Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Models and Approaches Dedicated to Study Lipid–Protein Interactions
2.1. Cell Imaging
2.1.1. Cell Imaging Approaches and Lipid Tools
2.1.2. Förster Resonance Energy Transfer
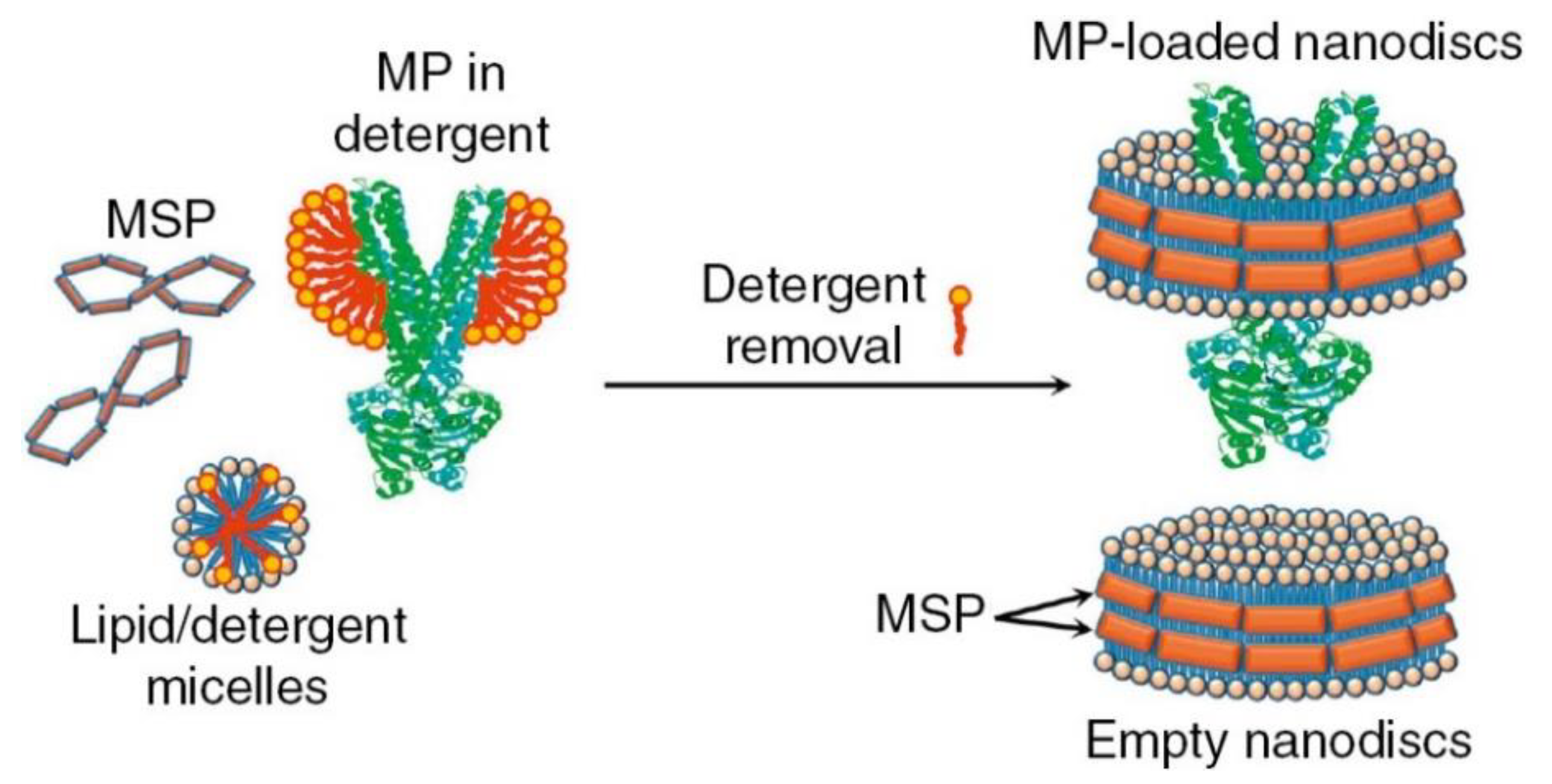
2.2. Reconstitution of Membrane Proteins in Artificial Environments
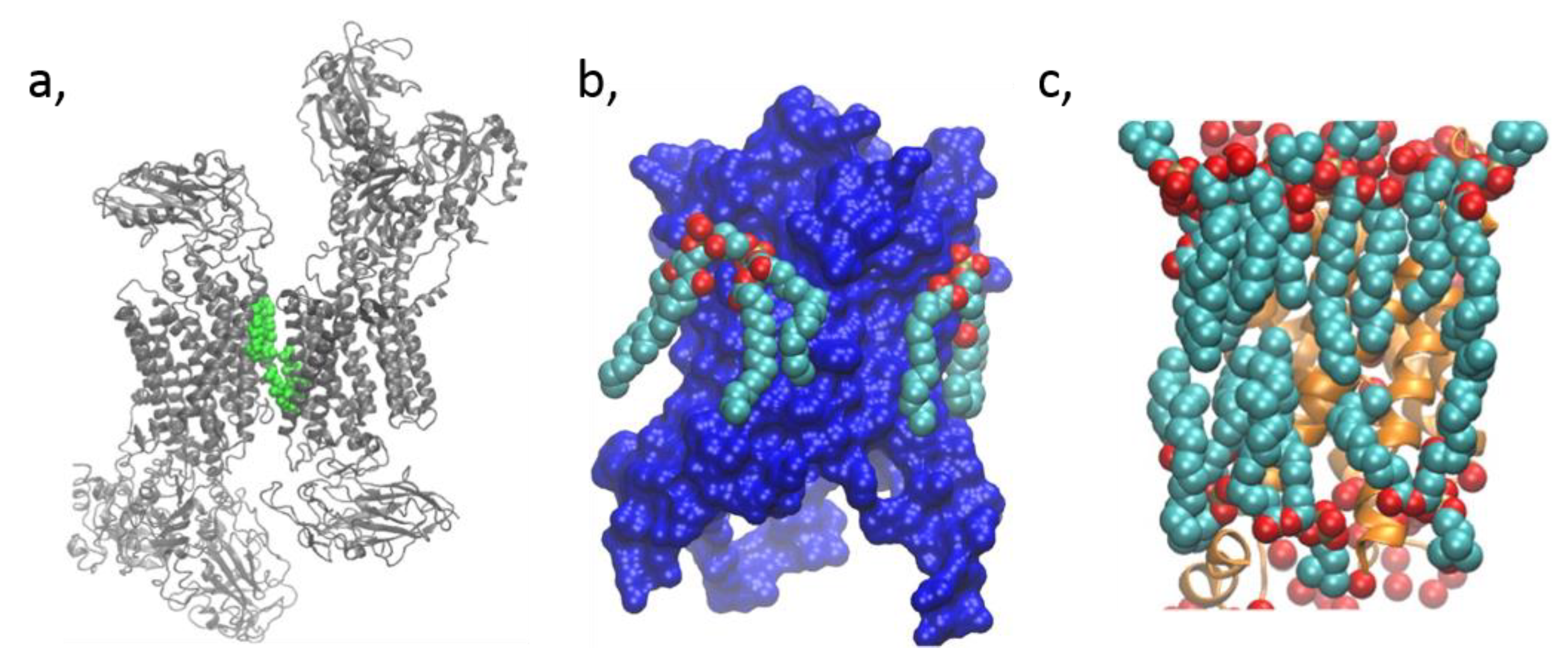
2.3. Membrane Protein Structural Biology and Mass Spectrometry
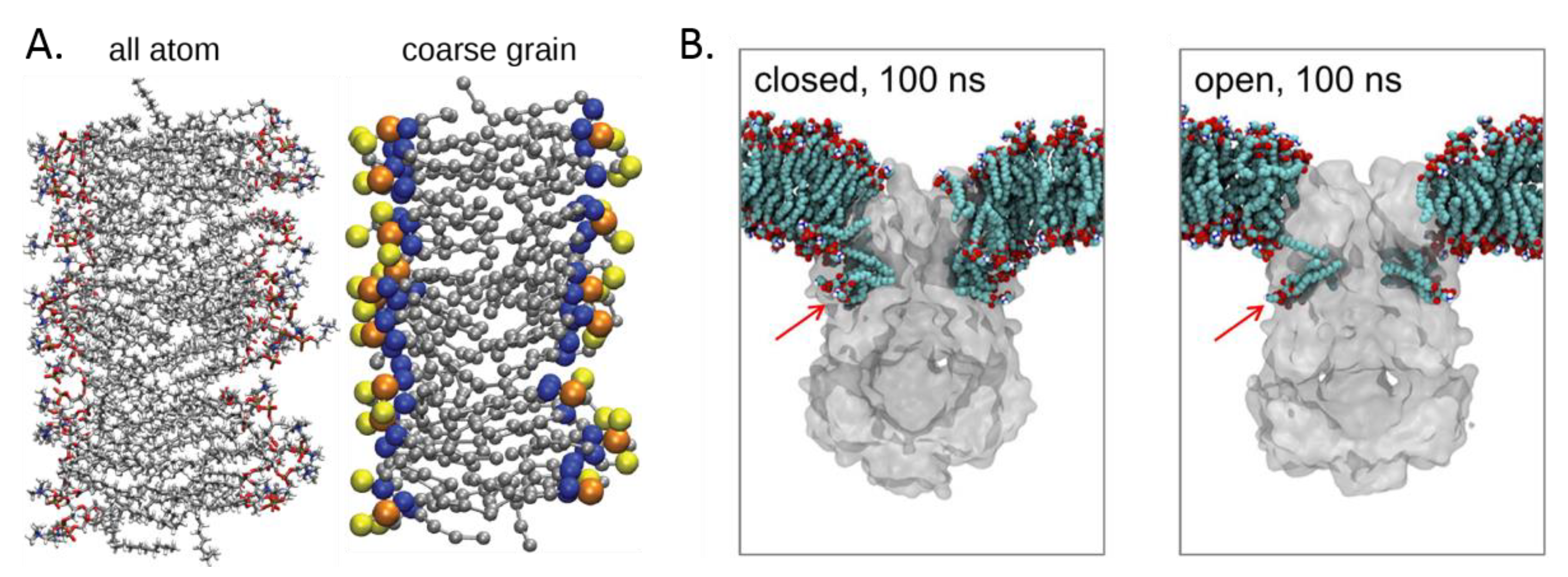
2.4. Molecular Simulation
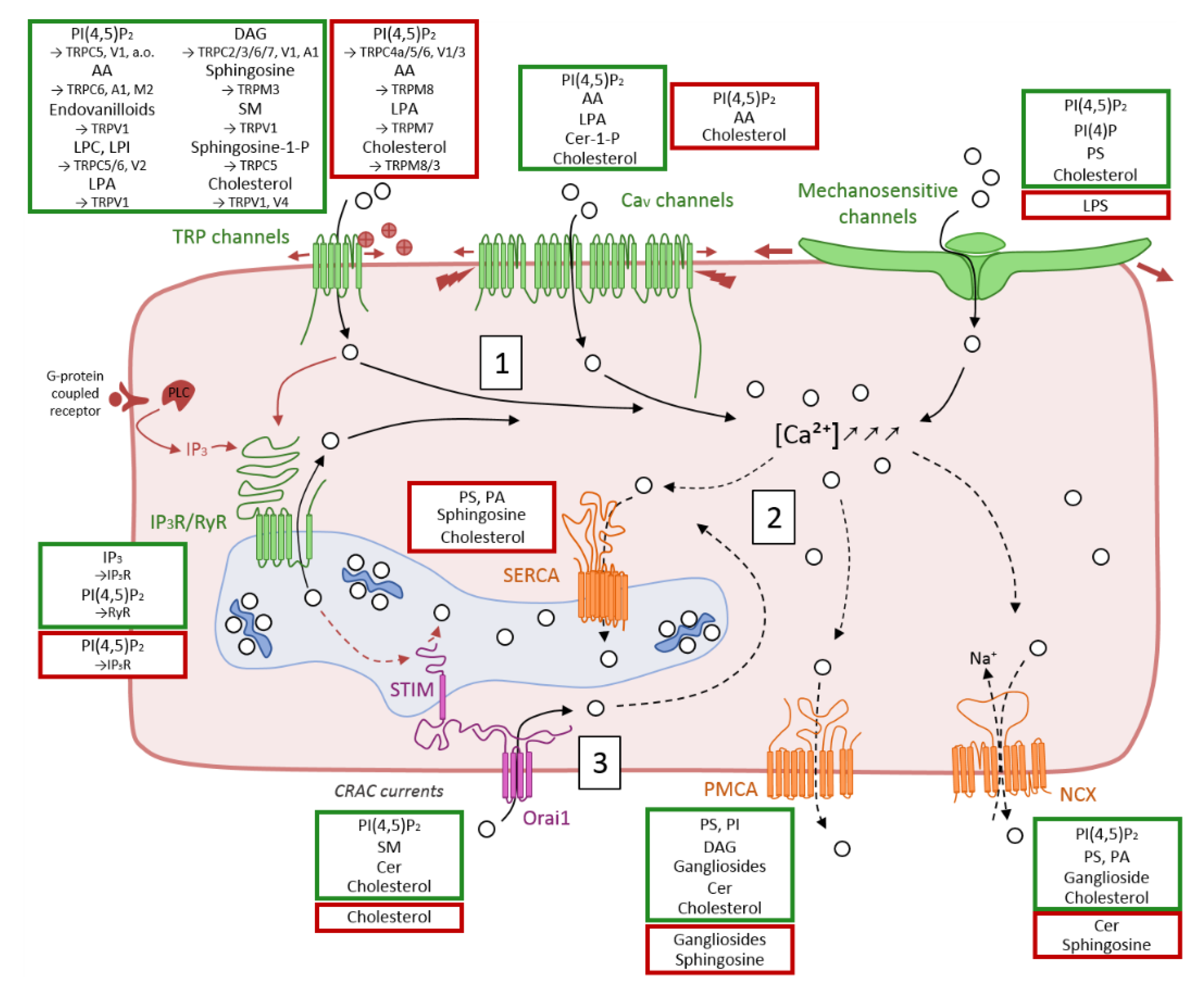
3. Overview of Membrane Ca2+ Transport Proteins and Evidences for Their Modulation by Lipids
3.1. Ca2+ Influx in the Cytosol through Cation Channels
3.1.1. Transient Receptor Potential Channels
Overview and Pathophysiological Implications
Regulation by Lipids
3.1.2. Voltage-Gated Channels
Overview and Pathophysiological Implications
Regulation by Lipids
3.1.3. Mechanosensitive Ion Channels
Overview and Pathophysiological Implications
Regulation by Lipids
3.1.4. Endo/Sarcoplasmic IP3R and RyR Channels
Overview and Pathophysiological Implications
Regulation by Lipids
3.1.5. Store-Operated Ca2+ Release-Activated Ca2+ (CRAC) Currents
Overview and Pathophysiological Implications
Regulation by Lipids
3.2. Ca2+ Efflux through Exchangers and Selective Pumps
3.2.1. Na+/Ca2+ Exchanger (NCX)
Overview and Pathophysiological Implications
Regulation by Lipids
3.2.2. Plasma Membrane Ca2+ ATPase (PMCA)
Overview and Pathophysiological Implications
Regulation by Lipids
3.2.3. Sarco/Endoplasmic Ca2+ ATPase (SERCA)
Overview and Pathophysiological Implications
Regulation by Lipids
4. Hypothetical Mechanisms for the Regulation of Membrane Ca2+ Transport Proteins by Lipids
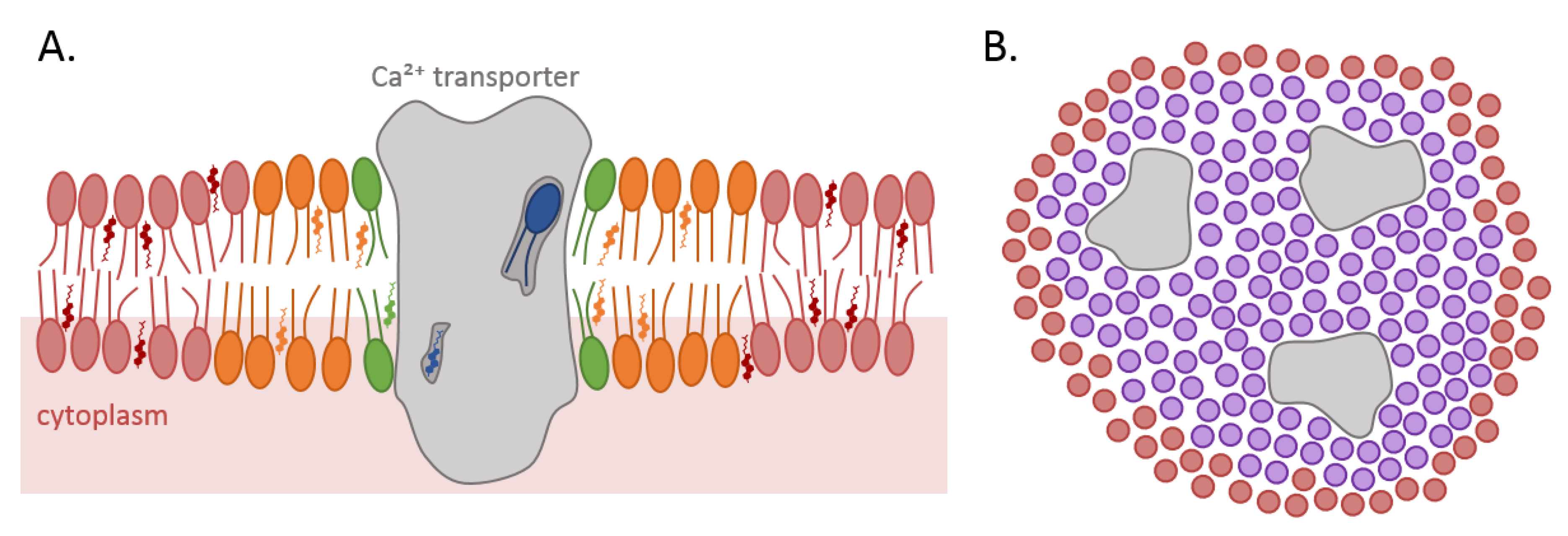
4.1. Direct Interaction with Non-Annular Lipids
4.2. Close Interaction with the First Shell of Annular Lipids
4.3. Regulation of Membrane Biophysical Properties Directly around the Protein through Annular Lipids
4.3.1. Membrane Lipid Packing
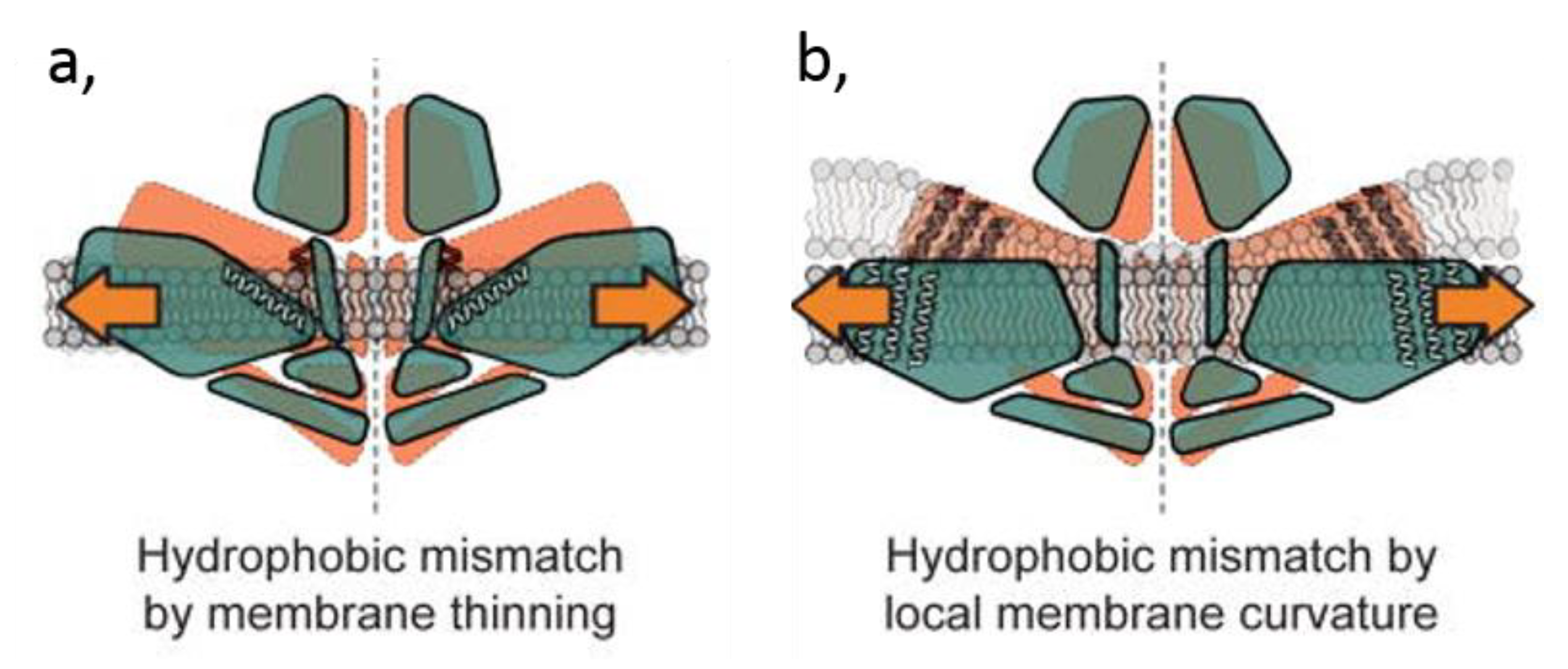
4.3.2. Membrane Thickness
4.3.3. Membrane Curvature
4.4. Contribution to Ca2+ Exchange/Signaling Inside Lipid Domains
5. Lipid and Ca2+ Transport Alterations in Physiological Aging and Diseases
6. Lipids as Therapeutic Targets in Ca2+ Transport-Related Diseases
7. Conclusions and Challenges for the Future
Funding
Conflicts of Interest
References
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar] [PubMed]
- Yu, S.P.; Choi, D.W. Na(+)-Ca2+ exchange currents in cortical neurons: Concomitant forward and reverse operation and effect of glutamate. Eur. J. Neurosci. 1997, 9, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Ziman, A.P.; Ward, C.W.; Rodney, G.G.; Lederer, W.J.; Bloch, R.J. Quantitative measurement of Ca(2)(+) in the sarcoplasmic reticulum lumen of mammalian skeletal muscle. Biophys. J. 2010, 99, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Bogdanova, A.; Makhro, A.; Wang, J.; Lipp, P.; Kaestner, L. Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 2013, 14, 9848–9872. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. Lipid-protein interactions in biological membranes: A structural perspective. Biochim. Biophys. Acta 2003, 1612, 1–40. [Google Scholar] [CrossRef]
- Marsh, D. Protein modulation of lipids, and vice-versa, in membranes. Biochim. Biophys. Acta 2008, 1778, 1545–1575. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.G.; Jacobson, K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 2002, 296, 1821–1825. [Google Scholar] [CrossRef]
- Koldso, H.; Sansom, M.S. Local Lipid Reorganization by a Transmembrane Protein Domain. J. Phys. Chem. Lett. 2012, 3, 3498–3502. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Vicidomini, G.; Ta, H.; Honigmann, A.; Mueller, V.; Clausen, M.P.; Waithe, D.; Galiani, S.; Sezgin, E.; Diaspro, A.; Hell, S.W.; et al. STED-FLCS: An Advanced Tool to Reveal Spatiotemporal Heterogeneity of Molecular Membrane Dynamics. Nano Lett. 2015, 15, 5912–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, M.B.; Shelby, S.A.; Veatch, S.L. Super-Resolution Microscopy: Shedding Light on the Cellular Plasma Membrane. Chem. Rev. 2017, 11, 7457–7477. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdes, M.; Sanchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Fujiwara, T.K.; Chadda, R.; Xie, M.; Tsunoyama, T.A.; Kalay, Z.; Kasai, R.S.; Suzuki, K.G. Dynamic organizing principles of the plasma membrane that regulate signal transduction: Commemorating the fortieth anniversary of Singer and Nicolson’s fluid-mosaic model. Annu. Rev. Cell Dev. Biol. 2012, 28, 215–250. [Google Scholar] [CrossRef] [PubMed]
- Carquin, M.; D’Auria, L.; Pollet, H.; Bongarzone, E.R.; Tyteca, D. Recent progress on lipid lateral heterogeneity in plasma membranes: From rafts to submicrometric domains. Prog. Lipid Res. 2016, 62, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Hammond, A.T.; Sengupta, P.; Hess, S.T.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA 2007, 104, 3165–3170. [Google Scholar] [CrossRef] [Green Version]
- Bernardino de la Serna, J.; Perez-Gil, J.; Simonsen, A.C.; Bagatolli, L.A. Cholesterol rules: Direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J. Biol. Chem. 2004, 279, 40715–40722. [Google Scholar] [CrossRef] [PubMed]
- Kahya, N.; Scherfeld, D.; Bacia, K.; Poolman, B.; Schwille, P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 2003, 278, 28109–28115. [Google Scholar] [CrossRef]
- Plasencia, I.; Norlen, L.; Bagatolli, L.A. Direct visualization of lipid domains in human skin stratum corneum’s lipid membranes: Effect of pH and temperature. Biophys. J. 2007, 93, 3142–3155. [Google Scholar] [CrossRef]
- Carquin, M.; Pollet, H.; Veiga-da-Cunha, M.; Cominelli, A.; Van Der Smissen, P.; N’Kuli, F.; Emonard, H.; Henriet, P.; Mizuno, H.; Courtoy, P.J.; et al. Endogenous sphingomyelin segregates into submicrometric domains in the living erythrocyte membrane. J. Lipid Res. 2014, 55, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Auria, L.; Fenaux, M.; Aleksandrowicz, P.; Van Der Smissen, P.; Chantrain, C.; Vermylen, C.; Vikkula, M.; Courtoy, P.J.; Tyteca, D. Micrometric segregation of fluorescent membrane lipids: Relevance for endogenous lipids and biogenesis in erythrocytes. J. Lipid Res. 2013, 54, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.A.; Tricerri, M.A.; Gratton, E. Laurdan generalized polarization fluctuations measures membrane packing micro-heterogeneity in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 7314–7319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carquin, M.; Conrard, L.; Pollet, H.; Van Der Smissen, P.; Cominelli, A.; Veiga-da-Cunha, M.; Courtoy, P.J.; Tyteca, D. Cholesterol segregates into submicrometric domains at the living erythrocyte membrane: Evidence and regulation. Cell. Mol. Life Sci. 2015, 72, 4633–4651. [Google Scholar] [CrossRef] [PubMed]
- Tyteca, D.; D’Auria, L.; Van Der Smissen, P.; Medts, T.; Carpentier, S.; Monbaliu, J.C.; de Diesbach, P.; Courtoy, P.J. Three unrelated sphingomyelin analogs spontaneously cluster into plasma membrane micrometric domains. Biochim. Biophys. Acta 2010, 1798, 909–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, J.N.; Bramkamp, M. Flotillins functionally organize the bacterial membrane. Mol. Microbiol. 2013, 88, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, G.; Opekarova, M.; Malinsky, J.; Weig-Meckl, I.; Tanner, W. Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. Embo J. 2007, 26, 1–8. [Google Scholar] [CrossRef]
- Gladwyn-Ng, I.; Cordon-Barris, L.; Alfano, C.; Creppe, C.; Couderc, T.; Morelli, G.; Thelen, N.; America, M.; Bessieres, B.; Encha-Razavi, F.; et al. Stress-induced unfolded protein response contributes to Zika virus-associated microcephaly. Nat. Neurosci. 2018, 21, 63–71. [Google Scholar] [CrossRef]
- Robison, P.; Caporizzo, M.A.; Ahmadzadeh, H.; Bogush, A.I.; Chen, C.Y.; Margulies, K.B.; Shenoy, V.B.; Prosser, B.L. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 2016, 352, aaf0659. [Google Scholar] [CrossRef]
- Neu, T.R.; Lawrence, J.R. Innovative techniques, sensors, and approaches for imaging biofilms at different scales. Trends Microbiol. 2015, 23, 233–242. [Google Scholar] [CrossRef]
- Kuerschner, L.; Ejsing, C.S.; Ekroos, K.; Shevchenko, A.; Anderson, K.I.; Thiele, C. Polyene-lipids: A new tool to image lipids. Nat. Methods 2005, 2, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.X.; Ernst, A.M.; Wieland, F.; Brugger, B. Specificity of intramembrane protein-lipid interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Contreras, F.X.; Ernst, A.M.; Haberkant, P.; Bjorkholm, P.; Lindahl, E.; Gonen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Levi, V.; Rossi, J.P.; Echarte, M.M.; Castello, P.R.; Gonzalez Flecha, F.L. Thermal stability of the plasma membrane calcium pump. Quantitative analysis of its dependence on lipid-protein interactions. J. Membr. Biol. 2000, 173, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Maeda, R.; Sato, T.; Okamoto, K.; Yanagawa, M.; Sako, Y. Lipid-Protein Interplay in Dimerization of Juxtamembrane Domains of Epidermal Growth Factor Receptor. Biophys. J. 2018, 114, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Manas, J.M.; Lakey, J.H.; Pattus, F. Brominated phospholipids as a tool for monitoring the membrane insertion of colicin A. Biochemistry 1992, 31, 7294–7300. [Google Scholar] [CrossRef] [PubMed]
- East, J.M.; Lee, A.G. Lipid selectivity of the calcium and magnesium ion dependent adenosinetriphosphatase, studied with fluorescence quenching by a brominated phospholipid. Biochemistry 1982, 21, 4144–4151. [Google Scholar] [CrossRef] [PubMed]
- Powl, A.M.; Carney, J.; Marius, P.; East, J.M.; Lee, A.G. Lipid interactions with bacterial channels: Fluorescence studies. Biochem. Soc. Trans. 2005, 33, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Sligar, S.G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 481–486. [Google Scholar] [CrossRef]
- Schuler, M.A.; Denisov, I.G.; Sligar, S.G. Nanodiscs as a new tool to examine lipid-protein interactions. Methods Mol. Biol. 2013, 974, 415–433. [Google Scholar]
- Bayburt, T.H.; Sligar, S.G. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Segrest, J.P.; Chung, B.H.; Chiovetti, R., Jr.; Weinstein, J.N. High-density lipoprotein recombinants: Evidence for a bicycle tire micelle structure obtained by neutron scattering and electron microscopy. FEBS Lett. 1979, 104, 231–235. [Google Scholar] [CrossRef]
- Zoghbi, M.E.; Altenberg, G.A. Membrane protein reconstitution in nanodiscs for luminescence spectroscopy studies. Nanotechnol. Rev. 2017, 6. [Google Scholar] [CrossRef]
- Civjan, N.R.; Bayburt, T.H.; Schuler, M.A.; Sligar, S.G. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. Biotechniques 2003, 35, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Cao, E.; Julius, D.; Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, E.E.; Kapanidis, A.N.; Tucker, S.J. Solution-Based Single-Molecule FRET Studies of K(+) Channel Gating in a Lipid Bilayer. Biophys. J. 2016, 110, 2663–2670. [Google Scholar] [CrossRef]
- Laursen, T.; Singha, A.; Rantzau, N.; Tutkus, M.; Borch, J.; Hedegard, P.; Stamou, D.; Moller, B.L.; Hatzakis, N.S. Single molecule activity measurements of cytochrome P450 oxidoreductase reveal the existence of two discrete functional states. Acs Chem. Biol. 2014, 9, 630–634. [Google Scholar] [CrossRef]
- Borch, J.; Roepstorff, P.; Moller-Jensen, J. Nanodisc-based co-immunoprecipitation for mass spectrometric identification of membrane-interacting proteins. Mol. Cell. Proteom. 2011, 10, O110 006775. [Google Scholar] [CrossRef]
- Borch, J.; Torta, F.; Sligar, S.G.; Roepstorff, P. Nanodiscs for immobilization of lipid bilayers and membrane receptors: Kinetic analysis of cholera toxin binding to a glycolipid receptor. Anal. Chem. 2008, 80, 6245–6252. [Google Scholar] [CrossRef]
- Marty, M.T.; Das, A.; Sligar, S.G. Ultra-thin layer MALDI mass spectrometry of membrane proteins in nanodiscs. Anal. Bioanal. Chem. 2012, 402, 721–729. [Google Scholar] [CrossRef]
- Knapp, E.W.; Fischer, S.F.; Zinth, W.; Sander, M.; Kaiser, W.; Deisenhofer, J.; Michel, H. Analysis of optical spectra from single crystals of Rhodopseudomonas viridis reaction centers. Proc. Natl. Acad. Sci. USA 1985, 82, 8463–8467. [Google Scholar] [CrossRef] [PubMed]
- Hunte, C.; Richers, S. Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 2008, 18, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Yeagle, P.L. Non-covalent binding of membrane lipids to membrane proteins. Biochim. Biophys. Acta 2014, 1838, 1548–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonen, T.; Cheng, Y.; Sliz, P.; Hiroaki, Y.; Fujiyoshi, Y.; Harrison, S.C.; Walz, T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature 2005, 438, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Norimatsu, Y.; Hasegawa, K.; Shimizu, N.; Toyoshima, C. Protein-phospholipid interplay revealed with crystals of a calcium pump. Nature 2017, 545, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Cryst. F Struct. Biol. Commun. 2015, 71, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.; Round, E.; Gushchin, I.; Polovinkin, V.; Balandin, T.; Kuzmichev, P.; Shevchenko, V.; Borshchevskiy, V.; Kuklin, A.; Round, A.; et al. Integral Membrane Proteins Can Be Crystallized Directly from Nanodiscs. Cryst. Growth Des. 2017, 17, 945–948. [Google Scholar] [CrossRef] [Green Version]
- Broecker, J.; Eger, B.T.; Ernst, O.P. Crystallogenesis of Membrane Proteins Mediated by Polymer-Bounded Lipid Nanodiscs. Structure 2017, 25, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R. Biomolecular solid state NMR: Advances in structural methodology and applications to peptide and protein fibrils. Annu. Rev. Phys. Chem. 2001, 52, 575–606. [Google Scholar] [CrossRef]
- Das, N.; Murray, D.T.; Cross, T.A. Lipid bilayer preparations of membrane proteins for oriented and magic-angle spinning solid-state NMR samples. Nat. Protoc. 2013, 8, 2256–2270. [Google Scholar] [CrossRef]
- Liang, B.; Tamm, L.K. NMR as a tool to investigate the structure, dynamics and function of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huster, D. Solid-state NMR spectroscopy to study protein-lipid interactions. Biochim. Biophys. Acta 2014, 1841, 1146–1160. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Wang, J.W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, A.; Merk, A.; Banerjee, S.; Matthies, D.; Wu, X.; Milne, J.L.; Subramaniam, S. 2.2 A resolution cryo-EM structure of beta-galactosidase in complex with a cell-permeant inhibitor. Science 2015, 348, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Mio, K.; Sato, C. Lipid environment of membrane proteins in cryo-EM based structural analysis. Biophys. Rev. 2018, 10, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.T.; Hoi, K.K.; Gault, J.; Robinson, C.V. Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs. Angew. Chem. Weinh. Bergstr. Ger. 2016, 128, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Bolla, J.R.; Agasid, M.T.; Mehmood, S.; Robinson, C.V. Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu. Rev. Biochem. 2019, 88, 85–111. [Google Scholar] [CrossRef]
- Gupta, K.; Li, J.; Liko, I.; Gault, J.; Bechara, C.; Wu, D.; Hopper, J.T.S.; Giles, K.; Benesch, J.L.P.; Robinson, C.V. Identifying key membrane protein lipid interactions using mass spectrometry. Nat. Protoc. 2018, 13, 1106–1120. [Google Scholar] [CrossRef]
- Lindahl, E.; Sansom, M.S. Membrane proteins: Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [Google Scholar] [CrossRef]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stansfeld, P.J.; Sansom, M.S. From Coarse Grained to Atomistic: A Serial Multiscale Approach to Membrane Protein Simulations. J. Chem. Theory Comput. 2011, 7, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Pluhackova, K.; Bockmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayton, G.S.; Noid, W.G.; Voth, G.A. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Awoonor-Williams, E.; Rowley, C.N. Molecular simulation of nonfacilitated membrane permeation. Biochim. Biophys. Acta 2016, 1858, 1672–1687. [Google Scholar] [CrossRef]
- Pliotas, C.; Dahl, A.C.; Rasmussen, T.; Mahendran, K.R.; Smith, T.K.; Marius, P.; Gault, J.; Banda, T.; Rasmussen, A.; Miller, S.; et al. The role of lipids in mechanosensation. Nat. Struct. Mol. Biol. 2015, 22, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.A.; Bond, P.J.; Ivetac, A.; Chetwynd, A.P.; Khalid, S.; Sansom, M.S. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 2008, 16, 621–630. [Google Scholar] [CrossRef]
- Wolf, M.G.; Hoefling, M.; Aponte-Santamaria, C.; Grubmuller, H.; Groenhof, G. g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 2010, 31, 2169–2174. [Google Scholar] [CrossRef] [Green Version]
- Fosso-Tande, J.; Black, C.G.; Aller, S.; Lu, L.; Lu, L.D.; Hills, R., Jr. Simulation of lipid-protein interactions with the CgProt force field. Aims Mol. Sci. 2017, 4, 352–369. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Ingolfsson, H.I.; Bockmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Koldso, H.; Shorthouse, D.; Helie, J.; Sansom, M.S. Lipid clustering correlates with membrane curvature as revealed by molecular simulations of complex lipid bilayers. PLoS Comput. Biol. 2014, 10, e1003911. [Google Scholar] [CrossRef] [PubMed]
- Ingolfsson, H.I.; Melo, M.N.; van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; de Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Lyman, E.; Hsieh, C.L.; Eggeling, C. From Dynamics to Membrane Organization: Experimental Breakthroughs Occasion a “Modeling Manifesto”. Biophys. J. 2018, 115, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.R.; Stansfeld, P.J.; Tucker, S.J.; Sansom, M.S. Simulation-based prediction of phosphatidylinositol 4,5-bisphosphate binding to an ion channel. Biochemistry 2013, 52, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Haas, E.; Stanley, D.W. Phospholipases. Xpharm: Compr. Pharmacol. Ref. 2007, 1–3. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Owsianik, G.; Nilius, B. TRP channels: An overview. Cell Calcium 2005, 38, 233–252. [Google Scholar] [CrossRef]
- Islam, M.S. TRP channels of islets. Adv. Exp. Med. Biol. 2011, 704, 811–830. [Google Scholar]
- Zeng, F.; Xu, S.Z.; Jackson, P.K.; McHugh, D.; Kumar, B.; Fountain, S.J.; Beech, D.J. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J. Physiol. 2004, 559, 739–750. [Google Scholar] [CrossRef]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 297–328. [Google Scholar] [CrossRef]
- Cui, M.; Honore, P.; Zhong, C.; Gauvin, D.; Mikusa, J.; Hernandez, G.; Chandran, P.; Gomtsyan, A.; Brown, B.; Bayburt, E.K.; et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J. Neurosci. 2006, 26, 9385–9393. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B. TRP channels in disease. Biochim. Biophys. Acta 2007, 1772, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Diaz, N.; Angelopoulos, I.; Cerda, O. A TR(i)P to Cell Migration: New Roles of TRP Channels in Mechanotransduction and Cancer. Front. Physiol. 2019, 10, 757. [Google Scholar] [CrossRef]
- Petho, Z.; Najder, K.; Bulk, E.; Schwab, A. Mechanosensitive ion channels push cancer progression. Cell Calcium 2019, 80, 79–90. [Google Scholar] [CrossRef]
- Levine, J.D.; Alessandri-Haber, N. TRP channels: Targets for the relief of pain. Biochim. Biophys. Acta 2007, 1772, 989–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szallasi, A.; Sheta, M. Targeting TRPV1 for pain relief: Limits, losers and laurels. Expert Opin. Investig. Drugs 2012, 21, 1351–1369. [Google Scholar] [CrossRef]
- Rohacs, T. Phosphoinositide regulation of TRP channels. Handb. Exp. Pharm. 2014, 223, 1143–1176. [Google Scholar]
- Otsuguro, K.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.X.; Zholos, A.V. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef]
- Doerner, J.F.; Hatt, H.; Ramsey, I.S. Voltage- and temperature-dependent activation of TRPV3 channels is potentiated by receptor-mediated PI(4,5)P2 hydrolysis. J. Gen. Physiol. 2011, 137, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trebak, M.; Lemonnier, L.; DeHaven, W.I.; Wedel, B.J.; Bird, G.S.; Putney, J.W., Jr. Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflug. Arch. 2009, 457, 757–769. [Google Scholar] [CrossRef]
- Cao, E.; Cordero-Morales, J.F.; Liu, B.; Qin, F.; Julius, D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 2013, 77, 667–679. [Google Scholar] [CrossRef]
- Ufret-Vincenty, C.A.; Klein, R.M.; Collins, M.D.; Rosasco, M.G.; Martinez, G.Q.; Gordon, S.E. Mechanism for phosphoinositide selectivity and activation of TRPV1 ion channels. J. Gen. Physiol. 2015, 145, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Ufret-Vincenty, C.A.; Klein, R.M.; Hua, L.; Angueyra, J.; Gordon, S.E. Localization of the PIP2 sensor of TRPV1 ion channels. J. Biol. Chem. 2011, 286, 9688–9698. [Google Scholar] [CrossRef]
- Holendova, B.; Grycova, L.; Jirku, M.; Teisinger, J. PtdIns(4,5)P2 interacts with CaM binding domains on TRPM3 N-terminus. Channels (Austin) 2012, 6, 479–482. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Hofmann, T.; Montell, C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol. Cell 2007, 25, 491–503. [Google Scholar] [CrossRef]
- Basora, N.; Boulay, G.; Bilodeau, L.; Rousseau, E.; Payet, M.D. 20-hydroxyeicosatetraenoic acid (20-HETE) activates mouse TRPC6 channels expressed in HEK293 cells. J. Biol. Chem. 2003, 278, 31709–31716. [Google Scholar] [CrossRef]
- Bandell, M.; Story, G.M.; Hwang, S.W.; Viswanath, V.; Eid, S.R.; Petrus, M.J.; Earley, T.J.; Patapoutian, A. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 2004, 41, 849–857. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Andersson, D.A.; Nash, M.; Bevan, S. Modulation of the cold-activated channel TRPM8 by lysophospholipids and polyunsaturated fatty acids. J. Neurosci. 2007, 27, 3347–3355. [Google Scholar] [CrossRef]
- Flemming, P.K.; Dedman, A.M.; Xu, S.Z.; Li, J.; Zeng, F.; Naylor, J.; Benham, C.D.; Bateson, A.N.; Muraki, K.; Beech, D.J. Sensing of lysophospholipids by TRPC5 calcium channel. J. Biol. Chem. 2006, 281, 4977–4982. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Di Marzo, V. Role of endocannabinoids and endovanilloids in Ca2+ signalling. Cell Calcium 2009, 45, 611–624. [Google Scholar] [CrossRef]
- Beech, D.J. Integration of transient receptor potential canonical channels with lipids. Acta Physiol. (Oxf.) 2012, 204, 227–237. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Colles, S.M.; Bhat, M.; Van Wagoner, D.R.; Birnbaumer, L.; Graham, L.M. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol. Biol. Cell 2008, 19, 3203–3211. [Google Scholar] [CrossRef]
- Monet, M.; Gkika, D.; Lehen’kyi, V.; Pourtier, A.; Vanden Abeele, F.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarsakaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta 2009, 1793, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Posadas, A.; Picazo-Juarez, G.; Llorente, I.; Jara-Oseguera, A.; Morales-Lazaro, S.; Escalante-Alcalde, D.; Islas, L.D.; Rosenbaum, T. Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat. Chem. Biol. 2011, 8, 78–85. [Google Scholar] [CrossRef]
- Runnels, L.W.; Yue, L.; Clapham, D.E. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat. Cell Biol. 2002, 4, 329–336. [Google Scholar] [CrossRef]
- Lucas, P.; Ukhanov, K.; Leinders-Zufall, T.; Zufall, F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: Mechanism of pheromone transduction. Neuron 2003, 40, 551–561. [Google Scholar] [CrossRef]
- Startek, J.B.; Boonen, B.; Talavera, K.; Meseguer, V. TRP Channels as Sensors of Chemically-Induced Changes in Cell Membrane Mechanical Properties. Int. J. Mol. Sci. 2019, 20, 371. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Albert, A.P.; Large, W.A. Synergism between inositol phosphates and diacylglycerol on native TRPC6-like channels in rabbit portal vein myocytes. J. Physiol. 2003, 552, 789–795. [Google Scholar] [CrossRef]
- Ahmmed, G.U.; Mehta, D.; Vogel, S.; Holinstat, M.; Paria, B.C.; Tiruppathi, C.; Malik, A.B. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J. Biol. Chem. 2004, 279, 20941–20949. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Zheng, F.; Gill, D.L. Regulation of canonical transient receptor potential (TRPC) channel function by diacylglycerol and protein kinase C. J. Biol. Chem. 2003, 278, 29031–29040. [Google Scholar] [CrossRef]
- Woo, D.H.; Jung, S.J.; Zhu, M.H.; Park, C.K.; Kim, Y.H.; Oh, S.B.; Lee, C.J. Direct activation of transient receptor potential vanilloid 1(TRPV1) by diacylglycerol (DAG). Mol. Pain 2008, 4, 42. [Google Scholar] [CrossRef]
- Ciardo, M.G.; Ferrer-Montiel, A. Lipids as central modulators of sensory TRP channels. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1615–1628. [Google Scholar] [CrossRef]
- Grimm, C.; Kraft, R.; Schultz, G.; Harteneck, C. Activation of the melastatin-related cation channel TRPM3 by D-erythro-sphingosine [corrected]. Mol. Pharm. 2005, 67, 798–805. [Google Scholar] [CrossRef]
- Hoffmann, A.; Grimm, C.; Kraft, R.; Goldbaum, O.; Wrede, A.; Nolte, C.; Hanisch, U.K.; Richter-Landsberg, C.; Bruck, W.; Kettenmann, H.; et al. TRPM3 is expressed in sphingosine-responsive myelinating oligodendrocytes. J. Neurochem. 2010, 114, 654–665. [Google Scholar] [CrossRef]
- Saghy, E.; Szoke, E.; Payrits, M.; Helyes, Z.; Borzsei, R.; Erostyak, J.; Janosi, T.Z.; Setalo, G., Jr.; Szolcsanyi, J. Evidence for the role of lipid rafts and sphingomyelin in Ca2+-gating of Transient Receptor Potential channels in trigeminal sensory neurons and peripheral nerve terminals. Pharm. Res. 2015, 100, 101–116. [Google Scholar] [CrossRef]
- Xu, S.Z.; Muraki, K.; Zeng, F.; Li, J.; Sukumar, P.; Shah, S.; Dedman, A.M.; Flemming, P.K.; McHugh, D.; Naylor, J.; et al. A sphingosine-1-phosphate-activated calcium channel controlling vascular smooth muscle cell motility. Circ. Res. 2006, 98, 1381–1389. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.; McNamara, J.O.; Williams, S.M. Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef]
- Iftinca, M.C.; Zamponi, G.W. Regulation of neuronal T-type calcium channels. Trends Pharm. Sci. 2009, 30, 32–40. [Google Scholar] [CrossRef]
- Langton, P.D. Calcium channel currents recorded from isolated myocytes of rat basilar artery are stretch sensitive. J. Physiol. 1993, 471, 1–11. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.C.; Li, Y.; Son, M.J.; Woo, S.H. Fluid pressure modulates L-type Ca2+ channel via enhancement of Ca2+-induced Ca2+ release in rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C966–C976. [Google Scholar] [CrossRef]
- Iribe, G.; Ward, C.W.; Camelliti, P.; Bollensdorff, C.; Mason, F.; Burton, R.A.; Garny, A.; Morphew, M.K.; Hoenger, A.; Lederer, W.J.; et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ. Res. 2009, 104, 787–795. [Google Scholar] [CrossRef]
- Bidaud, I.; Mezghrani, A.; Swayne, L.A.; Monteil, A.; Lory, P. Voltage-gated calcium channels in genetic diseases. Biochim. Biophys. Acta 2006, 1763, 1169–1174. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharm. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Kakimoto, Y.; Toda, K.; Naruse, K. Mechanobiology in cardiac physiology and diseases. J. Cell. Mol. Med. 2013, 17, 225–232. [Google Scholar] [CrossRef]
- Wu, L.; Bauer, C.S.; Zhen, X.G.; Xie, C.; Yang, J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 2002, 419, 947–952. [Google Scholar] [CrossRef]
- Suh, B.C.; Leal, K.; Hille, B. Modulation of high-voltage activated Ca(2+) channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron 2010, 67, 224–238. [Google Scholar] [CrossRef]
- de la Cruz, L.; Puente, E.I.; Reyes-Vaca, A.; Arenas, I.; Garduno, J.; Bravo-Martinez, J.; Garcia, D.E. PIP2 in pancreatic beta-cells regulates voltage-gated calcium channels by a voltage-independent pathway. Am. J. Physiol. Cell Physiol. 2016, 311, C630–C640. [Google Scholar] [CrossRef]
- Xie, B.; Nguyen, P.M.; Gucek, A.; Thonig, A.; Barg, S.; Idevall-Hagren, O. Plasma Membrane Phosphatidylinositol 4,5-Bisphosphate Regulates Ca(2+)-Influx and Insulin Secretion from Pancreatic beta Cells. Cell Chem. Biol. 2016, 23, 816–826. [Google Scholar] [CrossRef]
- Park, C.G.; Park, Y.; Suh, B.C. The HOOK region of voltage-gated Ca2+ channel beta subunits senses and transmits PIP2 signals to the gate. J. Gen. Physiol. 2017, 149, 261–276. [Google Scholar] [CrossRef]
- Rodriguez-Menchaca, A.A.; Adney, S.K.; Zhou, L.; Logothetis, D.E. Dual Regulation of Voltage-Sensitive Ion Channels by PIP(2). Front. Pharm. 2012, 3, 170. [Google Scholar] [CrossRef]
- Liu, L.; Barrett, C.F.; Rittenhouse, A.R. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am. J. Physiol. Cell Physiol. 2001, 280, C1293–C1305. [Google Scholar] [CrossRef]
- Barrett, C.F.; Liu, L.; Rittenhouse, A.R. Arachidonic acid reversibly enhances N-type calcium current at an extracellular site. Am. J. Physiol. Cell Physiol. 2001, 280, C1306–C1318. [Google Scholar] [CrossRef]
- Yang, L.; Andrews, D.A.; Low, P.S. Lysophosphatidic acid opens a Ca(++) channel in human erythrocytes. Blood 2000, 95, 2420–2425. [Google Scholar]
- Kaestner, L.; Steffen, P.; Nguyen, D.B.; Wang, J.; Wagner-Britz, L.; Jung, A.; Wagner, C.; Bernhardt, I. Lysophosphatidic acid induced red blood cell aggregation in vitro. Bioelectrochemistry 2012, 87, 89–95. [Google Scholar] [CrossRef]
- Combs, D.J.; Shin, H.G.; Xu, Y.; Ramu, Y.; Lu, Z. Tuning voltage-gated channel activity and cellular excitability with a sphingomyelinase. J. Gen. Physiol. 2013, 142, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Tornquist, K.; Blom, T.; Shariatmadari, R.; Pasternack, M. Ceramide 1-phosphate enhances calcium entry through voltage-operated calcium channels by a protein kinase C-dependent mechanism in GH4C1 rat pituitary cells. Biochem. J. 2004, 380, 661–668. [Google Scholar] [CrossRef]
- Arnadottir, J.; Chalfie, M. Eukaryotic mechanosensitive channels. Annu. Rev. Biophys. 2010, 39, 111–137. [Google Scholar] [CrossRef]
- Edwards, M.D.; Booth, I.R.; Miller, S. Gating the bacterial mechanosensitive channels: MscS a new paradigm? Curr. Opin. Microbiol. 2004, 7, 163–167. [Google Scholar] [CrossRef]
- Stokes, N.R.; Murray, H.D.; Subramaniam, C.; Gourse, R.L.; Louis, P.; Bartlett, W.; Miller, S.; Booth, I.R. A role for mechanosensitive channels in survival of stationary phase: Regulation of channel expression by RpoS. Proc. Natl. Acad. Sci. USA 2003, 100, 15959–15964. [Google Scholar] [CrossRef] [Green Version]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Saotome, K.; Murthy, S.E.; Kefauver, J.M.; Whitwam, T.; Patapoutian, A.; Ward, A.B. Structure of the mechanically activated ion channel Piezo1. Nature 2017, 554, 481–486. [Google Scholar] [CrossRef]
- Parpaite, T.; Coste, B. Piezo channels. Curr. Biol. Cb. 2017, 27, R250–R252. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Kuebler, W.M. Mechanotransduction by TRP channels: General concepts and specific role in the vasculature. Cell Biochem. Biophys. 2010, 56, 1–18. [Google Scholar] [CrossRef]
- Patel, A.; Sharif-Naeini, R.; Folgering, J.R.; Bichet, D.; Duprat, F.; Honore, E. Canonical TRP channels and mechanotransduction: From physiology to disease states. Pflug. Arch. 2010, 460, 571–581. [Google Scholar] [CrossRef]
- Albuisson, J.; Murthy, S.E.; Bandell, M.; Coste, B.; Louis-Dit-Picard, H.; Mathur, J.; Feneant-Thibault, M.; Tertian, G.; de Jaureguiberry, J.P.; Syfuss, P.Y.; et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 2013, 4, 1884. [Google Scholar] [CrossRef]
- Coste, B.; Houge, G.; Murray, M.F.; Stitziel, N.; Bandell, M.; Giovanni, M.A.; Philippakis, A.; Hoischen, A.; Riemer, G.; Steen, U.; et al. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc. Natl. Acad. Sci. USA 2013, 110, 4667–4672. [Google Scholar] [CrossRef] [Green Version]
- Borbiro, I.; Badheka, D.; Rohacs, T. Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci. Signal 2015, 8, ra15. [Google Scholar] [CrossRef]
- Rohacs, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Regul. 2013, 53, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Powl, A.M.; East, J.M.; Lee, A.G. Anionic phospholipids affect the rate and extent of flux through the mechanosensitive channel of large conductance MscL. Biochemistry 2008, 47, 4317–4328. [Google Scholar] [CrossRef]
- Chemin, J.; Patel, A.J.; Duprat, F.; Sachs, F.; Lazdunski, M.; Honore, E. Up- and down-regulation of the mechano-gated K(2P) channel TREK-1 by PIP (2) and other membrane phospholipids. Pflug. Arch. 2007, 455, 97–103. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Hara, Y.; Okuda, M.; Itoh, K.; Nishioka, R.; Shiomi, A.; Nagao, K.; Mori, M.; Mori, Y.; Ikenouchi, J.; et al. Cell surface flip-flop of phosphatidylserine is critical for PIEZO1-mediated myotube formation. Nat. Commun. 2018, 9, 2049. [Google Scholar] [CrossRef]
- Petersen, O.H.; Tepikin, A.V. Polarized calcium signaling in exocrine gland cells. Annu. Rev. Physiol. 2008, 70, 273–299. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Delmotte, P.; Bai, Y.; Perez-Zogbhi, J.F. Regulation of airway smooth muscle cell contractility by Ca2+ signaling and sensitivity. Proc. Am. Thorac. Soc. 2008, 5, 23–31. [Google Scholar] [CrossRef]
- Feske, S.; Giltnane, J.; Dolmetsch, R.; Staudt, L.M.; Rao, A. Gene regulation mediated by calcium signals in T lymphocytes. Nat. Immunol. 2001, 2, 316–324. [Google Scholar] [CrossRef]
- Malcuit, C.; Kurokawa, M.; Fissore, R.A. Calcium oscillations and mammalian egg activation. J. Cell. Physiol. 2006, 206, 565–573. [Google Scholar] [CrossRef]
- Sutko, J.L.; Airey, J.A.; Welch, W.; Ruest, L. The pharmacology of ryanodine and related compounds. Pharm. Rev. 1997, 49, 53–98. [Google Scholar]
- Galvan, D.L.; Borrego-Diaz, E.; Perez, P.J.; Mignery, G.A. Subunit oligomerization, and topology of the inositol 1,4, 5-trisphosphate receptor. J. Biol. Chem. 1999, 274, 29483–29492. [Google Scholar] [CrossRef]
- Fleischer, S. Personal recollections on the discovery of the ryanodine receptors of muscle. Biochem. Biophys. Res. Commun. 2008, 369, 195–207. [Google Scholar] [CrossRef]
- Ramos-Franco, J.; Galvan, D.; Mignery, G.A.; Fill, M. Location of the permeation pathway in the recombinant type 1 inositol 1,4,5-trisphosphate receptor. J. Gen. Physiol. 1999, 114, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; West, D.J.; Sitsapesan, R. Light at the end of the Ca(2+)-release channel tunnel: Structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q. Rev. Biophys. 2001, 34, 61–104. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Imai, S. Structure and function of inositol 1,4,5-trisphosphate receptor. Jpn. J. Pharm. 1997, 74, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Yang, D.; Lan, X.; Li, K.; Li, X.; Chen, J.; Zhang, Y.; Xiao, R.P.; Han, Q.; Cheng, H. Nuclear Ca2+ sparks and waves mediated by inositol 1,4,5-trisphosphate receptors in neonatal rat cardiomyocytes. Cell Calcium 2008, 43, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ljubojevic, S.; Bers, D.M. Nuclear calcium in cardiac myocytes. J. Cardiovasc. Pharm. 2015, 65, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Jaggar, J.H.; Stevenson, A.S.; Nelson, M.T. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am. J. Physiol. 1998, 274, C1755–C1761. [Google Scholar] [CrossRef]
- Durham, W.J.; Wehrens, X.H.; Sood, S.; Hamilton, S.L. Calcium Signalling and Disease; Carafoli, E., Brini, M., Eds.; Diseases associated with altered ryanodine receptor activity series; Springer, Dordrecht: Berlin, Germany, 2007; Volume 45, pp. 273–321. [Google Scholar]
- Bezprozvanny, I. Calcium Signalling and Disease; Carafoli, E., Brini, M., Eds.; Inositol 1,4,5-tripshosphate receptor, calcium signalling and Huntington’s disease series; Springer, Dordrecht: Berlin, Germany, 2007; Volume 45, pp. 323–335. [Google Scholar]
- Vanderheyden, V.; Devogelaere, B.; Missiaen, L.; De Smedt, H.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim. Biophys. Acta 2009, 1793, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Yule, D.I.; Betzenhauser, M.J.; Joseph, S.K. Linking structure to function: Recent lessons from inositol 1,4,5-trisphosphate receptor mutagenesis. Cell Calcium 2010, 47, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Choe, C.U.; Ehrlich, B.E. The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: Sometimes good and sometimes bad teamwork. Sci. Stke 2006, 2006, re15. [Google Scholar] [CrossRef] [PubMed]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef]
- Chu, A.; Stefani, E. Phosphatidylinositol 4,5-bisphosphate-induced Ca2+ release from skeletal muscle sarcoplasmic reticulum terminal cisternal membranes. Ca2+ flux and single channel studies. J. Biol. Chem. 1991, 266, 7699–7705. [Google Scholar] [PubMed]
- Lupu, V.D.; Kaznacheyeva, E.; Krishna, U.M.; Falck, J.R.; Bezprozvanny, I. Functional coupling of phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 1998, 273, 14067–14070. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. Cb. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Cahalan, M.D. STIMulating store-operated Ca(2+) entry. Nat. Cell Biol. 2009, 11, 669–677. [Google Scholar] [CrossRef]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca(2+) signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef]
- Smyth, J.T.; Dehaven, W.I.; Bird, G.S.; Putney, J.W., Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J. Cell Sci. 2008, 121, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Meraner, P.; Kwon, H.T.; Machnes, D.; Oh-hora, M.; Zimmer, J.; Huang, Y.; Stura, A.; Rao, A.; Hogan, P.G. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat. Struct. Mol. Biol. 2010, 17, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, X.; Mancarella, S.; Hendron, E.; Eguchi, S.; Soboloff, J.; Tang, X.D.; Gill, D.L. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 2010, 330, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.M.; Chvanov, M.; Haynes, L.P.; Petersen, O.H.; Tepikin, A.V.; Burgoyne, R.D. Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 2009, 425, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef]
- Bolotina, V.M. Orai, STIM1 and iPLA2beta: A view from a different perspective. J. Physiol. 2008, 586, 3035–3042. [Google Scholar] [CrossRef]
- Smani, T.; Zakharov, S.I.; Csutora, P.; Leno, E.; Trepakova, E.S.; Bolotina, V.M. A novel mechanism for the store-operated calcium influx pathway. Nat. Cell Biol. 2004, 6, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Combs, D.J.; Lu, Z. Sphingomyelinase D inhibits store-operated Ca2+ entry in T lymphocytes by suppressing ORAI current. J. Gen. Physiol. 2015, 146, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colina, C.; Flores, A.; Rojas, H.; Acosta, A.; Castillo, C.; Garrido Mdel, R.; Israel, A.; DiPolo, R.; Benaim, G. Ceramide increase cytoplasmic Ca2+ concentration in Jurkat T cells by liberation of calcium from intracellular stores and activation of a store-operated calcium channel. Arch. Biochem. Biophys. 2005, 436, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.; Dominguez, L.; Bohorquez-Hernandez, A.; Asanov, A.; Vaca, L. A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep. 2016, 6, 29634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lackner, B.; Absolonova, M.; et al. Cholesterol modulates Orai1 channel function. Sci. Signal 2016, 9, ra10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Naik, J.S.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reduced membrane cholesterol after chronic hypoxia limits Orai1-mediated pulmonary endothelial Ca(2+) entry. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H359–H369. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lytton, J. The cation/Ca(2+) exchanger superfamily: Phylogenetic analysis and structural implications. Mol. Biol. Evol. 2004, 21, 1692–1703. [Google Scholar] [CrossRef]
- Besserer, G.M.; Ottolia, M.; Nicoll, D.A.; Chaptal, V.; Cascio, D.; Philipson, K.D.; Abramson, J. The second Ca2+-binding domain of the Na+ Ca2+ exchanger is essential for regulation: Crystal structures and mutational analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 18467–18472. [Google Scholar] [CrossRef]
- Brini, M.; Di Leva, F.; Ortega, C.K.; Domi, T.; Ottolini, D.; Leonardi, E.; Tosatto, S.C.; Carafoli, E. Deletions and mutations in the acidic lipid-binding region of the plasma membrane Ca2+ pump: A study on different splicing variants of isoform 2. J. Biol. Chem. 2010, 285, 30779–30791. [Google Scholar] [CrossRef]
- Carafoli, E.; Santella, L.; Branca, D.; Brini, M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 107–260. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P.; Yeh, C.H.; Sensi, S.L.; Gwag, B.J.; Canzoniero, L.M.; Farhangrazi, Z.S.; Ying, H.S.; Tian, M.; Dugan, L.L.; Choi, D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997, 278, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Hilgemann, D.W.; Ball, R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 1996, 273, 956–959. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Feng, S.; Tong, Q.; Hilgemann, D.W.; Philipson, K.D. Interaction of PIP(2) with the XIP region of the cardiac Na/Ca exchanger. Am. J. Physiol. Cell Physiol. 2000, 278, C661–C666. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.P.; Condrescu, M. Ionic regulation of the cardiac sodium-calcium exchanger. Channels (Austin) 2008, 2, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Yaradanakul, A.; Feng, S.; Shen, C.; Lariccia, V.; Lin, M.J.; Yang, J.; Kang, T.M.; Dong, P.; Yin, H.L.; Albanesi, J.P.; et al. Dual control of cardiac Na+ Ca2+ exchange by PIP(2): Electrophysiological analysis of direct and indirect mechanisms. J. Physiol. 2007, 582, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, R.; Philipson, K.D. Phospholipid composition modulates the Na+-Ca2+ exchange activity of cardiac sarcolemma in reconstituted vesicles. Biochim. Biophys. Acta 1988, 937, 258–268. [Google Scholar] [CrossRef]
- Denning, E.J.; Beckstein, O. Influence of lipids on protein-mediated transmembrane transport. Chem. Phys. Lipids 2013, 169, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Condrescu, M.; Reeves, J.P. Inhibition of sodium-calcium exchange by ceramide and sphingosine. J. Biol. Chem. 2001, 276, 4046–4054. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Nuclear sphingolipids: Metabolism and signaling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef]
- Wu, G.; Xie, X.; Lu, Z.H.; Ledeen, R.W. Sodium-calcium exchanger complexed with GM1 ganglioside in nuclear membrane transfers calcium from nucleoplasm to endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2009, 106, 10829–10834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Lu, Z.H.; Wang, J.; Wang, Y.; Xie, X.; Meyenhofer, M.F.; Ledeen, R.W. Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: Protection with LIGA 20, a membrane-permeant analog of GM1. J. Neurosci. 2005, 25, 11014–11022. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Coletto, L.; Pinton, P.; Rizzuto, R.; Brini, M.; Carafoli, E. Inhibitory interaction of the 14-3-3 protein with isoform 4 of the plasma membrane Ca(2+)-ATPase pump. J. Biol. Chem. 2005, 280, 37195–37203. [Google Scholar] [CrossRef] [PubMed]
- Guerini, D.; Pan, B.; Carafoli, E. Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: Focus on calpain sensitivity. J. Biol. Chem. 2003, 278, 38141–38148. [Google Scholar] [CrossRef] [PubMed]
- Paszty, K.; Antalffy, G.; Hegedus, L.; Padanyi, R.; Penheiter, A.R.; Filoteo, A.G.; Penniston, J.T.; Enyedi, A. Cleavage of the plasma membrane Ca+ATPase during apoptosis. Ann. N. Y. Acad. Sci. 2007, 1099, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Schuh, K.; Cartwright, E.J.; Jankevics, E.; Bundschu, K.; Liebermann, J.; Williams, J.C.; Armesilla, A.L.; Emerson, M.; Oceandy, D.; Knobeloch, K.P.; et al. Plasma membrane Ca2+ ATPase 4 is required for sperm motility and male fertility. J. Biol. Chem. 2004, 279, 28220–28226. [Google Scholar] [CrossRef] [PubMed]
- Schuh, K.; Uldrijan, S.; Telkamp, M.; Rothlein, N.; Neyses, L. The plasmamembrane calmodulin-dependent calcium pump: A major regulator of nitric oxide synthase I. J. Cell Biol. 2001, 155, 201–205. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Filomatori, C.V.; Rega, A.F. On the mechanism of activation of the plasma membrane Ca2+-ATPase by ATP and acidic phospholipids. J. Biol. Chem. 2003, 278, 22265–22271. [Google Scholar] [CrossRef]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Filoteo, A.G.; Enyedi, A.; Penniston, J.T. The lipid-binding peptide from the plasma membrane Ca2+ pump binds calmodulin, and the primary calmodulin-binding domain interacts with lipid. J. Biol. Chem. 1992, 267, 11800–11805. [Google Scholar] [PubMed]
- Niggli, V.; Adunyah, E.S.; Penniston, J.T.; Carafoli, E. Purified (Ca2+-Mg2+)-ATPase of the erythrocyte membrane. Reconstitution and effect of calmodulin and phospholipids. J. Biol. Chem. 1981, 256, 395–401. [Google Scholar] [PubMed]
- Perez-Gordones, M.C.; Lugo, M.R.; Winkler, M.; Cervino, V.; Benaim, G. Diacylglycerol regulates the plasma membrane calcium pump from human erythrocytes by direct interaction. Arch. Biochem. Biophys. 2009, 489, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Bechtel, M.D.; Bean, J.L.; Winefield, R.; Williams, T.D.; Zaidi, A.; Michaelis, E.K.; Michaelis, M.L. Effects of gangliosides on the activity of the plasma membrane Ca2+-ATPase. Biochim. Biophys. Acta 2014, 1838, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Fan, X.; Yang, F.; Zhang, X. Gangliosides modulate the activity of the plasma membrane Ca(2+)-ATPase from porcine brain synaptosomes. Arch. Biochem. Biophys. 2004, 427, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Zhang, J.; Zhao, Y.; Yang, F.; Zhang, X. Ganglioside GM2 modulates the erythrocyte Ca2+-ATPase through its binding to the calmodulin-binding domain and its ’receptor’. Arch. Biochem. Biophys. 2006, 454, 155–159. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, Y.; Duan, J.; Yang, F.; Zhang, X. Gangliosides activate the phosphatase activity of the erythrocyte plasma membrane Ca2+-ATPase. Arch. Biochem. Biophys. 2005, 444, 1–6. [Google Scholar] [CrossRef]
- Colina, C.; Cervino, V.; Benaim, G. Ceramide and sphingosine have an antagonistic effect on the plasma-membrane Ca2+-ATPase from human erythrocytes. Biochem. J. 2002, 362, 247–251. [Google Scholar] [CrossRef]
- Guerrero-Hernandez, A.; Dagnino-Acosta, A.; Verkhratsky, A. An intelligent sarco-endoplasmic reticulum Ca2+ store: Release and leak channels have differential access to a concealed Ca2+ pool. Cell Calcium 2010, 48, 143–149. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Dalton, K.A.; East, J.M.; Mall, S.; Oliver, S.; Starling, A.P.; Lee, A.G. Interaction of phosphatidic acid and phosphatidylserine with the Ca2+-ATPase of sarcoplasmic reticulum and the mechanism of inhibition. Biochem. J. 1998, 329, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaim, G.; Pimentel, A.A.; Felibertt, P.; Mayora, A.; Colman, L.; Sojo, F.; Rojas, H.; De Sanctis, J.B. Sphingosine inhibits the sarco(endo)plasmic reticulum Ca(2+)-ATPase (SERCA) activity. Biochem. Biophys. Res. Commun. 2016, 473, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [PubMed]
- Brown, M.F.; Chawla, U.; Perera, S.M.D.C. Membrane Lipid-Protein Interactions. Biophys. Cell Membr. Springer Ser. Biophys. 2017, 19, 61–84. [Google Scholar]
- Hedger, G.; Sansom, M.S.P. Lipid interaction sites on channels, transporters and receptors: Recent insights from molecular dynamics simulations. Biochim. Biophys. Acta 2016, 1858, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Muallem, S.; Chung, W.Y.; Jha, A.; Ahuja, M. Lipids at membrane contact sites: Cell signaling and ion transport. Embo Rep. 2017, 18, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.C.; Hille, B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008, 37, 175–195. [Google Scholar] [CrossRef]
- Gamper, N.; Shapiro, M.S. Target-specific PIP(2) signalling: How might it work? J. Physiol. 2007, 582, 967–975. [Google Scholar] [CrossRef]
- Haag, M.; Schmidt, A.; Sachsenheimer, T.; Brugger, B. Quantification of Signaling Lipids by Nano-Electrospray Ionization Tandem Mass Spectrometry (Nano-ESI MS/MS). Metabolites 2012, 2, 57–76. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.M.; Contreras, F.X.; Brugger, B.; Wieland, F. Determinants of specificity at the protein-lipid interface in membranes. FEBS Lett. 2010, 584, 1713–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.G. Lipid-protein interactions. Biochem. Soc. Trans. 2011, 39, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marius, P.; de Planque, M.R.; Williamson, P.T. Probing the interaction of lipids with the non-annular binding sites of the potassium channel KcsA by magic-angle spinning NMR. Biochim. Biophys. Acta 2012, 1818, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Paila, Y.D.; Tiwari, S.; Chattopadhyay, A. Are specific nonannular cholesterol binding sites present in G-protein coupled receptors? Biochim. Biophys. Acta 2009, 1788, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
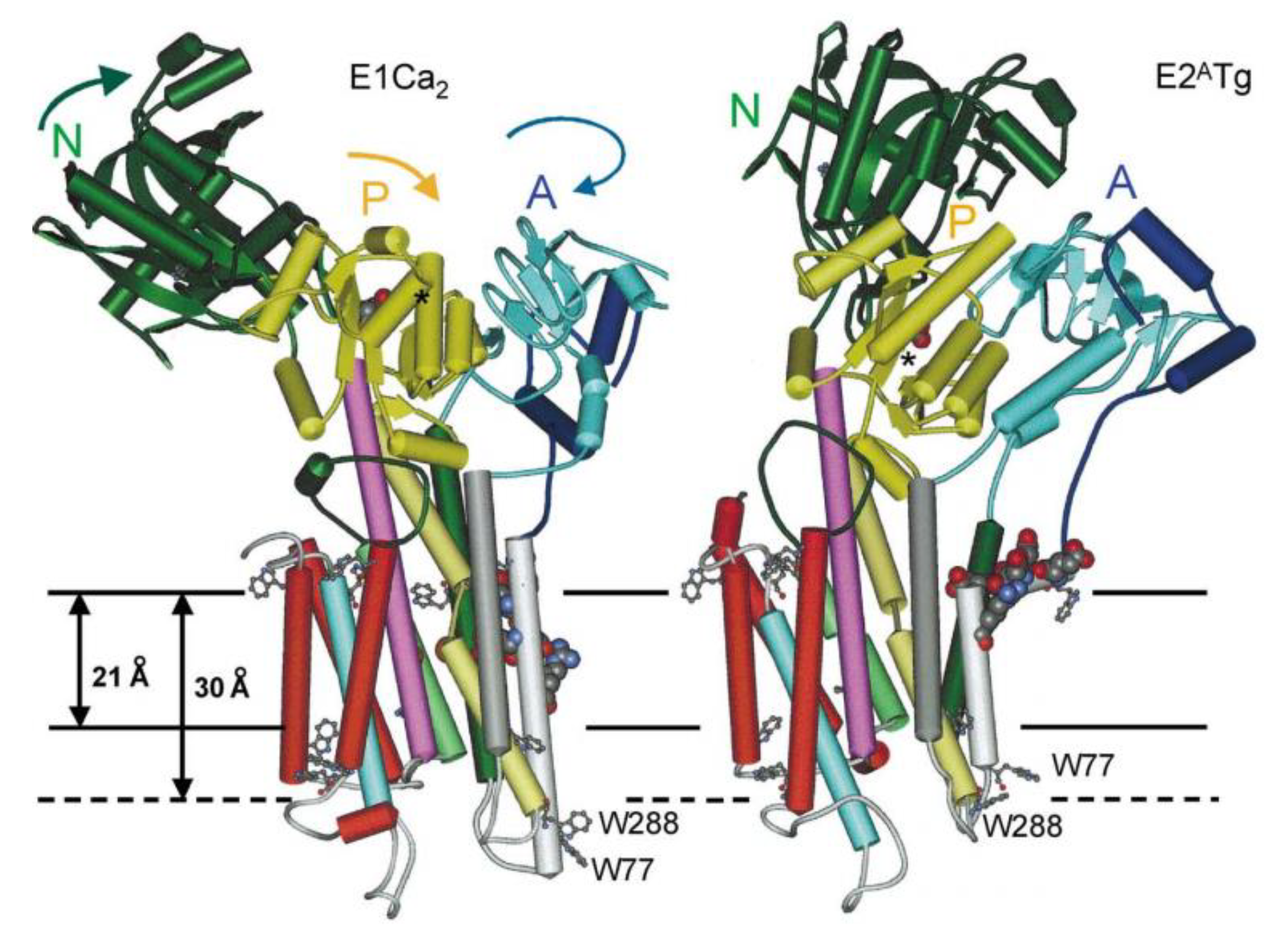
- Lee, A.G. Ca2+ -ATPase structure in the E1 and E2 conformations: Mechanism, helix-helix and helix-lipid interactions. Biochim. Biophys. Acta 2002, 1565, 246–266. [Google Scholar] [CrossRef]
- Autzen, H.E.; Siuda, I.; Sonntag, Y.; Nissen, P.; Moller, J.V.; Thogersen, L. Regulation of the Ca(2+)-ATPase by cholesterol: A specific or non-specific effect? Mol. Membr. Biol. 2015, 32, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Drachmann, N.D.; Olesen, C.; Moller, J.V.; Guo, Z.; Nissen, P.; Bublitz, M. Comparing crystal structures of Ca(2+) -ATPase in the presence of different lipids. FEBS J. 2014, 281, 4249–4262. [Google Scholar] [CrossRef]
- Espinoza-Fonseca, L.M. Probing the effects of nonannular lipid binding on the stability of the calcium pump SERCA. Sci. Rep. 2019, 9, 3349. [Google Scholar] [CrossRef] [Green Version]
- Deisenhofer, J.; Michel, H. The Photosynthetic Reaction Center from the Purple Bacterium Rhodopseudomonas viridis. Science 1989, 245, 1463–1473. [Google Scholar] [CrossRef]
- Ridder, A.N.; Morein, S.; Stam, J.G.; Kuhn, A.; de Kruijff, B.; Killian, J.A. Analysis of the role of interfacial tryptophan residues in controlling the topology of membrane proteins. Biochemistry 2000, 39, 6521–6528. [Google Scholar] [CrossRef] [PubMed]
- Luecke, H.; Schobert, B.; Richter, H.T.; Cartailler, J.P.; Lanyi, J.K. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999, 291, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Nett, J.H.; Trumpower, B.L.; Hunte, C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. Embo J. 2001, 20, 6591–6600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Klompenburg, W.; Nilsson, I.; von Heijne, G.; de Kruijff, B. Anionic phospholipids are determinants of membrane protein topology. Embo J. 1997, 16, 4261–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- von Heijne, G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; von Heijne, G. Do protein-lipid interactions determine the recognition of transmembrane helices at the ER translocon? Biochem. Soc. Trans. 2005, 33, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Newstead, S.; Hu, N.J.; Alguel, Y.; Nji, E.; Beis, K.; Yashiro, S.; Lee, C.; Leung, J.; Cameron, A.D.; et al. Benchmarking membrane protein detergent stability for improving throughput of high-resolution X-ray structures. Structure 2011, 19, 17–25. [Google Scholar] [CrossRef]
- Mangialavori, I.C.; Corradi, G.; Rinaldi, D.E.; de la Fuente, M.C.; Adamo, H.P.; Rossi, J.P. Autoinhibition mechanism of the plasma membrane calcium pump isoforms 2 and 4 studied through lipid-protein interaction. Biochem. J. 2012, 443, 125–131. [Google Scholar] [CrossRef]
- Koynova, R.; Caffrey, M. Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta 1998, 1376, 91–145. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Slotte, J.P. Biological functions of sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 2000, 182, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Kawai, F.; Shoda, M.; Harashima, R.; Sadaie, Y.; Hara, H.; Matsumoto, K. Cardiolipin domains in Bacillus subtilis marburg membranes. J. Bacteriol. 2004, 186, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Niemela, P.S.; Miettinen, M.S.; Monticelli, L.; Hammaren, H.; Bjelkmar, P.; Murtola, T.; Lindahl, E.; Vattulainen, I. Membrane proteins diffuse as dynamic complexes with lipids. J. Am. Chem. Soc. 2010, 132, 7574–7575. [Google Scholar] [CrossRef] [PubMed]
- Engelman, D.M. Membranes are more mosaic than fluid. Nature 2005, 438, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fernandes, D.; Mehta, N.; Bean, J.L.; Michaelis, M.L.; Zaidi, A. Partitioning of the plasma membrane Ca2+-ATPase into lipid rafts in primary neurons: Effects of cholesterol depletion. J. Neurochem. 2007, 102, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Dean, W.L.; Borchman, D.; Paterson, C.A. The influence of membrane lipid structure on plasma membrane Ca2+ -ATPase activity. Cell Calcium 2006, 39, 209–216. [Google Scholar] [CrossRef]
- Lange, Y. Disposition of intracellular cholesterol in human fibroblasts. J. Lipid Res. 1991, 32, 329–339. [Google Scholar]
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starling, A.P.; East, J.M.; Lee, A.G. Effects of phospholipid fatty acyl chain length on phosphorylation and dephosphorylation of the Ca(2+)-ATPase. Biochem. J. 1995, 310 Pt 3, 875–879. [Google Scholar] [CrossRef]
- Fajardo, V.A.; Trojanowski, N.; Castelli, L.M.; Miotto, P.M.; Amoye, F.; Ward, W.E.; Tupling, A.R.; LeBlanc, P.J. Saturation of SERCA’s lipid annulus may protect against its thermal inactivation. Biochem. Biophys. Res. Commun. 2017, 484, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.E.; Juranka, P.F.; Joos, B. Perturbed voltage-gated channel activity in perturbed bilayers: Implications for ectopic arrhythmias arising from damaged membrane. Prog. Biophys. Mol. Biol. 2012, 110, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Sen, L.; Bialecki, R.A.; Smith, E.; Smith, T.W.; Colucci, W.S. Cholesterol increases the L-type voltage-sensitive calcium channel current in arterial smooth muscle cells. Circ. Res. 1992, 71, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Xie, L.; Mihic, A.; Gao, X.; Chen, Y.; Gaisano, H.Y.; Tsushima, R.G. Inhibition of cholesterol biosynthesis impairs insulin secretion and voltage-gated calcium channel function in pancreatic beta-cells. Endocrinology 2008, 149, 5136–5145. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, I.S.; Brazer, S.C.; Liu, X.; Lockwich, T.; Singh, B. Plasma membrane localization of TRPC channels: Role of caveolar lipid rafts. Novartis Found. Symp. 2004, 258, 63–70. [Google Scholar] [PubMed]
- Szoke, E.; Borzsei, R.; Toth, D.M.; Lengl, O.; Helyes, Z.; Sandor, Z.; Szolcsanyi, J. Effect of lipid raft disruption on TRPV1 receptor activation of trigeminal sensory neurons and transfected cell line. Eur. J. Pharm. 2010, 628, 67–74. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, A.; Sardar, P.; Yadav, M.; Majhi, R.K.; Kumar, A.; Goswami, C. Influence of membrane cholesterol in the molecular evolution and functional regulation of TRPV4. Biochem. Biophys. Res. Commun. 2015, 456, 312–319. [Google Scholar] [CrossRef]
- Morenilla-Palao, C.; Pertusa, M.; Meseguer, V.; Cabedo, H.; Viana, F. Lipid raft segregation modulates TRPM8 channel activity. J. Biol. Chem. 2009, 284, 9215–9224. [Google Scholar] [CrossRef]
- Lakk, M.; Yarishkin, O.; Baumann, J.M.; Iuso, A.; Krizaj, D. Cholesterol regulates polymodal sensory transduction in Muller glia. Glia 2017, 65, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.O.; Massey, A.E.; Mata-Daboin, A.D.; Sierra-Valdez, F.J.; Chauhan, S.C.; Cordero-Morales, J.F.; Vasquez, V. Dietary fatty acids fine-tune Piezo1 mechanical response. Nat. Commun. 2019, 10, 1200. [Google Scholar] [CrossRef] [PubMed]
- Cantor, R.S. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 1999, 76, 2625–2639. [Google Scholar] [CrossRef]
- Brown, M.F. Soft Matter in Lipid-Protein Interactions. Annu. Rev. Biophys. 2017, 46, 379–410. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.A.; Engelman, D.M. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 1983, 166, 211–217. [Google Scholar] [CrossRef]
- Froud, R.J.; Earl, C.R.; East, J.M.; Lee, A.G. Effects of lipid fatty acyl chain structure on the activity of the (Ca2+ + Mg2+)-ATPase. Biochim. Biophys. Acta 1986, 860, 354–360. [Google Scholar] [CrossRef]
- Cornea, R.L.; Thomas, D.D. Effects of membrane thickness on the molecular dynamics and enzymatic activity of reconstituted Ca-ATPase. Biochemistry 1994, 33, 2912–2920. [Google Scholar] [CrossRef]
- Gustavsson, M.; Traaseth, N.J.; Veglia, G. Activating and deactivating roles of lipid bilayers on the Ca(2+)-ATPase/phospholamban complex. Biochemistry 2011, 50, 10367–10374. [Google Scholar] [CrossRef]
- Sonntag, Y.; Musgaard, M.; Olesen, C.; Schiott, B.; Moller, J.V.; Nissen, P.; Thogersen, L. Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nat. Commun. 2011, 2, 304. [Google Scholar] [CrossRef] [Green Version]
- Janmey, P.A.; Kinnunen, P.K. Biophysical properties of lipids and dynamic membranes. Trends Cell Biol. 2006, 16, 538–546. [Google Scholar] [CrossRef]
- Perozo, E.; Kloda, A.; Cortes, D.M.; Martinac, B. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 2002, 9, 696–703. [Google Scholar] [CrossRef]
- Phillips, R.; Ursell, T.; Wiggins, P.; Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature 2009, 459, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lewis, A.H.; Grandl, J. Touch, Tension, and Transduction–The Function and Regulation of Piezo Ion Channels. Trends Biochem. Sci. 2017, 42, 57–71. [Google Scholar] [CrossRef]
- Bavi, N.N.; Nikolaev, Y.A.; Bavi, O.; Ridone, P.; Martinac, A.D.; Nakayama, Y.; Cox, C.D.; Martinac, B. Principles of Mechanosensing at the Membrane Interface. Biophys. Cell Membr. Springer Ser. Biophys. 2017, 19, 85–119. [Google Scholar]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Cullis, P.R.; De Kruijff, B. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta (Bba)–Rev. Biomembr. 1979, 559, 399–420. [Google Scholar] [CrossRef]
- Sheetz, M.P.; Singer, S.J. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc. Natl. Acad. Sci. USA 1974, 71, 4457–4461. [Google Scholar] [CrossRef]
- Bavi, O.; Cox, C.D.; Vossoughi, M.; Naghdabadi, R.; Jamali, Y.; Martinac, B. Influence of Global and Local Membrane Curvature on Mechanosensitive Ion Channels: A Finite Element Approach. Membranes 2016, 6, 14. [Google Scholar] [CrossRef]
- Antonny, B. Mechanisms of membrane curvature sensing. Annu. Rev. Biochem. 2011, 80, 101–123. [Google Scholar] [CrossRef]
- Lewis, A.H.; Grandl, J. Mechanical sensitivity of Piezo1 ion channels can be tuned by cellular membrane tension. Elife 2015, 4. [Google Scholar] [CrossRef]
- Gauthier, N.C.; Masters, T.A.; Sheetz, M.P. Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 2012, 22, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Gnanasambandam, R.; Bae, C.; Gottlieb, P.A.; Sachs, F. Ionic Selectivity and Permeation Properties of Human PIEZO1 Channels. PLoS ONE 2015, 10, e0125503. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.A.; Bae, C.; Sachs, F. Gating the mechanical channel Piezo1: A comparison between whole-cell and patch recording. Channels (Austin) 2012, 6, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Moe, P.; Blount, P. Assessment of potential stimuli for mechano-dependent gating of MscL: Effects of pressure, tension, and lipid headgroups. Biochemistry 2005, 44, 12239–12244. [Google Scholar] [CrossRef] [PubMed]
- Sukharev, S. Purification of the small mechanosensitive channel of Escherichia coli (MscS): The subunit structure, conduction, and gating characteristics in liposomes. Biophys. J. 2002, 83, 290–298. [Google Scholar] [CrossRef]
- Sukharev, S. Mechanosensitive channels in bacteria as membrane tension reporters. FASEB J. 1999, 13, S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Cranfield, C.G.; Deplazes, E.; Owen, D.M.; Macmillan, A.; Battle, A.R.; Constantine, M.; Sokabe, M.; Martinac, B. Differential effects of lipids and lyso-lipids on the mechanosensitivity of the mechanosensitive channels MscL and MscS. Proc. Natl. Acad. Sci. USA 2012, 109, 8770–8775. [Google Scholar] [CrossRef] [Green Version]
- Léonard, C.; Alsteens, D.; Dumitru, A.C.; Mingeot-Leclercq, M.P.; Tyteca, D. Lipid Domains and Membrane (Re)Shaping: From Biophysics to Biology. Biophys. Cell Membr. 2017, 19, 121–175. [Google Scholar]
- Taverna, E.; Saba, E.; Rowe, J.; Francolini, M.; Clementi, F.; Rosa, P. Role of lipid microdomains in P/Q-type calcium channel (Cav2.1) clustering and function in presynaptic membranes. J. Biol. Chem. 2004, 279, 5127–5134. [Google Scholar] [CrossRef]
- Saheki, Y.; De Camilli, P. Endoplasmic Reticulum-Plasma Membrane Contact Sites. Annu. Rev. Biochem. 2017, 86, 659–684. [Google Scholar] [CrossRef]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Luo, X.; Wilkie, T.M.; Miller, L.J.; Peck, A.B.; Humphreys-Beher, M.G.; Muallem, S. Polarized expression of G protein-coupled receptors and an all-or-none discharge of Ca2+ pools at initiation sites of [Ca2+]i waves in polarized exocrine cells. J. Biol. Chem. 2001, 276, 44146–44156. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H. Ca(2)(+) signalling in the endoplasmic reticulum/secretory granule microdomain. Cell Calcium 2015, 58, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.; Li, J.; Milligan, C.J.; Zeng, F.; Sukumar, P.; Hou, B.; Sedo, A.; Yuldasheva, N.; Majeed, Y.; Beri, D.; et al. Pregnenolone sulphate- and cholesterol-regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ. Res. 2010, 106, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hui, K.; Qin, F. Thermodynamics of heat activation of single capsaicin ion channels VR1. Biophys. J. 2003, 85, 2988–3006. [Google Scholar] [CrossRef]
- Liu, M.; Huang, W.; Wu, D.; Priestley, J.V. TRPV1, but not P2X, requires cholesterol for its function and membrane expression in rat nociceptors. Eur. J. Neurosci. 2006, 24, 1–6. [Google Scholar] [CrossRef]
- Picazo-Juarez, G.; Romero-Suarez, S.; Nieto-Posadas, A.; Llorente, I.; Jara-Oseguera, A.; Briggs, M.; McIntosh, T.J.; Simon, S.A.; Ladron-de-Guevara, E.; Islas, L.D.; et al. Identification of a binding motif in the S5 helix that confers cholesterol sensitivity to the TRPV1 ion channel. J. Biol. Chem. 2011, 286, 24966–24976. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Andolfi, L.; Frattini, F.; Mayer, F.; Lazzarino, M.; Hu, J. Membrane stiffening by STOML3 facilitates mechanosensation in sensory neurons. Nat. Commun. 2015, 6, 8512–8525. [Google Scholar] [CrossRef]
- Balijepalli, R.C.; Foell, J.D.; Hall, D.D.; Hell, J.W.; Kamp, T.J. Localization of cardiac L-type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)-adrenergic regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7500–7505. [Google Scholar] [CrossRef]
- Calaghan, S.; White, E. Caveolae modulate excitation-contraction coupling and beta2-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc. Res. 2006, 69, 816–824. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Bi, Y.; Yang, W.; Guo, X.; Jiang, Y.; Wan, C.; Li, L.; Bai, Y.; Guo, J.; Wang, Y.; et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 2013, 493, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Visser, D.; Langeslag, M.; Kedziora, K.M.; Klarenbeek, J.; Kamermans, A.; Horgen, F.D.; Fleig, A.; van Leeuwen, F.N.; Jalink, K. TRPM7 triggers Ca2+ sparks and invadosome formation in neuroblastoma cells. Cell Calcium 2013, 54, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sameni, S.; Malacrida, L.; Tan, Z.; Digman, M.A. Alteration in Fluidity of Cell Plasma Membrane in Huntington Disease Revealed by Spectral Phasor Analysis. Sci. Rep. 2018, 8, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolobkova, Y.A.; Vigont, V.A.; Shalygin, A.V.; Kaznacheyeva, E.V. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef]
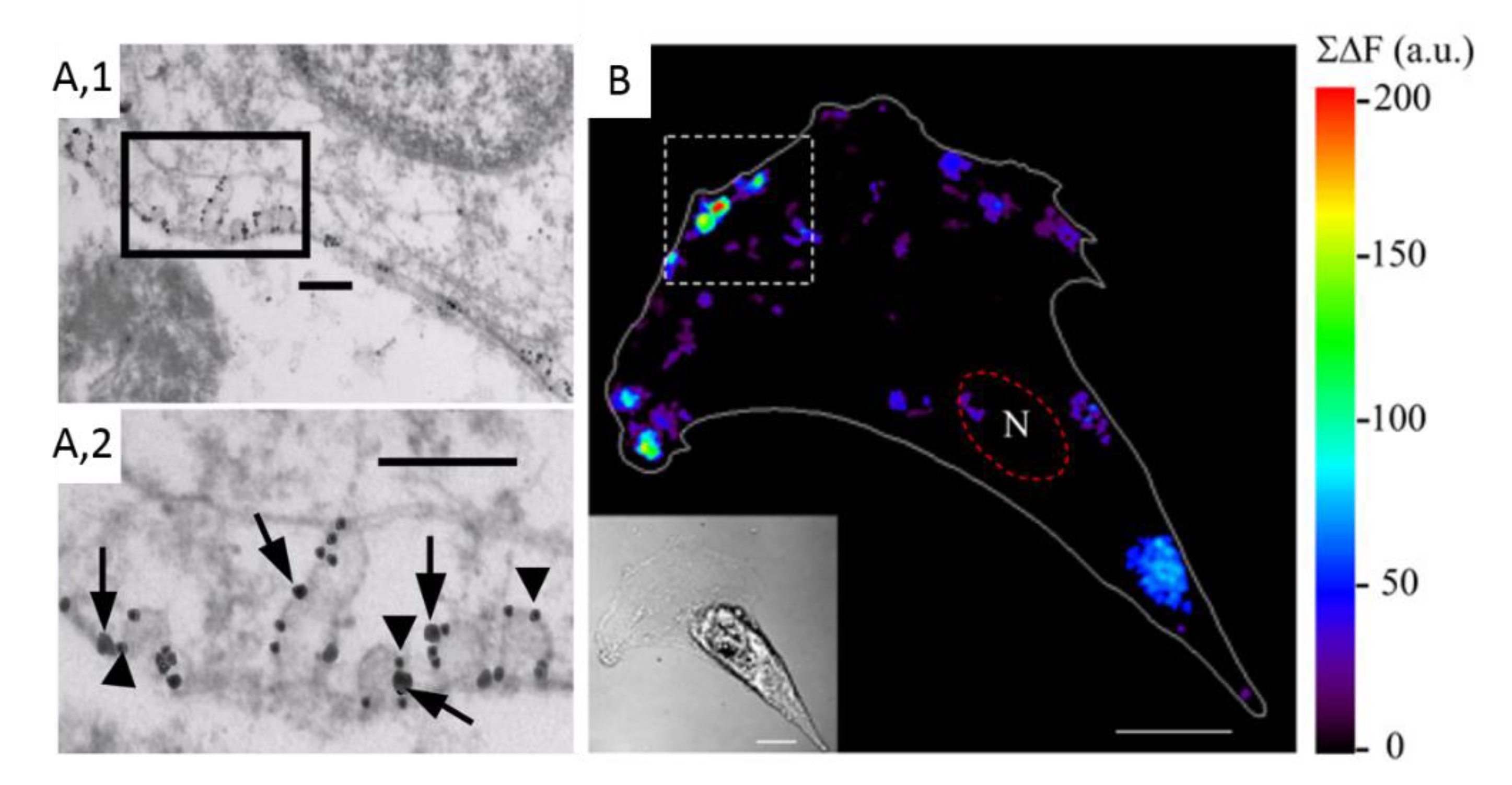
- Conrard, L.; Stommen, A.; Cloos, A.S.; Steinkuhler, J.; Dimova, R.; Pollet, H.; Tyteca, D. Spatial Relationship and Functional Relevance of Three Lipid Domain Populations at the Erythrocyte Surface. Cell Physiol. Biochem. 2018, 51, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.L.; Bigelow, D.J.; Schoneich, C.; Williams, T.D.; Ramonda, L.; Yin, D.; Huhmer, A.F.; Yao, Y.; Gao, J.; Squier, T.C. Decreased plasma membrane calcium transport activity in aging brain. Life Sci. 1996, 59, 405–412. [Google Scholar] [CrossRef]
- Berrocal, M.; Corbacho, I.; Vazquez-Hernandez, M.; Avila, J.; Sepulveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 2015, 1852, 1465–1476. [Google Scholar] [CrossRef]
- Yamamoto, N.; Matsubara, T.; Sato, T.; Yanagisawa, K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid beta-protein fibrillogenesis. Biochim. Biophys. Acta 2008, 1778, 2717–2726. [Google Scholar] [CrossRef]
- Zaidi, A. Plasma membrane Ca-ATPases: Targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 2010, 1, 271–280. [Google Scholar] [CrossRef]
- Taberner, F.J.; Fernandez-Ballester, G.; Fernandez-Carvajal, A.; Ferrer-Montiel, A. TRP channels interaction with lipids and its implications in disease. Biochim. Biophys. Acta 2015, 1848, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Garattini, S. Long-chain n-3 fatty acids in lipid rafts: Implications for anti-inflammatory effects. J. Cardiovasc Med. (Hagerstown) 2007, 8, S30–S33. [Google Scholar] [CrossRef]
- Yaqoob, P.; Shaikh, S.R. The nutritional and clinical significance of lipid rafts. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 156–166. [Google Scholar] [CrossRef]
- Corradi, V.; Mendez-Villuendas, E.; Ingolfsson, H.I.; Gu, R.X.; Siuda, I.; Melo, M.N.; Moussatova, A.; DeGagne, L.J.; Sejdiu, B.I.; Singh, G.; et al. Lipid-Protein Interactions Are Unique Fingerprints for Membrane Proteins. Acs Cent. Sci. 2018, 4, 709–717. [Google Scholar] [CrossRef]



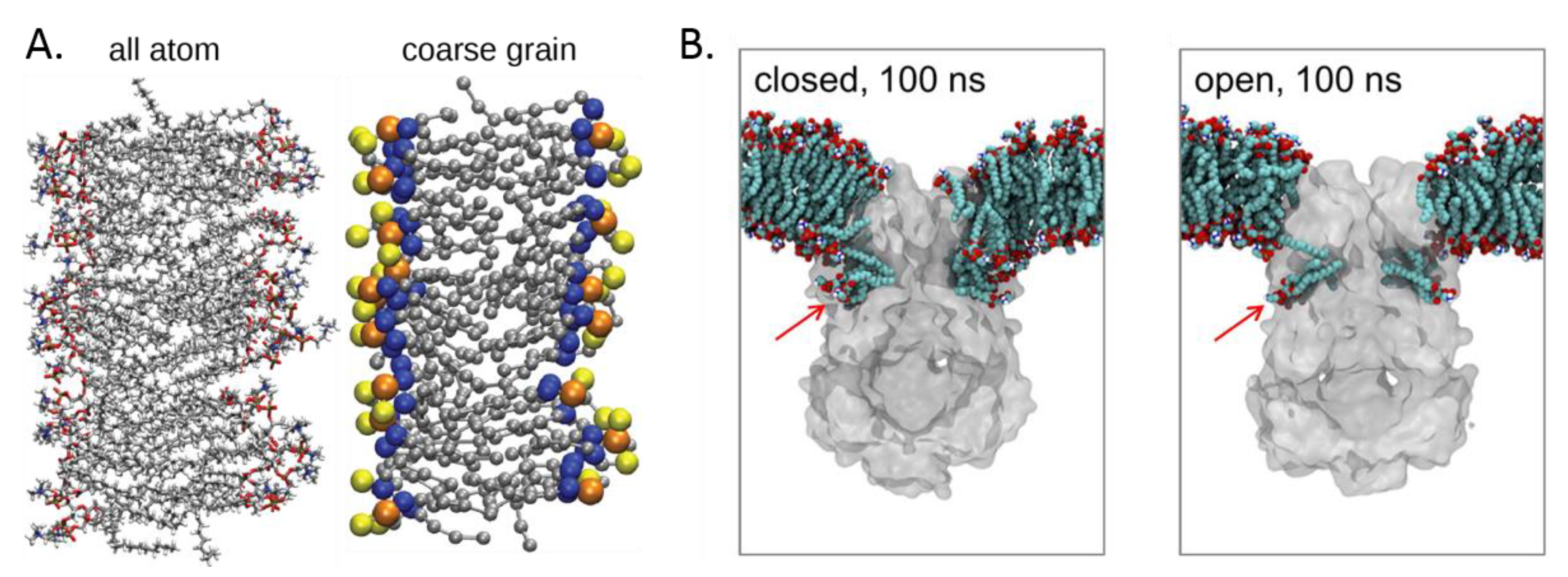
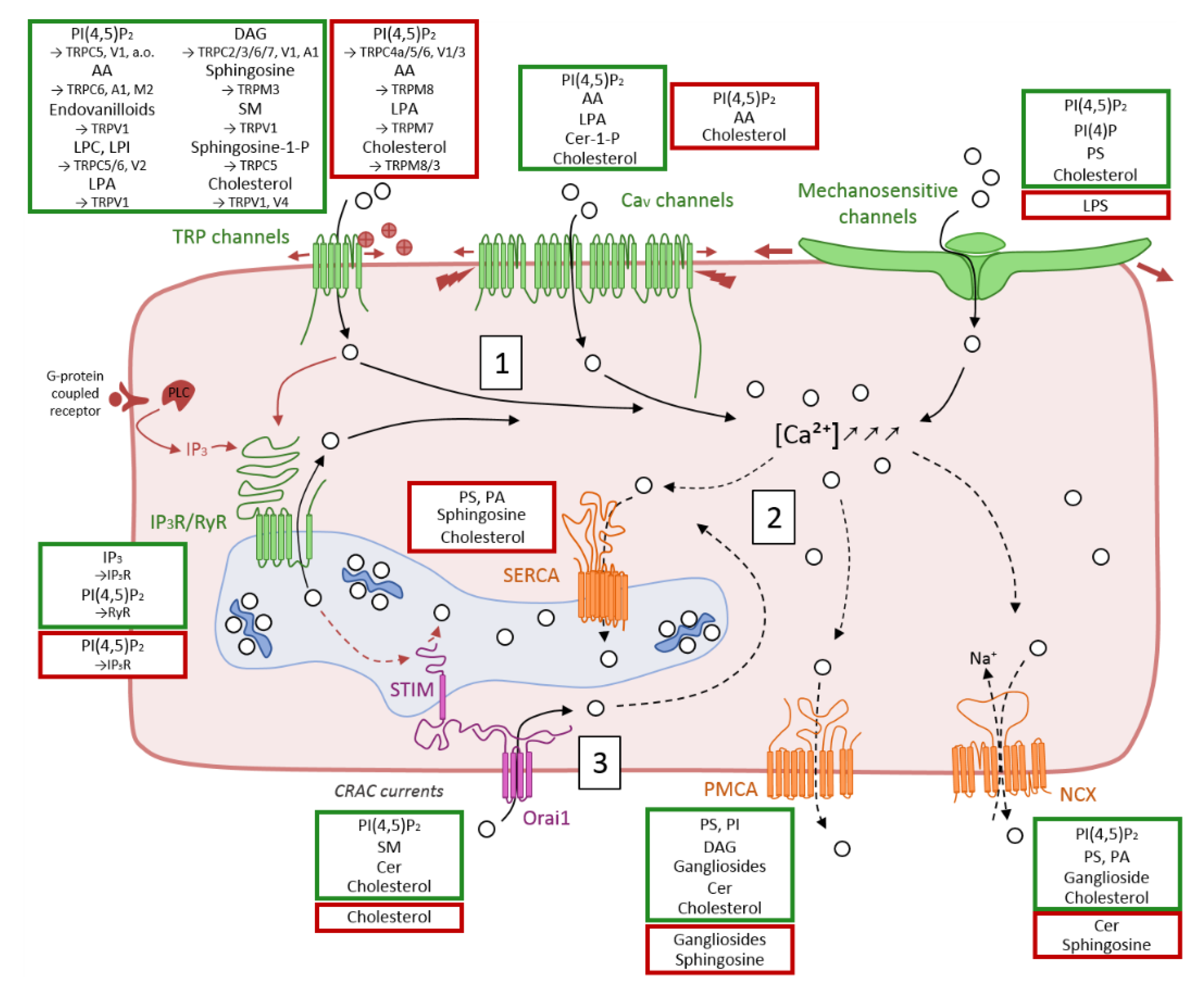

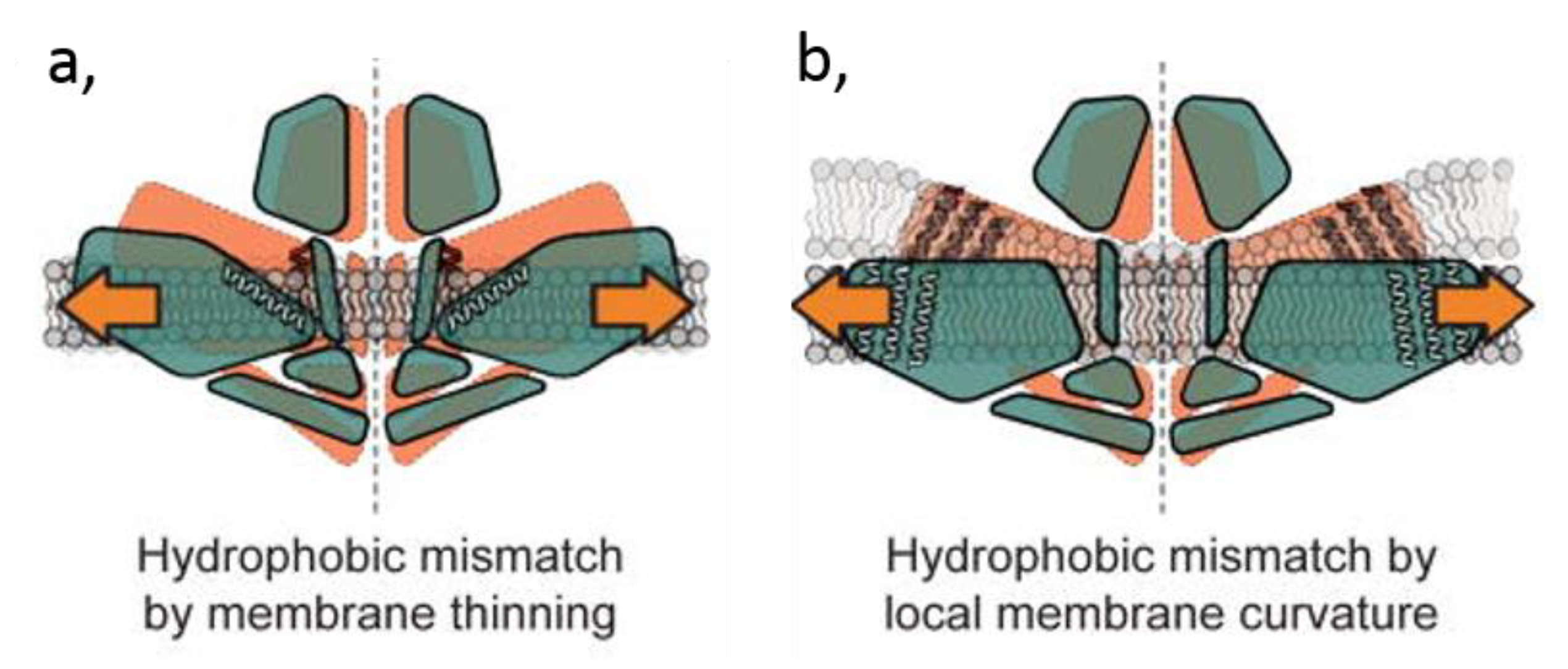

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conrard, L.; Tyteca, D. Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment. Biomolecules 2019, 9, 513. https://doi.org/10.3390/biom9100513
Conrard L, Tyteca D. Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment. Biomolecules. 2019; 9(10):513. https://doi.org/10.3390/biom9100513
Chicago/Turabian StyleConrard, Louise, and Donatienne Tyteca. 2019. "Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment" Biomolecules 9, no. 10: 513. https://doi.org/10.3390/biom9100513
APA StyleConrard, L., & Tyteca, D. (2019). Regulation of Membrane Calcium Transport Proteins by the Surrounding Lipid Environment. Biomolecules, 9(10), 513. https://doi.org/10.3390/biom9100513