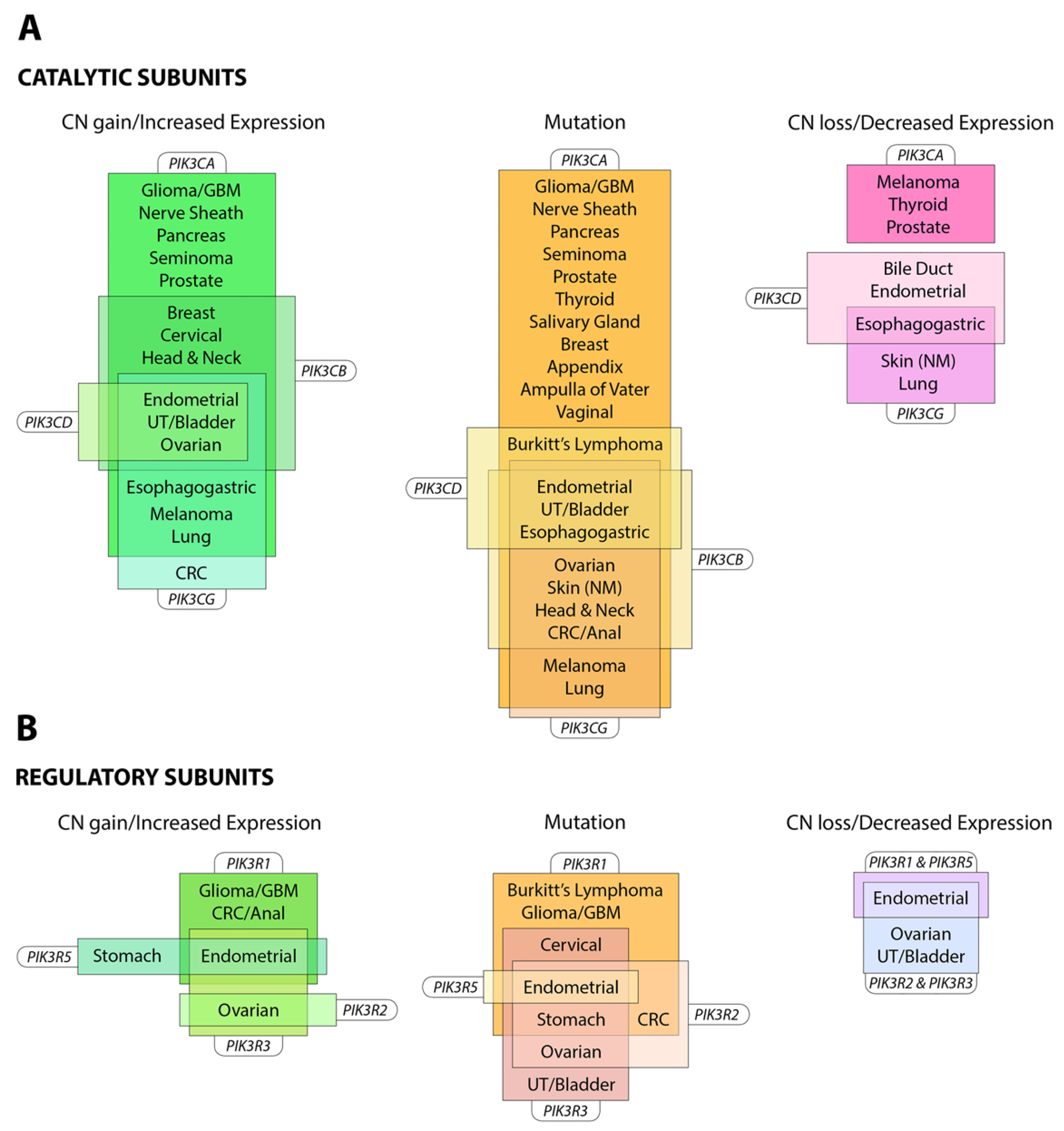

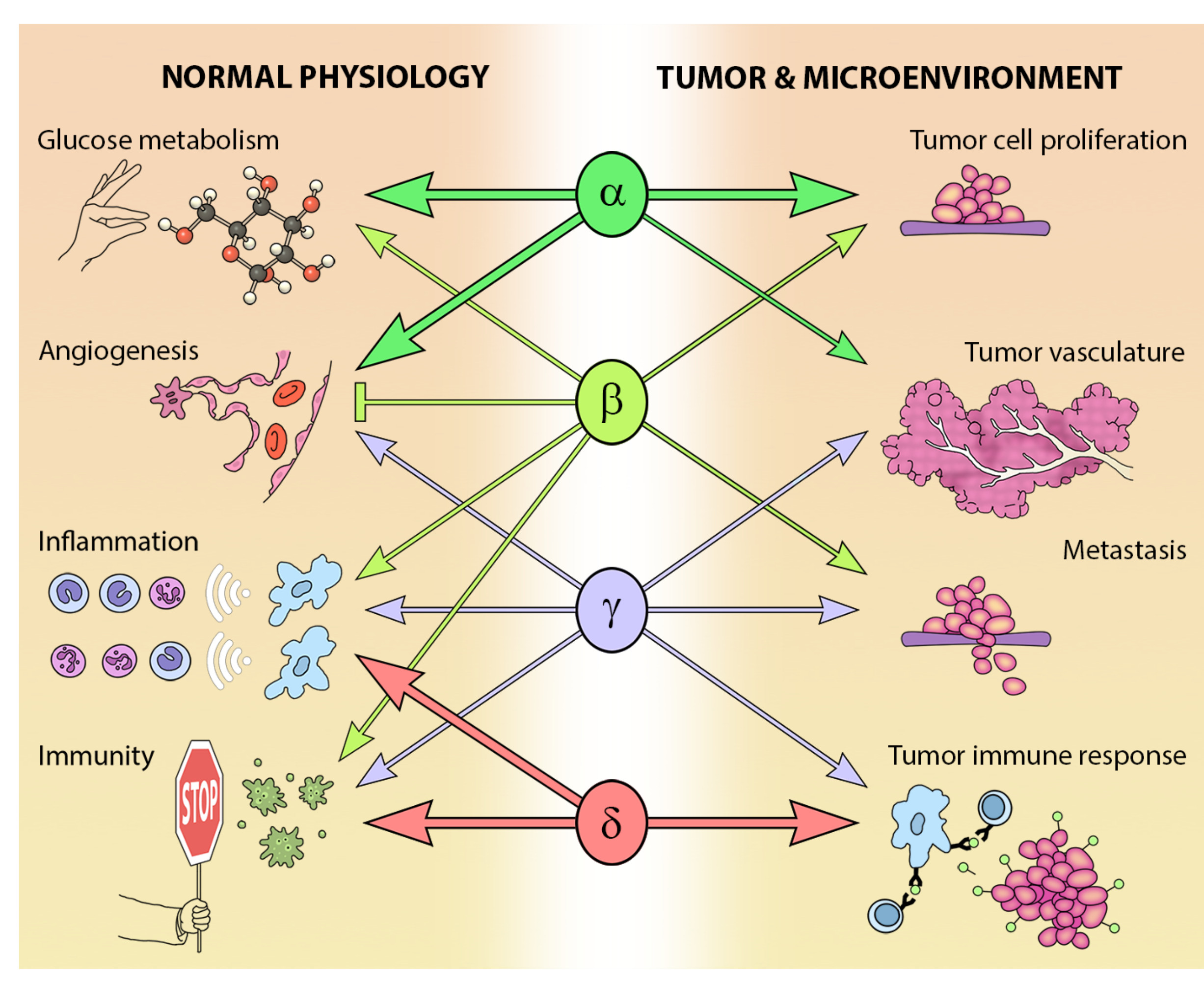{kind=link}
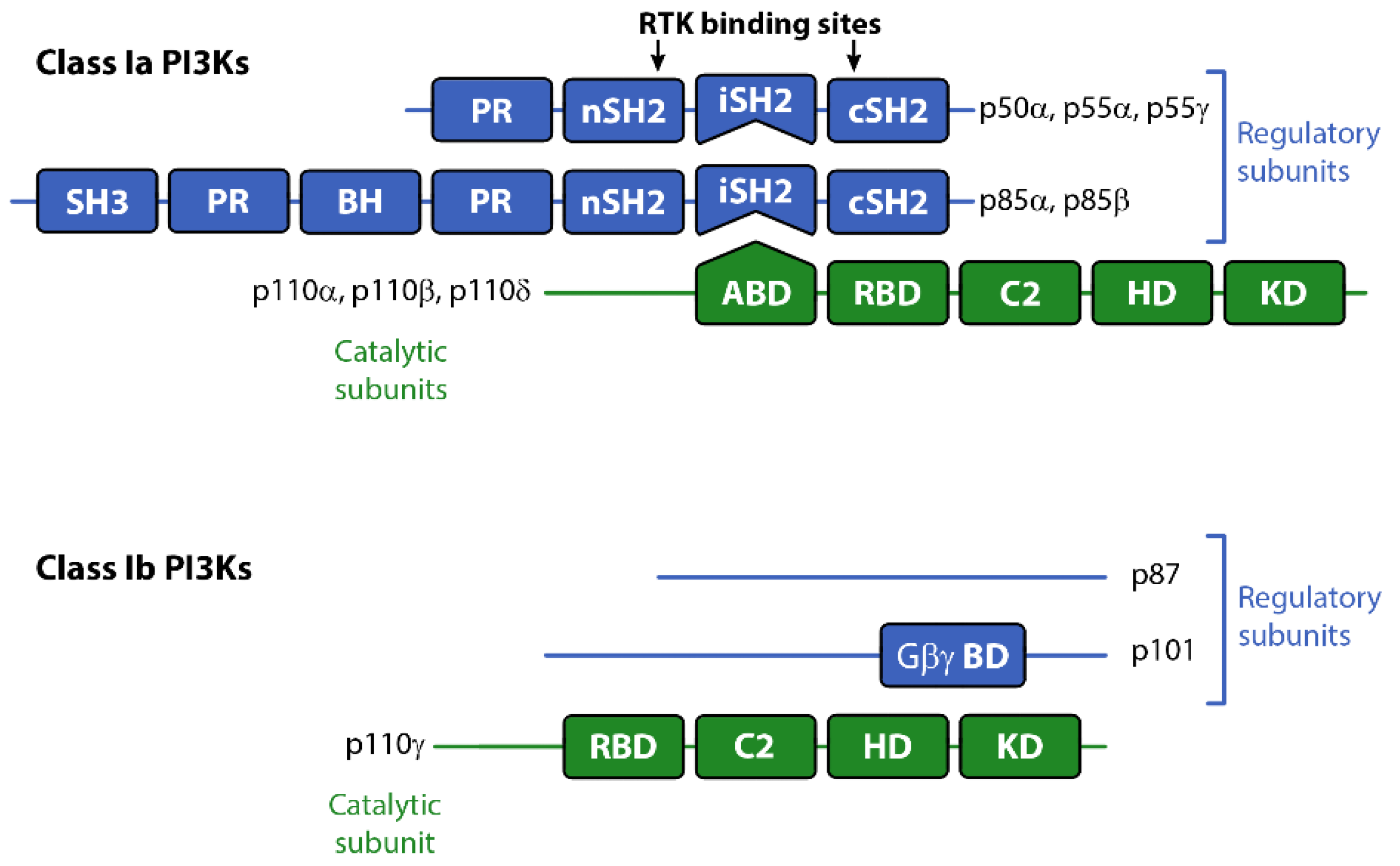{kind=link}
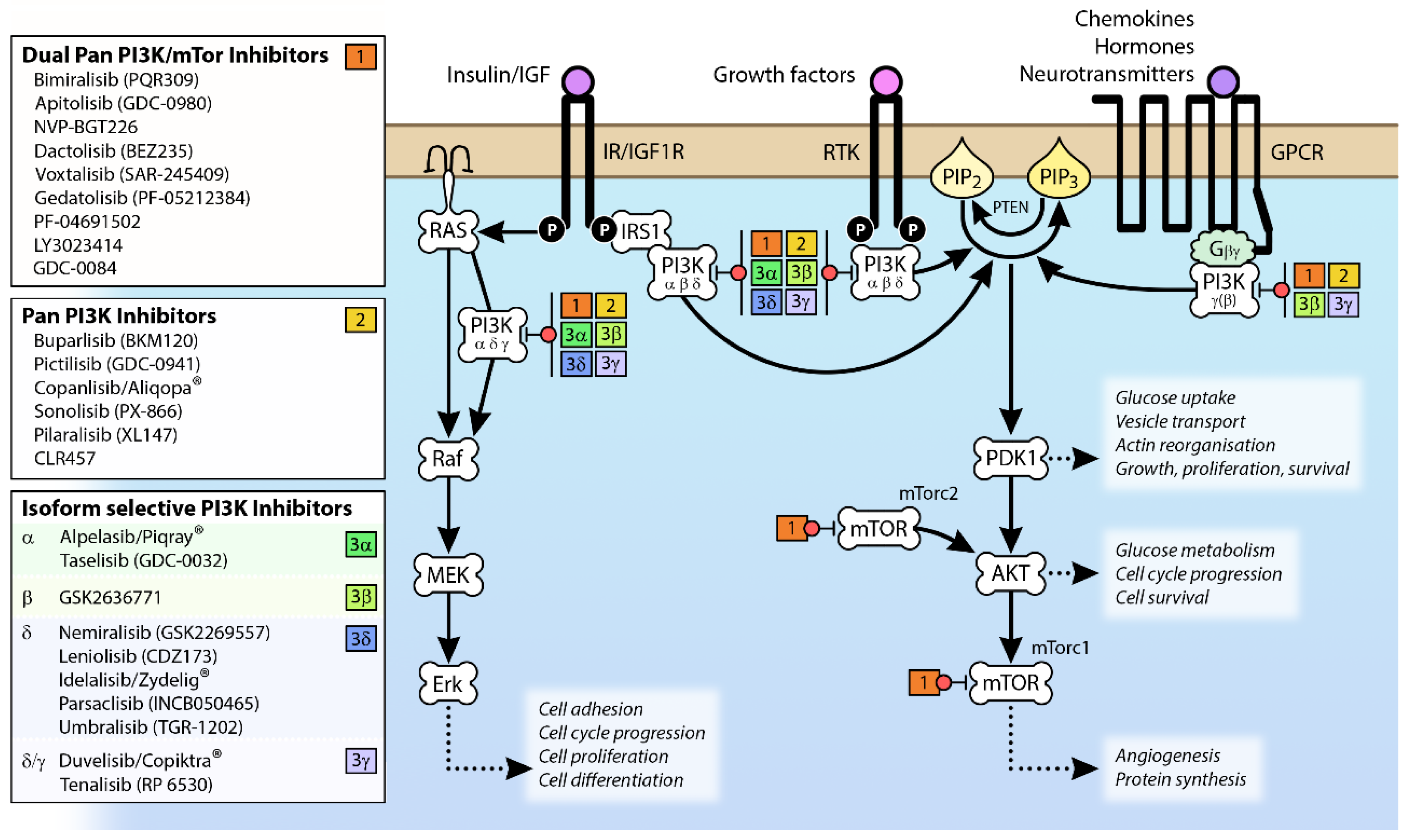{kind=link}
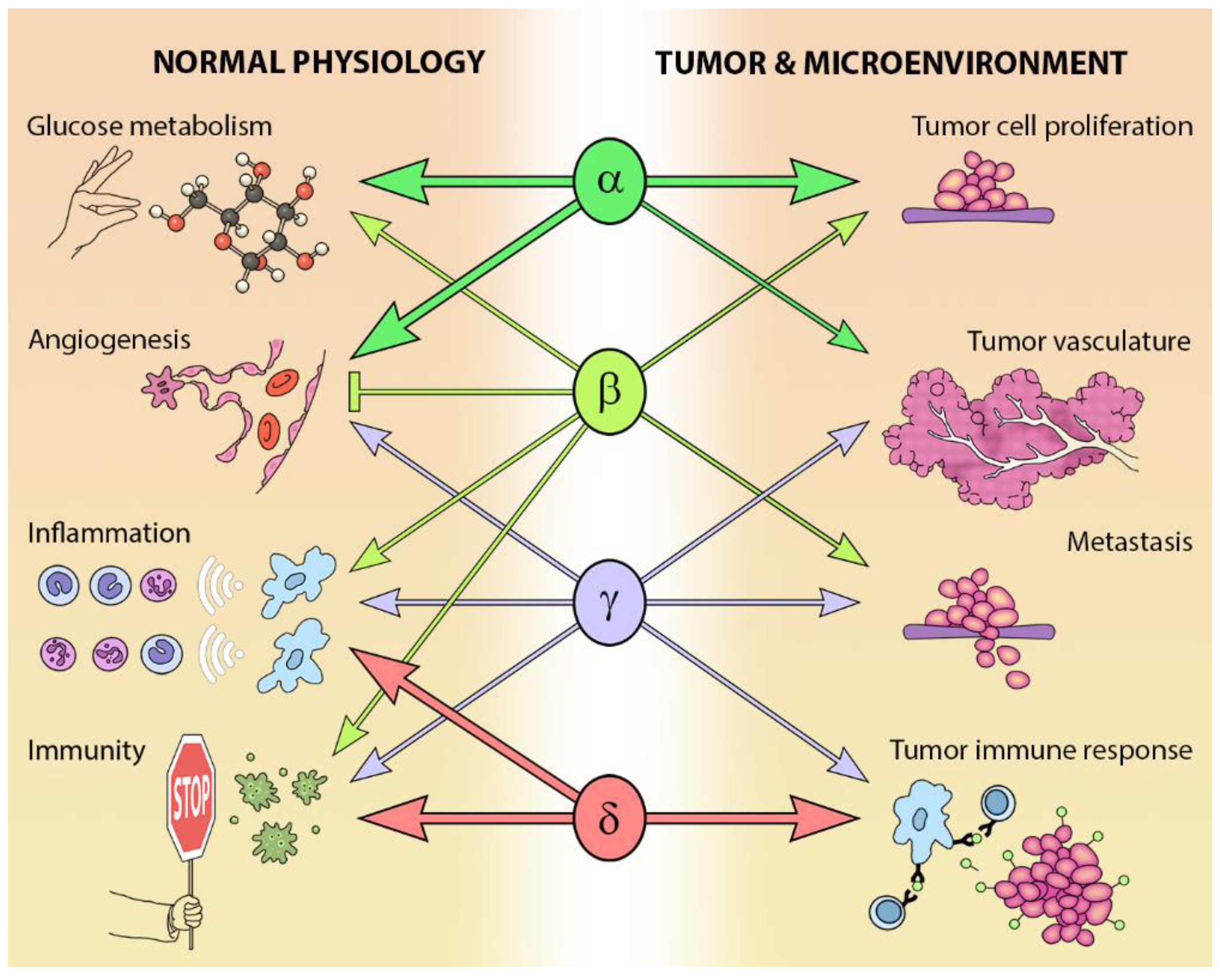{kind=link}
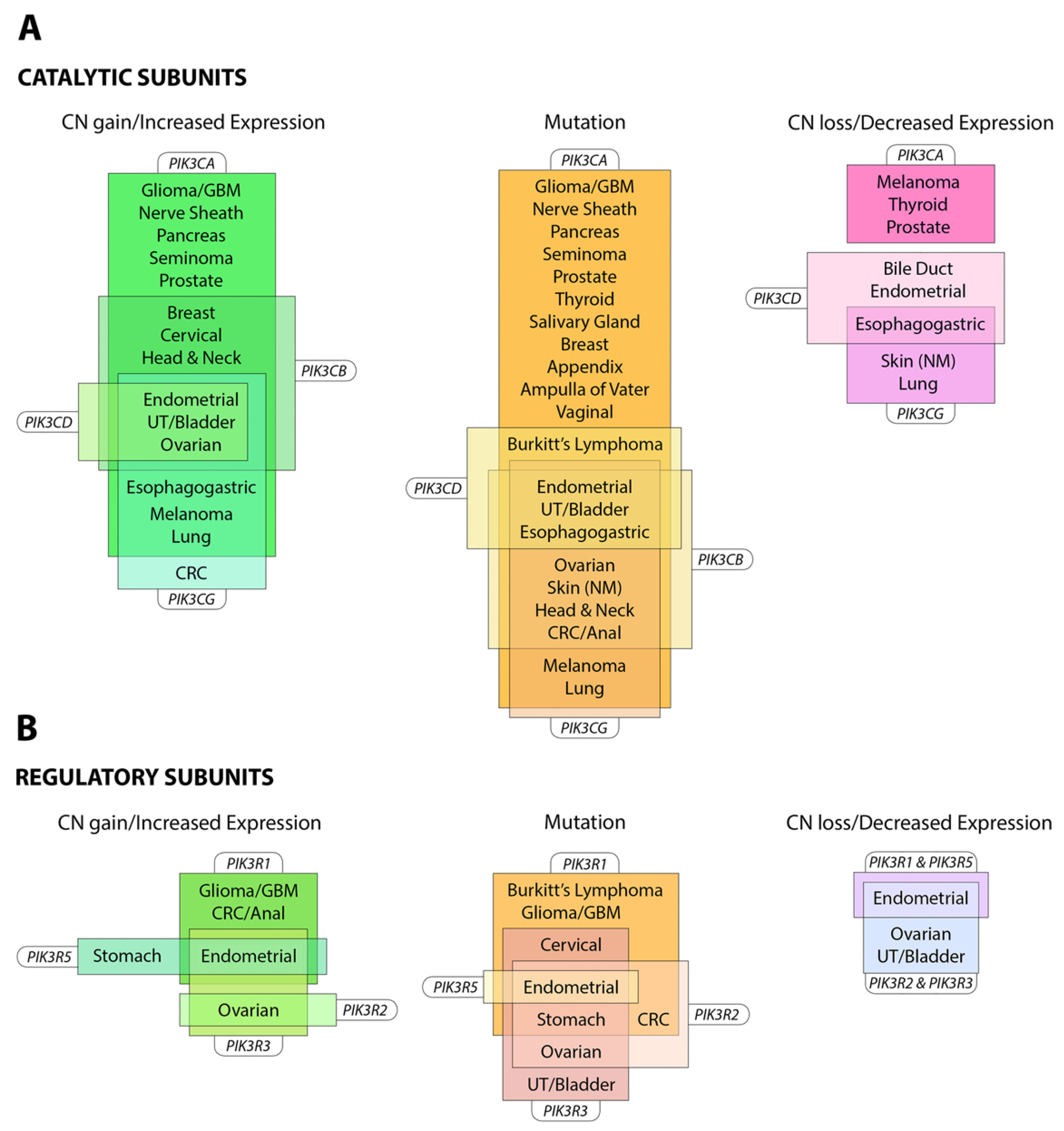{kind=link}
Table 1.
Gene and protein names of catalytic and regulatory subunits that make up the Class Ia and Class Ib phosphoinositide 3-kinases (PI3K) heterodimers. Also shown are the Class II and III phosphoinositide 3-kinases (PI3Ks) members, tissue distribution and the reaction catalyzed by the different classes of PI3K.
Table 1.
Gene and protein names of catalytic and regulatory subunits that make up the Class Ia and Class Ib phosphoinositide 3-kinases (PI3K) heterodimers. Also shown are the Class II and III phosphoinositide 3-kinases (PI3Ks) members, tissue distribution and the reaction catalyzed by the different classes of PI3K.
| Class | Catalytic Subunits | Regulatory Subunits | Tissue Distribution | Catalytic Reaction | ||
|---|---|---|---|---|---|---|
| Protein | Gene | Protein | Gene | |||
| Ia | p110α | PIK3CA | p85α | PIK3R1 | Ubiquitous | PI(4,5)P2→PI(3,4,5)P3 |
| p110β | PIK3CB | p85-β | PIK3R2 | Ubiquitous | ||
| p110δ | PIK3CD | p55-γ | PIK3R3 | Leukocytes, Neurons | ||
| p55-α | PIK3R1 | |||||
| p50-α | PIK3R1 | |||||
| Ib | p110γ | PIK3CG | p101 | PIK3R5 | Leukocytes, Cardiac myocytes, Endothelium | PI(4,5)P2→PI(3,4,5)P3 |
| p84/p87PIKRAP | PIK3R6 | |||||
| II | PI3K-C2α | PIK3C2A | Epithelium, Endothelium | PI→PI3P and PI4P→PI(3,4)P2 | ||
| PI3K-C2β | PIK3C2B | Ubiquitous | ||||
| PI3K-C2γ | PIK3C2G | Hepatocytes | ||||
| III | Vps34 | PIK3C3 | Ubiquitous | PI→PI3P | ||
Table 2.
Clinical toxicities associated with PI3K inhibitor use. The most likely physiological target for the named toxicity is also provided.
Table 2.
Clinical toxicities associated with PI3K inhibitor use. The most likely physiological target for the named toxicity is also provided.
| Kinase Target | Clinical Toxicities | Physiological Target |
|---|---|---|
| Pan PI3K Pan PI3K/mTOR PI3Kα/β/δ/γ | Colitis/diarrhea | Gut |
| Pan PI3K Pan PI3K/mTOR PI3Kα | Hyperglycemia | Glucose metabolism |
| Pan PI3K Pan PI3K/mTOR PI3Kα/β/δ | Fatigue | Energy metabolism, Neurological |
| Pan PI3K | Mood alterations | Neurological |
| Pan PI3K Pan PI3K/mTOR PI3Kα/β/δ | Nausea/vomiting | Gut |
| Pan PI3K Pan PI3K/mTOR PI3Kα/β | Decreased appetite | Brain, Gut |
| Pan PI3K PI3Kδ | Liver dysfunction | Liver |
| Pan PI3K Pan PI3K/mTOR PI3Kδ | Rash | Skin |
| PI3Kδ | Pneumonitis/pneumonia | Airways |
| PI3Kδ/γ | Hematologic toxicities: anemia, neutropenia, thrombocytopenia | Hematopoietic system |
| PI3Kδ | Pyrexia (fever) | Unspecified |
| Pan PI3K | Dysgeusia | Unspecified |
Table 3.
Acute and chronic metabolic effects of PI3K abrogation in a preclinical setting.
| Acute Administration of PI3K Inhibitors (Single Dose) [86] | Chronic Administration of PI3K Inhibitors (22 Days Dosing) [85] |
| Insulin resistant; increased gluconeogenesis, decreased glucose disposal | Insulin sensitive; normal gluconeogenesis, normal glycaemia |
| Decreased food intake | No effect on food intake, but decreased weight gain, fat mass, bone volume and bone strength |
| Decreased movement | Decreased movement |
| Young PI3Kα deficient mice [84] | Aged PI3Kα deficient mice [83] |
| Insulin resistant; hyperinsulinemic, hyperleptinemic, decreased glucose disposal | Insulin sensitive; normal glycaemia, increased longevity |
| Increased food intake but smaller; lower weight, length, skeletal mass, increased white adipose | No effect on food intake but males remained smaller; leaner, reduced adiposity |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Buchanan, C.M.; Lee, K.L.; Shepherd, P.R. For Better or Worse: The Potential for Dose Limiting the On-Target Toxicity of PI 3-Kinase Inhibitors. Biomolecules 2019, 9, 402. https://doi.org/10.3390/biom9090402
AMA Style
Buchanan CM, Lee KL, Shepherd PR. For Better or Worse: The Potential for Dose Limiting the On-Target Toxicity of PI 3-Kinase Inhibitors. Biomolecules. 2019; 9(9):402. https://doi.org/10.3390/biom9090402
Chicago/Turabian StyleBuchanan, Christina M., Kate L. Lee, and Peter R. Shepherd. 2019. "For Better or Worse: The Potential for Dose Limiting the On-Target Toxicity of PI 3-Kinase Inhibitors" Biomolecules 9, no. 9: 402. https://doi.org/10.3390/biom9090402
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.