Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism


Université de Strasbourg/CNRS, UMR7177, Institut de Chimie, 4, rue Blaise Pascal, 67070 Strasbourg, France
*
Author to whom correspondence should be addressed.
Biomolecules 2018, 8(2), 18; https://doi.org/10.3390/biom8020018
Submission received: 29 March 2018
/
Revised: 13 April 2018
/
Accepted: 16 April 2018
/
Published: 18 April 2018
(This article belongs to the Special Issue Antimicrobial Peptides: Development, Conjugation, and Beyond)
Abstract
:Biophysical and structural investigations are presented with a focus on the membrane lipid interactions of cationic linear antibiotic peptides such as magainin, PGLa, LL37, and melittin. Observations made with these peptides are distinct as seen from data obtained with the hydrophobic peptide alamethicin. The cationic amphipathic peptides predominantly adopt membrane alignments parallel to the bilayer surface; thus the distribution of polar and non-polar side chains of the amphipathic helices mirror the environmental changes at the membrane interface. Such a membrane partitioning of an amphipathic helix has been shown to cause considerable disruptions in the lipid packing arrangements, transient openings at low peptide concentration, and membrane disintegration at higher peptide-to-lipid ratios. The manifold supramolecular arrangements adopted by lipids and peptides are represented by the ‘soft membranes adapt and respond, also transiently’ (SMART) model. Whereas molecular dynamics simulations provide atomistic views on lipid membranes in the presence of antimicrobial peptides, the biophysical investigations reveal interesting details on a molecular and supramolecular level, and recent microscopic imaging experiments delineate interesting sequences of events when bacterial cells are exposed to such peptides. Finally, biophysical studies that aim to reveal the mechanisms of synergistic interactions of magainin 2 and PGLa are presented, including unpublished isothermal titration calorimetry (ITC), circular dichroism (CD) and dynamic light scattering (DLS) measurements that suggest that the peptides are involved in liposome agglutination by mediating intermembrane interactions. A number of structural events are presented in schematic models that relate to the antimicrobial and synergistic mechanism of amphipathic peptides when they are aligned parallel to the membrane surface.
1. Introduction
Antimicrobial peptides (AMPs) are effectors of the innate immune system which provide a first line of defense against a multitude of pathogenic microorganisms. Higher organisms release AMPs immediately when infections by bacteria or fungi occur [1,2]. They have been found in a wide variety of species from the plant and animal kingdom, including humans [3]. Furthermore, many peptides produced by microorganisms have been identified and investigated [4,5]. After their first discovery decades ago [6,7,8], many new sequences have been added to the corresponding data bases [9,10] and they have been investigated by a wide variety of techniques to better understand their mechanisms of action. Since the worldwide re-emergence of infectious diseases and a rapid increase in multi-resistant pathogens [11] they bear great promise to lead the way to new classes of antibiotics capable of counteracting the continuously increasing threat by resistant microorganisms; thus the declining number of effective pharmaceutical agents can be complemented. Whereas many natural peptides have potential topical applications, they are considered unsuitable for oral intake because of their fast degradation by proteases. However, peptides can be modified and made unavailable during transport by incorporation into nanostructures or by fixation to surfaces [12,13,14], thus they are protected and able to reach their target. Alternatively, understanding the mechanism of action of the natural sequences paves the way to designing molecules with favorable properties that mirror the essential characteristics of the template compounds. Therefore, the study of antimicrobial peptides (AMPs), which have evaded bacterial resistance during millions of years of evolution [2], promises to reveal novel strategies for the development of new lines of antibiotics.
The physico-chemical characteristics of antimicrobial peptides discussed in this paper in combination with a multitude of investigations indicates that they interact with lipid bilayers and interfere with the barrier function of bacterial membranes. In contrast, molecules that specifically target proteinaceous receptors can be made inefficient by mutagenesis of one or a few sites, and it is much less likely that bacteria develop resistance to compounds whose primary target is the destruction of the physico-chemical properties of the lipid membrane [15]. Membrane-active peptides exhibit a wide range of structural features some being helical in their bilayer-associated state [16,17], others forming cyclic [18,19,20,21] and/or β-sheet arrangements [22,23,24,25,26]. Indeed, following the insights gained from the studies of cationic amphipathic antimicrobial peptides a number of small amphipathic molecules [27,28], pseudopeptides [21,29,30,31,32,33,34,35,36], and polymers [37] have been designed and investigated, and found to also exhibit potent antimicrobial activities.
Here, some of the underlying research efforts shall be presented that have led to important mechanistic insights and the design of new compounds. The review focuses on linear cationic peptides such as magainins [38], cecropins and designed peptides [39,40,41] (amino acid sequences are provided in Table 1), antimicrobial peptides which have been described and investigated early on. In the following, insights with these AMPs provided guidelines for the design of new compounds and initiated the search for, and investigation of, related sequences. Despite decades of research, new structural and dynamic features of membrane-associated AMPs are continuously being discovered [26,42] because these peptides can adopt a large diversity of conformations and topologies whose exchanges and interactions are governed by multiple equilibria [43]. Finally, the synergistic interactions between PGLa and magainins will be discussed because the covalent or non-covalent combination of compounds provides an alternative strategy to enhance their efficiency and further reduces their susceptibility to bacterial resistance.
Peptides from frogs and insects have already been described in the 1960s [7,44], while magainins and cecropins, which act as specific antimicrobial compounds, have been discovered with some delay [2,45]. These sequences are linear, highly cationic, and form amphipathic helices when interacting with membranes. They are thought to specifically disrupt the integrity of bacterial and fungal membranes by insertion, thereby inhibiting the growth of microorganisms and/or enter into the cell interior [46]. Thereby, they constitute a first line of defense when infections occur [2,45]. By also modulating the immune response of the host organisms, their efficiency is considerably enhanced, and because of this extension of functionalities, it has been suggested to rename them as ‘host defense peptides’ [47,48,49,50]. Indeed, metabolomics studies reveal complex reactions by the bacterial cells when exposed to AMPs, many being unique to a specific sequence [51,52].
Magainins and derivatives thereof have been extensively studied by biophysical approaches (e.g., [53,54,55,56,57]), with sometimes unexpected results, and the insights thus obtained have formed the basis for suggesting novel mechanisms of action of these peptides [43,58]. When added to membranes, magainins and cecropins were found to exhibit lytic activities. In some electrophysiological experiments, they also showed discrete multi-level conductivities [59,60,61,62], which in analogy to models that had been proposed for the hydrophobic alamethicin peptide or for large helical channel proteins, was taken as an indicator for transmembrane helical bundle formation [63]. However, it is important to emphasize that unlike the alamethicin channels, those recorded in the presence of magainins or cecropins are less well defined, erratic, and characterized by large variations [59,60,61]. These pronounced differences in electrophysiological recordings reflect distinct physico-chemical properties of these sequences, such as number of charges, hydrophobicity, and hydrophobic moment (Table 1).
Furthermore, magainin pore formation was investigated on a macroscopic scale by following the kinetics of calcein release from individual giant unilamellar vesicles (GUV) made from defined mixture of membrane lipids, dioleoylphosphatidylcholine/dioleoylphosphatidylglycerol DOPC/DOPG at different molar ratios [64]. These experiments indicate that after addition of the peptide, it takes minutes before the release of fluorophores sets in. Once the pores have been established, the vesicles, which are several micrometers in size, empty within 30 s. Because the rate-limiting step is the formation of the pore, rather than diffusion through the pore, magainin pore formation in GUVs follows an all-or-nothing mechanism. This observation agrees with calcein release experiments from suspensions of large unilamellar vesicles made from palmitoyl-oleoyl-phosphatidylcholine (POPC) and palmitoyl-oleoyl-phosphatidylglycerol (POPG) lipid membranes [65]. Conversely, for magainin 2, an all-or-nothing mechanism dominates in the presence of 50 mol % phosphatidylglycerol (PG) a more graded release is observed when the PG content is reduced to 20 mol % [65]. These experiments showed no indications of peptide oligomerization in either state [65]. The subsequent fluorophore release is a two-stage process [66]. An initial fast release has been associated with magainin interacting with only the outside monolayer, which causes an unbalance. Equilibration of the peptide concentration between the outer and inner leaflets results in the transient formation of very large pores [66]. Thereafter, smaller pores assure a slower release of fluorophore, but even these persistent openings are large enough to allow for the passage of molecules with a hydrodynamic radius of 3 nm (equivalent to a globular protein of molecular weight (MW) > 20 kDa) [66].
Recently, microscopic imaging techniques were introduced into the field, which reveal the spatio-temporal binding of antimicrobial peptides to live bacteria and the related membrane permeabilization events. For example, the human peptide LL37 preferentially attacks septating Escherichia coli cells where the peptide is found associated with the septum and the curved regions of the outer membrane [67]. In non-septating cells, it prefers to bind to one of the endcaps. Influx of the AMPs to the periplasmic space results in cell shrinking, probably via an osmotic effect. After permeabilization of the outer membrane, there is a short delay before cytoplasmatic membrane permeabilization occurs. These openings of the outer and cytoplasmatic membranes are localized and persistent, rather than global and transient [68]. Notably, whereas many events observed on this cellular level resemble each other, the exact details vary with the antimicrobial compound when cationic polymers, longer or shorter peptides such as LL37, cecropin A, or melittin are compared to each other [69]. Furthermore, the events that happen with E. coli cells that are grown either under aerobic or anaerobic conditions have been compared to each other and correlated with mutagenesis experiments [70]. This data suggests that LL37 specifically affects the electron transport chain [70]. Notably, the permeabilization in the presence of alamethicin follows a different series of events, even though the data do not rule out a chaotic pore or a carpet mechanisms for this hydrophobic peptide [71]. Whereas a chaotic pore structure is shown in Figure 1A,B a peptide carpet is illustrated in reference [43].
Structural investigations show that the random coil structure of magainins in aqueous solution becomes helical once the peptide inserts into membrane environments [72]. This conformational transition has been identified to be a driving force of membrane association [78,79]. Importantly, both circular dichroism (CD) and solid-state nuclear magnetic resonance (NMR) spectroscopy on uniaxially oriented membranes indicate that the magainin helix is oriented parallel to the membrane surface, which results in membrane association being reversible [38]. The in-planar alignment has been confirmed for magainin 2 in membranes of different composition [38,54], for magainin analogues [80,81] and for a number of other linear cationic antimicrobial peptides [82,83,84,85,86]. In contrast, alamethicin with its much different characteristics in both electrophysiological recordings and physico-chemical properties (Table 1), has been found to form well-defined channels (reviewed e.g., in [17,87]) and to adopt stable transmembrane helical alignments in canonical dimyristoyl-phosphatidylcholine (DMPC) and (POPC) membranes [88,89,90,91,92]. However, even this peptide adopts in-plane alignments under certain conditions [42,93] emphasizing the dynamic nature of peptide-lipid interactions involving multiple equilibria [43]. CD spectroscopy has also been used to study the association kinetics of magainin to whole cells and lipopolysaccharides [94].
When investigated in more detail, the topology of magainin 2 has been found to be parallel to the membrane surface regardless of the lipid composition [38,54]. In contrast, a much wider range of alignments has been observed for its relative, PGLa [95,96] (Table 1). Interestingly, the difference becomes only apparent in membranes where both fatty acyl chains are saturated. For example, in DMPC, PGLa changes its tilt angle by up to 30 degrees upon an increase in peptide concentration [97]. The transition occurs at 0.5 to 2 mol % depending on the membrane hydration conditions [95,97]. A continuous range of tilt angles was observed when saturated phosphatidylcholine (PC) bilayers with decreasing hydrophobic thickness where investigated [95,96]. These studies reveal helical tilt angles that are suggestive of transmembrane orientations when thin phosphatidylcholine bilayers (C10 or C12 fatty acyl chains) are investigated [95]. When PGLa is studied in phospholipid bilayers carrying unsaturations (such as palmitoyl-oleoyl-phospholipids), the peptide remains stably aligned along the surface [38,95,98].
When an amphipathic peptide such as magainin resides in the bilayer interface, it pushes apart the lipids at the level of the head group and glycerol regions [54], which loosens the packing of the hydrophobic region (disordering effect, Figure 1A) [99,100]. The accompanying compensation by the membrane lipids results in a reduction of the membrane thickness [57,101]. Deuterium solid-state NMR measurements have indeed revealed a decrease of the order parameters in the bilayer interior upon addition of magainin 2, PGLa, and other amphipathic peptides [99,102,103,104]. Notably, the bilayer disruptive properties of such peptides have been estimated to cover a 50 Å radius [105,106].
The question arises for how in-plane oriented peptides can promote the passage of water, ions, and fluorescence dyes across the lipid bilayer. Molecular dynamics simulations have provided atomistic views on how this may be possible. They show not only the method by which hydrophobic peptides insert into the membrane to form peptide channels made from transmembrane helical arrangements [63], but also how in-plane oriented helices deform the lipid bilayer, and how their side chains reach to the opposite bilayer leaflet of the membrane, thus resulting in the formation of water-filled openings [73,107,108].
Lazaridis and co-workers report on 5–9 µs all-atom molecular dynamics simulations, starting from tetrameric transmembrane helical bundles of magainin or PGLa in 80–120 lipids of DMPC or DMPC/DMPG 3/1 [108]. During the simulations, the peptides lose their transmembrane orientation and adopt tilted configurations where magainin also occurs as an antiparallel dimer (illustrated in Figure 1B).
Furthermore, Vacha and coworkers present coarse-grained molecular dynamics (MD) of schematic amphipathic peptides and find evidence for a novel double belt arrangement where peptides oriented parallel to the bilayer plane form defined membrane openings (Figure 1C) [73]. The exact topology depends on the length of the peptide and its hydrophobicity distribution, but also on the membrane thickness [109]. All-atom 100 ns simulations in palmitoyl-oleoyl-phosphatidylethanolamine/ palmitoyl-oleoyl-phosphatidylglycerol (POPE/POPG) 3/1 of one, two, or eight peptides and 512 lipids show a stable in-plane topology of magainin and pleurocidin, some oligomerization, but no pore or supramolecular rearrangement within this time frame [110]. Finally, recent simulation work investigating magainin 2-lipopolysaccharide and ion interactions shall be mentioned [111].
Taken together, the molecular dynamics simulations provide a rather heterogeneous view of the magainin membrane interactions, where, despite some peptide-peptide interactions, pores form through stochastic rearrangements of peptides and lipids rather than through well-defined channel structures (Figure 1B). Although this view is in good agreement with electrophysiological recordings [59,60,61,62] the comparatively small size of the membrane patches and their relatively short duration does not yet capture the membrane lytic nature of the peptides, or the large pores that become apparent in dye release experiments [65,66].
Based on the GxxxG amino acid motif, which has been shown to be a dimer recognition sequence for transmembrane helical domains [112], a symmetric antiparallel dimer of PGLa has been assembled and then simulated by all-atom MD for up to 2 µs [113]. Although this time frame is too short to follow larger supramolecular rearrangements or the dissociation of preformed oligomers, the simulation provides interesting images of possible arrangements of PGLa in lipid bilayers. Unfortunately, to our knowledge so far, experimental proof for dimer formation in membrane environments such as solid-state NMR distance measurements is still missing, thus the exact reason for the change in topology when the peptide concentration increases (cf. ultra) [95,96,97] remains a matter of speculation. An interesting question in this context is the local environment of the GxxxG motif, which to our knowledge has only been shown to drive dimerization within the hydrophobic core of the membrane [112]. Dimerization thus assures that the polar backbone atoms of helical glycines are shielded from the hydrophobic surroundings. Along this line, dimer formation is suggestive of a deep membrane penetration of the PGLa helix, placing the two glycines in a non-polar environment.
The bilayer disruptive properties of amphipathic helices that are aligned along the membrane interface, with some of the helical cross-section interacting with the hydrophobic fatty acyl chain and the opposite face exposed to the polar head group, can be rationalized by the molecular shape concept that has been originally introduced to explain the phase behavior of lipids [114,115]. Geometrical considerations are used to explain why the cylindrical PC lipids arrange into extended bilayers, the cone shaped phosphatidylethanolamine (PE) have a tendency to adopt HII phases and detergents with an inverted cone shape assemble into micelles. When compared to these lipids, surfactin, a cyclic peptide with a long fatty acyl chain [20], or the magainin 2 in-planar interfacial helix, use up much more space in the head group than the hydrophobic core region of the membrane, thereby resembling detergents (Figure 1A) [115]. A predictive model for the activities of linear cationic peptides, based on this and previous considerations [58,72,115], has recently been elaborated for magainin 2 and melittin [116].
The wide variety of observations made with magainins and other cationic amphipathic antimicrobial peptides has resulted in a number of seemingly contradictory models for their interactions and supramolecular arrangement in bacterial membranes. These include toroidal pores [117,118], the ‘carpet’ model where peptides cover the membrane surface at alignments parallel to the surface (Figure 1B) [119], or random aggregates within the membrane [120]. Furthermore, in electrophysiological recordings, channel-like events have been observed [59,60,61], whereas at high peptide concentration, the formation of worm like structures, disk-shaped particles, or micelles have been shown to occur [5,8,102,121]. A model should provide explanations for all of such a wide variety of features.
It is important to note that the peptides are flexible and highly dynamic, and can adjust their conformation and topology to the environment (e.g., [122,123]). In a related manner, lipid bilayers are soft, can change shape and thickness, and are capable of adjusting to the presence of peptides or to other environmental factors. To take into account the flexibility and dynamics of both the peptides and the lipid membrane, the SMART model has been introduced, where ‘Soft Membranes Adapt and Respond, also Transiently’, in the presence of antimicrobial peptides (or other external stimuli). As suggested by its name the model takes into account that lipid membranes can adapt to some extent to the disruptive properties of the peptides, but undergo macroscopic phase transitions at higher peptide concentrations, locally (Figure 1B) or globally. Notably, such phase changes can be transient; for example, during the peptides crossing the membrane in order to equilibrate concentration gradients between the outer and the inner leaflet of the membrane [66] (cf. above). Transient openings also occur because the peptides diffuse laterally, thereby stochastic fluctuations in the local peptide-to-lipid ratio occur locally [72]. Furthermore, phase diagrams are a convenient way to represent different modes of interactions between the peptides and lipids where the supramolecular morphologies—such as bilayer, wormholes, tubular structures, bicelle, micelle, or hexagonal phases—depend on the peptide-to-lipid ratio, the detailed membrane composition, temperature, hydration, salt, pH, etc. [58]. For example, for a number of peptides, in-plane or transmembrane topologies have been observed depending on pH, hydration, peptide concentration, and lipid composition [42,124,125,126]. In a recent investigation using dual polarization interferometry, surface plasmon resonance and atomic force microscopy, membrane disordering, associated mass, and structural changes were followed in real-time revealing a number of intermediate states including the lysis and recovery of membranes in the presence of magainin 2 [127]. Furthermore, changes in line tension have been suggested to be a common mechanism for a wide variety of AMPs, observations that are in good agreement with the ideas of the SMART model [127,128].
Within this model at low peptide concentrations, the bilayer structure is maintained, where only transient and local openings may appear. At higher peptide concentrations, an increasing strain on the membrane packing results in openings [65] and macroscopic phase transitions of the peptide-lipid assembly [115]. Thereby, the phase boundaries represent threshold concentrations where the peptides change their level of activities. Thus, in-planar helix orientations agree with both the disruption of the bilayer integrity at higher peptide-to-lipid ratios, as suggested by the ‘carpet model’ [119], and the stochastic and transient rupture and closure of the membrane as revealed by electrophysiological recordings when the peptide concentration is low [59,60,61].
In this context, it is noteworthy that magainins, carrying several positive charges, have been shown to interact better with membranes carrying a negative surface charge from anionic lipids or lipopolysaccharides. Indeed, such preferential association forms part of the explanation why these peptides kill bacteria or tumor cells, which expose negative charges to the outside, and are non-toxic to healthy eukaryotic cells (which are charge-neutral at their outer membrane leaflet) [129,130,131,132,133]. This preferential association can be dissected into an attractive electrostatic interaction that causes a number of orders of magnitude of increase in local surface concentration along the negatively charged surface, and a hydrophobic insertion characterized by partitioning coefficients that are of similar order of magnitude for all membranes investigated (around 1000 M−1) [131,134]. Changes in electrostatic interactions upon membrane association of multicationic antimicrobial peptides have also been suggested to result in the release of peripheral membrane proteins, thereby exerting antimicrobial activities [135].
Beyond increases in the apparent association constants through electrostatic interactions, modulation of pore-forming and antimicrobial activities arises from the effect of anionic lipids on the helical penetration depth and/or the topological equilibrium of the cationic peptides [136]. In addition, the formation of domains enriched in cationic peptides and acidic phospholipids has been postulated from 2H solid-state NMR experiments using selectively deuterated lipids [137,138]. Electrostatics also play an important role for the interactions between peptides in lipid-mediated mesophase-like arrangements along the membrane surface (Figure 1E) [74], an observation that requires further investigation.
According to the SMART model, other cationic amphipathic molecules have the potential to also exhibit antibacterial activities. Designed antimicrobial compounds should accumulate at the surface of negatively charged bacteria and intercalate into their membranes at the level of the phospholipid headgroups due to hydrophobic interactions. Interactions with healthy eukaryotic cells and toxicity should be avoided by tuning the composition to an overall moderate hydrophobicity. Furthermore, the compounds should not insert too deeply into the lipid bilayer or span the lipid membranes, but they should exhibit interfacial partitioning. Indeed, compounds with such features have been designed and exhibit potent antimicrobials activities. These include short peptide sequences [139,140,141,142,143,144,145], peptide mimetics [29,30,31,32,33,34,35,36], amphipathic polymers [37], or organic molecules encompassing an aromatic ring system, a hydrophobic chain, and cationic functional groups [27].
2. Synergistic Enhancement of the Activities of Antimicrobial Peptides
The efficiency of antimicrobial compounds can sometimes be potentiated by applying them in combination [50,146]. For example, mixtures of peptides with conventional antibiotics [37,145,147,148,149,150] or ions [151] show synergistic enhancement. Whereas this enhancement can in some instances be explained by one compound paving the way for the active antimicrobial ingredient [152], other combinations, of e.g., dermaseptins or of bacteriocins, seem to interact more specifically to exhibit synergistic activity [50,153]. Synergistic interactions involving magainin 2 have been detected with PGLa [154], or with the cyclic beta-sheet peptide tachyplesin I [155].
The combination of magainin 2 and PGLa does not only show increased killing efficiency of bacteria, but also a more efficient release of calcein from liposomes made from phospholipid bilayers [75,156,157]. It is interesting to note that these peptides are naturally stored as a cocktail in the skin of Xenopus laevis frogs. Thus, it seems that the naturally active synergism has been initially destroyed by the standard analytical processes which involve separating the peptides from the complex mixture and investigating each of them individually. In an early investigation on synergism, Masuzaki et al. suggested that the pore formation rate of magainin is slower, but the pores are more stable than those of PGLa [157]. In the mixture, synergism is a consequence of combining fast pore formation and moderate stability. More recent work by Heerklotz and co-workers suggest that synergistic vesicle leakage is a result of optimizing the size of the pores and their distribution among the liposomes [158]. They should be large enough, but at the same time sufficient in number to cause dye release from all vesicles in the suspension. The propositions by both laboratories are related to the heterogeneity of the peptide distribution in the membranes, and thereby related to the size of the vesicles or of bacterial cells [158]. An additional ingredient to be considered is the possibility that one factor of the synergistic mixture solubilizes the second one, thus increasing its availability [158].
In equimolar peptide mixtures, PGLa and magainin have both been found to exhibit an alignment parallel to the membrane surface, provided that the membranes carry at least one unsaturation per phospholipid [95,159,160], such as in E. coli lipid extracts [161] (Figure 1E–G). This helix topology resembles those when magainin or PGLa are investigated individually by solid-state NMR spectroscopy [38,95]. Contrasting data are obtained from lipids all carrying only saturated fatty acyl chains where magainin remains oriented along the membrane surface, but PGLa, which is present in the same mixtures, flips into transmembrane alignments [95,159]. Notably, in DMPC and in the presence of PGLa, a 30° deviation from perfectly in-planar topology has also been observed for magainin 2 [162]. Because fully saturated lipid mixtures do not represent the physiological membrane composition well, it seems reasonable to assume an in-planar membrane topology for both peptides (Figure 1E–G), and to use the context of the SMART model when mechanistic explanations for the synergistic antibacterial activities are elaborated [43].
In order to develop a structural model of a supposed synergistic complex, experiments were designed to delineate possible interactions sites between magainin 2 and PGLa. For example, modified peptide sequences have been investigated. Early on, during dye release experiments from egg PC/PG (1:1) liposomes, the F16W and E19Q mutants of magainin 2 were shown to exhibit reduced synergistic activity, whereas the F5W mutation did not exhibit any effect [157]. It should be noted however, that a quantitative comparison of synergism is hampered by variations that are observed when different bacterial strains are compared to each other or to model membranes [75,163,164,165]. Importantly in a recent investigation Leber et al. showed that the synergism of calcein release activities from liposomes is not only a function of the absolute peptide concentration, but also of the membrane intrinsic curvature and thus lipid composition [75]. Synergism is most pronounced for membranes with high negative intrinsic curvature involving POPE lipids, and only apparent at peptide concentrations ≥ 0.4 µM [75]. The synergistic factors were much reduced and even abolished for calcein release experiments from POPC/POPG 3/1 where the peptides alone exhibit a high activity [75]. In this context, it is interesting to note that in recent investigations, the antimicrobial activity of magainin and PGLa derivatives leveled out at minimum inhibitory concentrations (MICs) of about 1 µM [161,77]. Therefore, when only the synergistic factor is considered smaller values are obtained for peptides that exhibit already high antimicrobial activity when investigated alone [161]. Similarly, when an α-helical sequence was modified by the insertion of prolines, the synergy with conventional antibiotics increased as the antibacterial effectiveness of the peptides decreased [166].
An extensive mutagenesis study showed that the synergistic activity is abolished successively when removing/inverting the negative charges at E19 and the carboxy terminus of magainin 2 [164]. As for PGLa the positively charged K15 and K19 sites had a favorable effect on the synergistic enhancement [164]. Whereas making the hydrophobic face more hydrophobic increased the antimicrobial efficiency of magainin 2, it had no effect on synergism [165]. Recently, the PGLa residues G7, G11, and L18 have been found to be important for the synergistic enhancement of activities between the two peptides [164]. Although it is interesting that glycines 7 and 11 form a GxxxG motif, which has been shown to drive dimerization of transmembrane helical sequences in highly apolar environments, the PGLa helix is localized at the interface rather than the hydrophobic interior of the membrane [95,161]. Furthermore, there is no GxxxG motif on magainin 2 which could serve as a counterpart for the formation of a magainin-PGLa heterodimer (Table 1). Therefore, to better understand the role of the two glycines and of L18 in promoting synergistic activities, further structural investigations are required.
Coarse-grain MD simulations over several microseconds encompassing 24 peptides (PGLa and magainin) and 512 dilauroylphosphatidylcholine (DLPC; C12:0) lipids were also performed [167]. These simulations confirmed a tilting and deeper penetration of PGLa into the membrane, without adopting a transmembrane orientation, whereas magainin stays on the bilayer surface. The simulations reveal a clustering of the peptides by electrostatic interactions, concomitant with a parallel alignment of the two helices, albeit without the explicit formation of pores [167]. An all-atom MD simulation of the peptide mixture in DMPC and DMPG membranes, starting from transmembrane tetramers was performed. The simulations suggest antiparallel helix arrangements in the 1:1 heterotetramer with stronger interactions in the heterodimer than in the homodimer. Plausible interactions could occur between the S8 and E19 residues of magainin and K12 and K19 of PGLa [108]. Though in the mixture of both peptides the tilt angle of PGLa is reduced when compared to PGLa alone, a large variety of helix alignments relative to the normal membrane still persists [108].
Fluorescence spectra were used to derive constants for the membrane association of magainin and its interactions with PGLa in its membrane. Favorable PGLa-magainin interaction energies were obtained when investigated in egg-PG membranes, where the exact value depends on the assumed numbers of peptides involved in the process [157]. Energies for homo- and heterodimer formation based on the midpoints of the concentration-dependent transition of PGLa from an in-planar to a transmembrane alignment were extracted in a later investigation [168]. Notably, this transition only occurs in fully saturated membranes, but not in the presence of lipid unsaturation, as they occur in biological membranes. One should also be aware that these quantities are associated with a specifically chosen interaction model (cf. reference [157]). Furthermore, they represent a multitude of interaction terms that change during the topological transition; thus the energies involved include the transfer of residues in between polar and non-polar membrane environments, and energies associated with disordering the lipids or local changes in membrane phase, direct interactions between the peptide and the lipid as well as between peptides [104,125]. In this context, it should be noted that fluorescence resonance energy transfer (FRET) experiments did not reveal a strong interaction between magainin 2 and PGLa when associated to POPE/POPG 3/1 or POPC/POPS 3/1 membranes [77].
Isothermal Titration Calorimetry (ITC) has already provided valuable insights into the thermodynamics or membrane association of magainin 2 and PGLa [56,131,134,169,170,171]. In order to further explore possible interactions with membranes, large unilamellar vesicles (LUVs) made of POPE/POPG 3/1 at pH 7 were prepared as a model system for bacterial membranes. In this context, the interactions of both peptides individually and as a mixture were investigated (Figure 2). Interestingly, only endothermic enthalpies (ΔH) were observed when each peptide was titrated into the lipid suspension individually (Figure 2A,B) while the peptide mixture revealed a considerably more complex time trace of reaction enthalpies (Figure 2C), suggesting additional modes of interaction. Additional exothermic enthalpies are observed for peptide to lipid molar ratios > 1.5%, i.e. for times of injections t > 1000 s. When compared to previous investigations with 30 nm small unilamellar vesicles (SUVs) [131,134,169,170], the reaction enthalpies of magainin 2 and PGLa with 100 nm LUVs, also used here, are relatively small [171]. A quantitative analysis of the ITC traces (Figure 2D,E) reveals enthalpies of 3–4 kcal/mol for PGLa and magainin, respectively, entropies of 40 cal·mol−1·K−1, and apparent membrane association constants in the 106 M−1 range (apparent stoichiometry P/L ≈ 1.7 mol %). Although different experimental conditions have been chosen for the experiments shown in Figure 2 when compared to previous investigations, a closely related stoichiometry becomes apparent [134]. By comparing the enthalpy produced from titration of the mixed peptide solution with the combined data from the individual injections, a ΔH in the range of −2 kcal/mol remains for additional processes in the peptide mixture (Figure 2F).
Circular dichroism (CD) analysis (not shown) combined with Dynamic light scattering (DLS) measurements were performed under the same conditions (Figure 3). Both peptides adopt largely α-helical conformations while they interact with the membrane. At the same time, large supramolecular structures form, suggesting the flocculation of the vesicles in the simultaneous presence of both peptides, similar to observations made with a designed model antimicrobial peptide [76,172]. Previously, a reduction in bilayer repeat distance of mechanically oriented membranes in the presence of magainin and magainin/PGLa, but not PGLa alone, has been observed [103]. It seems possible that these previous observations are related to the interbilayer interactions observed in our ITC and DLS experiments (Figure 2 and Figure 3). Figure 1F schematically illustrates the possible role of peptide–peptide interactions during such processes. Membrane pore formation by the AMP mastoparan-X and micellation at much higher P/L ratio was previously reported from ITC data [173].
In order to further explore possible interactions between membrane-associated PGLa and magainin 2, cross linking experiments have been performed with peptides carrying a GGC extensions [174]. This work shows that when added to egg PC/PG(1/1) lipid membranes, parallel dimers preferentially form [174]. Based on this data, covalent dimers linked through C-terminal GGC extensions were prepared, and all the (PGLa-GGC)2 and (magainin-GGC)2 homodimers, as well as the magainin-GGC/PGLa-GGC heterodimer were more active in calcein release from POPE/POPG 3/1 liposomes, than with the same amount of unmodified peptides in a mixture [75]. However, when investigated in POPC/cholesterol 3/1 mixtures, quite different results were obtained because only the PGLa-homodimer and the PGLa-magainin heterodimer, but not the individual peptides or their mixture, showed significant release activities [75]. Thus, the increased activities of the dimers seem not to be related to a particular structure formed by the combination of PGLa and magainin 2, but rather they reflect the increased membrane-perturbing properties of larger peptide aggregates [175,176,177]. Notably, the comparison of dimer and monomer antibacterial activities of wild type magainin and PGLa and their derivatives are complicated by the fact that they are already increased by the GGC extensions [161,163].
There are only few biophysical investigations that elucidate the mechanisms of the synergism observed for membrane-associated amphipathic peptides. Nevertheless, our view has already moved from models of heterooligomeric transmembrane bundles to helices that somehow play together when being oriented at the membrane surface. Thereby the situation resembles early research on cationic amphipathic peptides which had been found to reside at the membrane interface rather than forming transmembrane helical bundles [178,179]. From there the field has developed [46,58,180,181] and after years of research still bears surprises [74]. It can be expected that our views on synergism will evolve in a related manner [75,95,158,159].
Recently it has been shown that synergism is most pronounced when the peptides alone have not reached their optimum [75,161]. In good agreement with the SMART model [43], the peptide sequence and the membrane lipid composition are both important elements of synergy [75,161]. Thus, synergistic enhancements of calcein release only becomes apparent in membranes with intrinsic negative curvature and negative surface charge [75]. It is suggested that PGLa preconditions the more densely packed POPE membranes by softening up its interface, thus magainin, being more amphipathic, can penetrate deeper and be more active (Figure 1E) [75]. Furthermore, it is of interest for our understanding of the mechanism of action of antimicrobial peptides per se and the synergism they develop in their membrane associated state, that fluorescence quenching experiments are indicative of mesophase arrangements of both peptides along the membrane interface and a more densely packed supramolecular arrangement when both peptides are present in equimolar quantities [74,77] (Figure 1D,G).
Acknowledgments
We gratefully acknowledge the many co-workers and colleagues from our own team and from outside that over many years have contributed to this work. In particular, the collaborative efforts and discussion with Karl Lohner, Georg Pabst, Martin Hof, Mariana Amaro, Marina Rautenbach, Robert Vácha, Jarbas Resende, Rodrigo Verly, and their teams are much appreciated. The financial contributions of the Agence Nationale de la Recherche (projects TRANSPEP 07-PCV-0018, ProLipIn 10-BLAN-731, membraneDNP 12-BSV5-0012, MemPepSyn 14-CE34-0001-01, InMembrane 15-CE11-0017-01, Biosupramol 17-CE18-0033-3 and the LabEx Chemistry of Complex Systems 10-LABX-0026_CSC), the IRTG Soft Matter Science (Freiburg, Strasbourg), the Marie-Curie Research and Training Network 33439 of the European Commission BIOCONTROL, the University of Strasbourg, the CNRS, the Région Alsace and the RTRA International Center of Frontier Research in Chemistry, and the French Foundation for Medical Research (FRM) are gratefully acknowledged. BB is grateful to the Institut Universitaire de France for providing additional time to be dedicated to research.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Abbreviations
| Aib | α-aminobutyric acid |
| AMP | antimicrobial peptide |
| CD | circular dichroism |
| DLPC | 1, 2-lauroyl-sn-glycero-3-phosphocholine |
| DLS | dynamic light scattering |
| DMPC | 1, 2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPG | 1, 2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| DOPC | 1, 2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPG | 1, 2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| GUV | giant unilamellar vesicle |
| IP | in-plane |
| ITC | isothermal titration calorimetry |
| LUV | large unilamellar vesicle |
| MD | molecular dynamics |
| NMR | nuclear magnetic resonance |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PG | phosphatidylglycerol |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
| POPG | 1-palmitoyl-2-oleoyl -sn-glycero-3- phospho-(1′-rac-glycerol) |
| POPS | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine |
| SMART | Soft Membranes Adapt and Respond, also Transiently |
| TM | transmembrane |
References
- Boman, H.G. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Agerberth, B.; Gunne, H.; Odeberg, J.; Kogner, P.; Boman, H.G.; Gudmundsson, G.H. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc. Natl. Acad. Sci. USA 1995, 92, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vagvolgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J.; Hotchkiss, R.D. The production of bactericidal substances by aerobic sporulating bacilli. J. Exp. Med. 1941, 73, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.; Michl, H. Öber das Giftsekret der Gelbbauchunke Bombina variegata L. Toxicon 1962, 1, 33–39. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Sievert, D.M.; Hageman, J.C.; Boulton, M.L.; Tenover, F.C.; Downes, F.P.; Shah, S.; Rudrik, J.T.; Pupp, G.R.; Brown, W.J.; et al. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N. Engl. J. Med. 2003, 348, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, E.; Karakecili, A. Antibacterial activity on electrospun poly(lactide-co-glycolide) based membranes via Magainin II grafting. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zou, R.; Zhu, Y.; Liu, B.; Yao, D.; Jiang, J.; Wu, J.; Tian, H. Magainin II modified polydiacetylene micelles for cancer therapy. Nanoscale 2014, 6, 14772–14783. [Google Scholar] [CrossRef] [PubMed]
- Reijmar, K.; Edwards, K.; Andersson, K.; Agmo Hernandez, V. characterizing and controlling the loading and release of cationic amphiphilic peptides onto and from PEG-stabilized lipodisks. Langmuir 2016, 32, 12091–12099. [Google Scholar] [CrossRef] [PubMed]
- Rollins-Smith, L.A.; Doersam, J.K.; Longcore, J.E.; Taylor, S.K.; Shamblin, J.C.; Carey, C.; Zasloff, M.A. Antimicrobial peptide defenses against pathogens associated with global amphibian declines. Dev. Comp. Immunol. 2002, 26, 63–72. [Google Scholar] [CrossRef]
- Sansom, M.S.P. The Biophysics of Peptide Models of Ion Channels. Prog. Biophys. Mol. Biol. 1991, 55, 139–235. [Google Scholar] [CrossRef]
- Bechinger, B. Structure and Functions of Channel-Forming Polypeptides: Magainins, Cecropins, Melittin and Alamethicin. J. Membr. Biol. 1997, 156, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, L.S.; Elmore, J.M.; Dang, U.T.; Wallace, M.J.; Marreddy, R.; Lee, R.B.; Tan, G.T.; Hurdle, J.G.; Lee, R.E.; Sun, D. Solid-Phase Synthesis and Antibacterial Activity of Cyclohexapeptide Wollamide B Analogs. ACS Comb. Sci. 2018, 20, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yang, Y.; Uche, F.I.; Hart, S.R.; Li, W.W.; Yuan, C. Coupling Plant-Derived Cyclotides to Metal Surfaces: An Antibacterial and Antibiofilm Study. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Xue, Y.; Gao, W.; Li, J.; Zu, X.; Fu, D.; Bai, X.; Zuo, Y.; Hu, Z.; Zhang, F. Bacillaceae-derived peptide antibiotics since 2000. Peptides 2018, 101, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Laurencin, M.; Simon, M.; Fleury, Y.; Baudy-Floc’h, M.; Bondon, A.; Legrand, B. Selectivity Modulation and Structure of alpha/aza-beta(3) Cyclic Antimicrobial Peptides. Chemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A.; Dathe, M.E. Antifungal membranolytic activity of the tyrocidines against filamentous plant fungi. Biochimie 2016, 130, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Sychev, S.V.; Sukhanov, S.V.; Panteleev, P.V.; Shenkarev, Z.O.; Ovchinnikova, T.V. Marine antimicrobial peptide arenicin adopts a monomeric twisted beta-hairpin structure and forms low conductivity pores in zwitterionic lipid bilayers. Biopolymers 2017. [Google Scholar] [CrossRef]
- Salnikov, E.; Aisenbrey, C.; Balandin, S.V.; Zhmak, M.N.; Ovchinnikova, A.Y.; Bechinger, B. Structure and alignment of the membrane-associated antimicrobial peptide arenicin by oriented solid-state NMR spectroscopy. Biochemistry 2011, 50, 3784–3795. [Google Scholar] [CrossRef] [PubMed]
- Usachev, K.S.; Kolosova, O.A.; Klochkova, E.A.; Yulmetov, A.R.; Aganov, A.V.; Klochkov, V.V. Oligomerization of the antimicrobial peptide Protegrin-5 in a membrane-mimicking environment. Structural studies by high-resolution NMR spectroscopy. Eur. Biophys. J. 2017, 46, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Su, Y. Structure and dynamics of cationic membrane peptides and proteins: Insights from solid-state NMR. Protein Sci. 2011, 20, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Manjunath, G.B.; Akkapeddi, P.; Yarlagadda, V.; Hoque, J.; Uppu, D.S.S.M.; Konai, M.M.; Haldar, J. Small Molecular Antibacterial Peptoid Mimics: The Simpler the Better! J. Med. Chem. 2014, 57, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Arnusch, C.J.; Albada, H.B.; van Vaardegem, M.; Liskamp, R.M.J.; Sahl, H.G.; Shadkchan, Y.; Osherov, N.; Shai, Y. Trivalent Ultrashort Lipopeptides are Potent pH Dependent Antifungal Agents. J. Med. Chem. 2012, 55, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: In Vitro and In Vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef] [PubMed]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial beta-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; DeGrado, W.F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 2005, 127, 4128–4129. [Google Scholar] [CrossRef] [PubMed]
- Violette, A.; Fournel, S.; Lamour, K.; Chaloin, O.; Frisch, B.; Briand, J.P.; Monteil, H.; Guichard, G. Mimicking helical antibacterial peptides with nonpeptidic folding oligomers. Chem. Biol. 2006, 13, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.F.; Kuroda, K. Structural determinants of antimicrobial activity in polymers which mimic host defense peptides. Appl. Microbiol. Biotechnol. 2010, 87, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.W.; DeGrado, W.F.; Tew, G.N. De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 2008, 19, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Rank, L.A.; Walsh, N.M.; Liu, R.; Lim, F.Y.; Bok, J.W.; Huang, M.; Keller, N.P.; Gellman, S.H.; Hull, C.M. A Cationic Polymer that Shows High Antifungal Activity against Diverse Human Pathogens. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Insights into the mechanisms of action of host defence peptides from biophysical and structural investigations. J. Pept Sci. 2011, 17, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.D.; Wasserman, Z.R.; DeGrado, W.F. Synthetic amphiphilic peptide models for protein ion channels. Science 1988, 240, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Killian, J.A.; de Planque, M.R.R.; van der Wel, P.C.A.; Salemink, I.; De Kruijff, B.; Greathouse, D.V.; Koeppe, R.E. Modulation of membrane structure and function by hydrophobic mismatch between proteins and lipids. Pure Appl. Chem. 1998, 70, 75–82. [Google Scholar]
- Harzer, U.; Bechinger, B. The alignment of lysine-anchored membrane peptides under conditions of hydrophobic mismatch: A CD, 15N and 31P solid-state NMR spectroscopy investigation. Biochemistry 2000, 39, 13106–13114. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.; Aisenbrey, C.; Vidovic, V.; Bechinger, B. Solid-state NMR approaches to measure topological equilibria and dynamics of membrane polypeptides. Biochim. Biophys. Acta 2010, 1798, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The SMART Model: Soft Membranes Adapt and Respond, also Transiently, to External Stimuli. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Habermann, E.; Jentsch, J. Sequenzanalyse Des Melittins Aus Den Tryptischen Und Peptischen Spatstucken. Hoppe Seyler’s Z. Physiol. Chem. 1967, 348, 37–50. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Roversi, D.; Luca, V.; Aureli, S.; Park, Y.; Mangoni, M.L.; Stella, L. How Many AMP Molecules Kill a Bacterium? Spectroscopic Determination of PMAP-23 Binding to E. coli. ACS Chem. Biol. 2014, 9, 2003–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Holzl, M.A.; Hofer, J.; Steinberger, P.; Pfistershammer, K.; Zlabinger, G.J. Host antimicrobial proteins as endogenous immunomodulators. Immunol. Lett. 2008, 119, 4–11. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, D.G.; Cudic, P.; Yu, M.K.; Behenna, D.C.; Kruger, R. Synergy and duality in peptide antibiotic mechanisms. Curr. Opin. Chem. Biol. 1999, 3, 672–680. [Google Scholar] [CrossRef]
- Kozlowska, J.; Vermeer, L.S.; Rogers, G.B.; Rehnnuma, N.; Amos, S.B.; Koller, G.; McArthur, M.; Bruce, K.D.; Mason, A.J. Combined systems approaches reveal highly plastic responses to antimicrobial peptide challenge in Escherichia coli. PLoS Pathog. 2014, 10, e1004104. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; de Almeida, K.C.; Candido, E.S.; Murad, A.M.; Dias, S.C.; Franco, O.L. Comparative NanoUPLC-MS(E) analysis between magainin I-susceptible and -resistant Escherichia coli strains. Sci. Rep. 2017, 7, 4197. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Zasloff, M.; Opella, S.J. Structure and Dynamics of the Antibiotic Peptide PGLa in Membranes by Multidimensional Solution and Solid-State NMR Spectroscopy. Biophys. J. 1998, 74, 981–987. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Tokuda, H.; Funakoshi, S.; Fujii, N.; Miyajima, K. Orientational and Aggregational States of Magainin 2 in Phospholipid Bilayers. Biochemistry 1994, 33, 3342–3349. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Wieprecht, T.; Apostolov, O.; Seelig, J. Binding of the antibacterial peptide magainin 2 amide to small and large unilamellar vesicles. Biophys. Chem. 2000, 85, 187–198. [Google Scholar] [CrossRef]
- Ludtke, S.; He, K.; Huang, H. Membrane thinning caused by magainin 2. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like action of linear cationic membrane-active antibiotic peptides. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Duclohier, H.; Molle, G.; Spach, G. Antimicrobial Peptide Magainin I from Xenopus Skin Forms Anion-Permeable Channels in Planar Lipid Bilayers. Biophys. J. 1989, 56, 1017–1021. [Google Scholar] [CrossRef]
- Cruciani, R.A.; Barker, J.L.; Zasloff, M.; Chen, H.C.; Colamonici, O. Antibiotic magainins exert cytolytic activity transformed cell lines through channel formation. Proc. Natl. Acad. Sci. USA 1991, 88, 3792–3796. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Fink, J.; Merrifield, R.B.; Mauzerall, D. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 5072–5076. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kawano, R. Channel Current Analysis for Pore-forming Properties of an Antimicrobial Peptide, Magainin 1, Using the Droplet Contact Method. Anal. Sci. 2016, 32, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, D.P.; Hess, B.; Sansom, M.S. Analysis and evaluation of channel models: Simulations of alamethicin. Biophys. J. 2002, 83, 2393–2407. [Google Scholar] [CrossRef]
- Islam, M.Z.; Alam, J.M.; Tamba, Y.; Karal, M.A.S.; Yamazaki, M. The single GUV method for revealing the functions of antimicrobial, pore-forming toxin, and cell-penetrating peptides or proteins. Phys. Chem. Chem. Phys. 2014, 16, 15752–15767. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Cavenaugh, A.; Journigan, V.; Pokorny, A.; Almeida, P.F.F. A quantitative model for the all-or-none permeabilization of phospholipid vesicles by the antimicrobial peptide cecropin A. Biophys. J. 2008, 94, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Tamba, Y.; Ariyama, H.; Levadny, V.; Yamazaki, M. Kinetic Pathway of Antimicrobial Peptide Magainin 2-Induced Pore Formation in Lipid Membranes. J. Phys. Chem. B 2010, 114, 12018–12026. [Google Scholar] [CrossRef] [PubMed]
- Barns, K.J.; Weisshaar, J.C. Real-time attack of LL-37 on single Bacillus subtilis cells. Biochim. Biophys. Acta 2013, 1828, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, N.; Bakshi, S.; Weisshaar, J.C. Localized permeabilization of E. coli membranes by the antimicrobial peptide Cecropin A. Biochemistry 2013, 52, 6584–6594. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Choi, H.; Weisshaar, J.C. Melittin-Induced Permeabilization, Re-sealing, and Re-permeabilization of E. coli Membranes. Biophys. J. 2018, 114, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Yang, Z.; Weisshaar, J.C. Oxidative stress induced in E. coli by the human antimicrobial peptide LL-37. PLoS Pathog. 2017, 13, e1006481. [Google Scholar] [CrossRef] [PubMed]
- Barns, K.J.; Weisshaar, J.C. Single-cell, time-resolved study of the effects of the antimicrobial peptide alamethicin on Bacillus subtilis. Biochim. Biophys. Acta 2016, 1858, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Vacha, R.; Frenkel, D. Simulations suggest possible novel membrane pore structure. Langmuir 2014, 30, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, C.; Bechinger, B. Molecular Packing of Amphipathic Peptides on the Surface of Lipid Membranes. Langmuir 2014, 30, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Leber, R.; Pachler, M.; Kabelka, I.; Svoboda, I.; Enkoller, D.; Vácha, R.; Lohner, K.; Pabst, G. Synergism of Antimicrobial Frog Peptides Couples to Membrane Intrinsic Curvature Strain. Biophys. J. 2018, in press. [Google Scholar]
- Marquette, A.; Lorber, B.; Bechinger, B. Reversible liposome association induced by LAH4: A peptide with potent antimicrobial and nucleic acid transfection activities. Biophys. J. 2010, 98, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Salnikov, E.; Glattard, E.; Aisenbrey, C.; Bechinger, B. Magainin 2-PGLa interactions in membranes—Two peptides that exhibit synergistic enhancement of antimicrobial activity. Curr. Top. Med. Chem. 2015, 16, 65–75. [Google Scholar] [CrossRef]
- Klocek, G.; Schulthess, T.; Shai, Y.; Seelig, J. Thermodynamics of melittin binding to lipid bilayers. Aggregation and pore formation. Biochemistry 2009, 48, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, A.; Thennarasu, S.; Lee, D.K.; Tan, A.; Maloy, L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006, 91, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.J.; Moussaoui, W.; Abdelrhaman, T.; Boukhari, A.; Bertani, P.; Marquette, A.; Shooshtarizaheh, P.; Moulay, G.; Boehm, N.; Guerold, B.; et al. Structural determinants of antimicrobial and antiplasmodial activity and selectivity in histidine rich amphipathic cationic peptides. J. Biol. Chem. 2009, 284, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.A.; Separovic, F. Antimicrobial Peptide Structures: From Model Membranes to Live Cells. Chemistry 2018, 24, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.M.; Goldberg, G.K.; Ferguson, B.M.; Schoeneck, M.W.; Libardo, M.D.; Mayeux, S.E.; Shrestha, A.; Bogardus, K.A.; Hammer, J.; Pryshchep, S.; et al. Complementary Effects of Host Defense Peptides Piscidin 1 and Piscidin 3 on DNA and Lipid Membranes: Biophysical Insights into Contrasting Biological Activities. J. Phys. Chem. B 2015, 119, 15235–15246. [Google Scholar] [CrossRef] [PubMed]
- Resende, J.M.; Verly, R.M.; Aisenbrey, C.; Amary, C.; Bertani, P.; Pilo-Veloso, D.; Bechinger, B. Membrane interactions of Phylloseptin-1, -2, and -3 peptides by oriented solid-state NMR spectroscopy. Biophys. J. 2014, 107, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Resende, J.M.; Moraes, C.M.; Munhoz, V.H.D.O.; Aisenbrey, C.; Verly, R.M.; Bertani, P.; Cesar, A.; Pilo-Veloso, D.; Bechinger, B. Membrane structure and conformational changes of the antibiotic heterodimeric peptide distinctin by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 16639–16644. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Sansom, M.S. Alamethicin and related peptaibols—Model ion channels. Eur. Biophys. J. 1993, 22, 105–124. [Google Scholar] [CrossRef] [PubMed]
- North, C.L.; Barranger-Mathys, M.; Cafiso, D.S. Membrane orientation of the N-terminal segment of alamethicin determined by solid-state 15N-NMR. Biophys. J. 1995, 69, 2392–2397. [Google Scholar] [CrossRef]
- Bak, M.; Bywater, R.P.; Hohwy, M.; Thomsen, J.K.; Adelhorst, K.; Jakobsen, H.J.; Sorensen, O.W.; Nielsen, N.C. Conformation of alamethicin in oriented phospholipid bilayers determined by N-15 solid-state nuclear magnetic resonance. Biophys. J. 2001, 81, 1684–1698. [Google Scholar] [CrossRef]
- Salnikov, E.S.; Friedrich, H.; Li, X.; Bertani, P.; Reissmann, S.; Hertweck, C.; O’Neil, J.D.; Raap, J.; Bechinger, B. Structure and alignment of the membrane-associated peptaibols ampullosporin A and alamethicin by oriented 15N and 31P solid-state NMR spectroscopy. Biophys. J. 2009, 96, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Raya, J.; De Zotti, M.; Zaitseva, E.; Peggion, C.; Ballano, G.; Toniolo, C.; Raap, J.; Bechinger, B. Alamethicin supramolecular organization in lipid membranes from 19F solid-state NMR. Biophys. J. 2016, 111, 2450–2459. [Google Scholar] [CrossRef] [PubMed]
- Milov, A.D.; Samoilova, R.I.; Tsvetkov, Y.D.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Handgraaf, J.W.; Raap, J. Structure of Self-Aggregated Alamethicin in ePC Membranes Detected by Pulsed Electron-Electron Double Resonance and Electron Spin Echo Envelope Modulation Spectroscopies. Biophys. J. 2009, 96, 3197–3209. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Ludtke, S.J.; Heller, W.T.; Huang, H.W. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 1996, 71, 2669–2679. [Google Scholar] [CrossRef]
- Avitabile, C.; D’Andrea, L.D.; Romanelli, A. Circular Dichroism studies on the interactions of antimicrobial peptides with bacterial cells. Sci. Rep. 2014, 4, 4293. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.; Bechinger, B. Lipid-controlled peptide topology and interactions in bilayers: Structural insights into the synergistic enhancement of the antimicrobial activities of PGLa and magainin 2. Biophys. J. 2011, 100, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Tremouilhac, P.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 2006, 281, 32089–32094. [Google Scholar] [CrossRef] [PubMed]
- Tremouilhac, P.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid state 2H-NMR. Biochim. Biophys. Acta 2006, 1758, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Tiltak, D.; Ehni, S.; Wadhwani, P.; Ulrich, A.S. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim. Biophys. Acta 2012, 1818, 1764–1776. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Mason, A.J.; Bechinger, B. Membrane order perturbation in the presence of antimicrobial peptides by 2H solid-state NMR spectroscopy. Biochimie 2009, 91, 743. [Google Scholar] [CrossRef]
- Bortolus, M.; Dalzini, A.; Toniolo, C.; Hahm, K.S.; Maniero, A.L. Interaction of hydrophobic and amphipathic antimicrobial peptides with lipid bicelles. J. Pept. Sci. 2014, 20, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Spano, J.; Park, E.K.; Wi, S. Evidence of pores and thinned lipid bilayers induced in oriented lipid membranes interacting with the antimicrobial peptides, magainin-2 and aurein-3.3. Biochim. Biophys. Acta 2009, 1788, 1482–1496. [Google Scholar] [CrossRef] [PubMed]
- Hallock, K.J.; Lee, D.K.; Omnaas, J.; Mosberg, H.I.; Ramamoorthy, A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys. J. 2002, 83, 1004–1013. [Google Scholar] [CrossRef]
- Grage, S.L.; Afonin, S.; Kara, S.; Buth, G.; Ulrich, A.S. Membrane Thinning and Thickening Induced by Membrane-Active Amphipathic Peptides. Front. Cell Dev. Biol. 2016, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Harmouche, N.; Pachler, M.; Lohner, K.; Pabst, G.; Bechinger, B. Lipid-mediated interactions between the amphipathic antimicrobial peptides magainin 2 and PGLa in phospholipid bilayers. Preparation submitted for publication. 2018. [Google Scholar]
- Chen, F.Y.; Lee, M.T.; Huang, H.W. Evidence for membrane thinning effect as the mechanism for Peptide-induced pore formation. Biophys. J. 2003, 84, 3751–3758. [Google Scholar] [CrossRef]
- Mecke, A.; Lee, D.K.; Ramamoorthy, A.; Orr, B.G.; Banaszak Holl, M.M. Membrane thinning due to antimicrobial peptide binding: An atomic force microscopy study of MSI-78 in lipid bilayers. Biophys. J. 2005, 89, 4043–4050. [Google Scholar] [CrossRef] [PubMed]
- Farrotti, A.; Bocchinfuso, G.; Palleschi, A.; Rosato, N.; Salnikov, E.S.; Voievoda, N.; Bechinger, B.; Stella, L. Molecular Dynamics Methods to Predict Peptide Location in Membranes: LAH4 as a Stringent Test Case. Biochim. Biophys. Acta 2015, 1848, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Pino-Angeles, A.; Leveritt, J.M., III; Lazaridis, T. Pore Structure and Synergy in Antimicrobial Peptides of the Magainin Family. PLoS Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef] [PubMed]
- Kabelka, I.; Vacha, R. Optimal conditions for opening of membrane pore by amphiphilic peptides. J. Chem. Phys. 2015, 143, 243115. [Google Scholar] [CrossRef] [PubMed]
- Amos, S.T.; Vermeer, L.S.; Ferguson, P.M.; Kozlowska, J.; Davy, M.; Bui, T.T.; Drake, A.F.; Lorenz, C.D.; Mason, A.J. Antimicrobial Peptide Potency is Facilitated by Greater Conformational Flexibility when Binding to Gram-negative Bacterial Inner Membranes. Sci. Rep. 2016, 6, 37639. [Google Scholar] [CrossRef] [PubMed]
- Smart, M.; Rajagopal, A.; Liu, W.K.; Ha, B.Y. Opposing effects of cationic antimicrobial peptides and divalent cations on bacterial lipopolysaccharides. Phys. Rev. E 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Smith, J.C.; Ulmschneider, M.B.; Ulrich, A.S.; Strandberg, E. Reorientation and Dimerization of the Membrane-Bound Antimicrobial Peptide PGLa from Microsecond All-Atom MD Simulations. Biophys. J. 2012, 103, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Marcelja, S.; Horn, R.G. Physical principles of membrane organization. Q. Rev. Biophys. 1980, 13, 121–200. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Rationalizing the membrane interactions of cationic amphipathic antimicrobial peptides by their molecular shape. Curr. Opin. Colloid Interface Sci. 2009, 14, 349–355. [Google Scholar] [CrossRef]
- Paterson, D.J.; Tassieri, M.; Reboud, J.; Wilson, R.; Cooper, J.M. Lipid topology and electrostatic interactions underpin lytic activity of linear cationic antimicrobial peptides in membranes. Proc. Natl. Acad. Sci. USA 2017, 114, E8324–E8332. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1998, 1376, 391–400. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion, and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Aisenbrey, C.; Harmouche, N.; Raya, J.; Bertani, P.; Voievoda, N.; Süss, R.; Bechinger, B. pH-dependent membrane interactions of the histidine-rich cell penetrating peptide LAH4-L1. Biophys. J. 2017, 113, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliot, M.; Hancock, R.E.W.; Straus, S.K. Effect of Membrane Composition on Antimicrobial Peptides Aurein 2.2 and 2.3 From Australian Southern Bell Frogs. Biophys. J. 2009, 96, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta 2011, 1808, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Wu, Y. Lipid-alamethicin interactions influence alamethicin orientation. Biophys. J. 1991, 60, 1079–1087. [Google Scholar] [CrossRef]
- Bechinger, B. Towards membrane protein design: pH-sensitive topology of histidine-containing polypeptides. J. Mol. Biol. 1996, 263, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim. Biophys. Acta 2006, 1758, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Lee, T.H.; Mechler, A.I.; Swann, M.J.; Aguilar, M.I. Real-time measurement of membrane conformational states induced by antimicrobial peptides: Balance between recovery and lysis. Sci. Rep. 2014, 4, 5479. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Waring, A.J.; Separovic, F.; Lee, K.Y.C. Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J. 2016, 111, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. Membrane-lytic peptides. Crit. Rev. Plant Sci. 2004, 23, 271–292. [Google Scholar] [CrossRef]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wenk, M.; Seelig, J. Magainin 2 amide interaction with lipid membranes: Calorimetric detection of peptide binding and pore formation. Biochemistry 1998, 37, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Harada, M.; Funakoshi, S.; Fujii, N.; Miyajima, K. Physicochemical Determinants for the Interactions of Magainins 1 and 2 with Acidic Lipid Bilayers. Biochim. Biophys. Acta 1991, 1063, 162–170. [Google Scholar] [CrossRef]
- Klocek, G.; Seelig, J. Melittin interaction with sulfated cell surface sugars. Biochemistry 2008, 47, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Beyermann, M.; Seelig, J. Binding of antibacterial magainin peptides to electrically neutral membranes: Thermodynamics and structure. Biochemistry 1999, 38, 10377–10378. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Kramer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, E1409–E1418. [Google Scholar] [CrossRef] [PubMed]
- Perrone, B.; Miles, A.J.; Salnikov, E.S.; Wallace, B.; Bechinger, B. Lipid-interactions of the LAH4, a peptide with antimicrobial and nucleic transfection activities. Eur. Biophys. J. 2014, 43, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.J.; Martinez, A.; Glaubitz, C.; Danos, O.; Kichler, A.; Bechinger, B. The antibiotic and DNA-transfecting peptide LAH4 selectively associates with, and disorders, anionic lipids in mixed membranes. FASEB J. 2006, 20, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Voievoda, N. Biophysical Investigations of the Membrane and Nucleic Acids Interactions of the Transfection Peptide LAH4-L1. Ph.D. Thesis, University of Strasbourg, Strasbourg, France, 2014. [Google Scholar]
- Kindrachuk, J.; Napper, S. Structure-activity relationships of multifunctional host defence peptides. Mini Rev. Med. Chem. 2010, 10, 596–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Zhou, L.; Lakshminarayanan, R.; Beuerman, R.W. Multivalent Antimicrobial Peptides as Therapeutics: Design Principles and Structural Diversities. Int. J. Pept. Res. Ther. 2010, 16, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Hadley, E.B.; Hancock, R.E. Strategies for the Discovery and Advancement of Novel Cationic Antimicrobial Peptides. Curr. Top. Med. Chem. 2010, 10, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Oyston, P.C.; Fox, M.A.; Richards, S.J.; Clark, G.C. Novel peptide therapeutics for treatment of infections. J. Med. Microbiol. 2009, 58 (Pt 8), 977–987. [Google Scholar] [CrossRef]
- Ahn, M.; Gunasekaran, P.; Rajasekaran, G.; Kim, E.Y.; Lee, S.J.; Bang, G.; Cho, K.; Hyun, J.K.; Lee, H.J.; Jeon, Y.H.; et al. Pyrazole derived ultra-short antimicrobial peptidomimetics with potent anti-biofilm activity. Eur. J. Med. Chem. 2017, 125, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Shao, C.; Wang, J.; Shan, A.; Xu, L.; Dong, N.; Li, Z. Short, multiple-stranded beta-hairpin peptides have antimicrobial potency with high selectivity and salt resistance. Acta Biomater. 2016, 30, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Acar, J.F. Antibiotic synergy and antagonism. Med. Clin. N. Am. 2000, 84, 1391–1406. [Google Scholar] [CrossRef]
- Kim, E.Y.; Rajasekaran, G.; Shin, S.Y. LL-37-derived short antimicrobial peptide KR-12-a5 and its d-amino acid substituted analogs with cell selectivity, anti-biofilm activity, synergistic effect with conventional antibiotics, and anti-inflammatory activity. Eur. J. Med. Chem. 2017, 136, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.E.; Dubois, A.V.; Ingram, R.J.; Weldon, S.; Taggart, C.C.; Elborn, J.S.; Tunney, M.M. Activity of innate antimicrobial peptides and ivacaftor against clinical cystic fibrosis respiratory pathogens. Int. J. Antimicrob. Agents 2017, 50, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sakoulas, G.; Kumaraswamy, M.; Kousha, A.; Nizet, V. Interaction of Antibiotics with Innate Host Defense Factors against Salmonella enterica Serotype Newport. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Bolosov, I.A.; Kalashnikov, A.A.; Panteleev, P.V.; Ovchinnikova, T.V. Analysis of Synergistic Effects of Antimicrobial Peptide Arenicin-1 and Conventional Antibiotics. Bull. Exp. Biol. Med. 2017, 162, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Walkenhorst, W.F.; Sundrud, J.N.; Laviolette, J.M. Additivity and synergy between an antimicrobial peptide and inhibitory ions. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Del Prete, M.S.; Paggi, A.M.; D’Errico, M.M.; Scalise, G. Combination studies between polycationic peptides and clinically used antibiotics against Gram-positive and Gram-negative bacteria. Peptides 2000, 21, 1155–1160. [Google Scholar] [CrossRef]
- Mor, A.; Hani, K.; Nicolas, P. The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J. Biol. Chem. 1994, 269, 31635–31641. [Google Scholar] [PubMed]
- Vaz Gomes, A.; de Waal, A.; Berden, J.A.; Westerhoff, H.V. Electric Potentiation, Cooperativity, and Synergism of Magainin Peptides in Protein-Free Liposomes. Biochemistry 1993, 32, 5365–5372. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Hirakura, Y.; Matsuzaki, K. Bacteria-selective synergism between the antimicrobial peptides alpha-helical magainin 2 and cyclic beta-sheet tachyplesin I: Toward cocktail therapy. Biochemistry 2001, 40, 14330–14335. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V.; Zasloff, M.; Rosner, J.L.; Hendler, R.W.; de Waal, A.; Vaz, G.; Jongsma, P.M.; Riethorst, A.; Juretic, D. Functional synergism of the magainins PGLa and magainin-2 in Escherichia coli, tumor cells and liposomes. Eur. J. Biochem. 1995, 228, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Mitani, Y.; Akada, K.; Murase, O.; Yoneyama, S.; Zasloff, M.; Miyajima, K. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry 1998, 37, 15144–15153. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Huynh, Q.; Barlehner, D.; Heerklotz, H. Additive and Synergistic Membrane Permeabilization by Antimicrobial (Lipo)Peptides and Detergents. Biophys. J. 2014, 106, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. Synergistic Insertion of Antimicrobial Magainin-Family Peptides in Membranes Depends on the Lipid Spontaneous Curvature. Biophys. J. 2013, 104, L09–L11. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, E.S.; Aisenbrey, C.; Aussenac, F.; Ouari, O.; Sarrouj, H.; Reiter, C.; Tordo, P.; Engelke, F.; Bechinger, B. Membrane topologies of the PGLa antimicrobial peptide and a transmembrane anchor sequence by Dynamic Nuclear Polarization/solid-state NMR spectroscopy. Sci. Rep. 2016, 6, 20895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glattard, E.; Salnikov, E.S.; Aisenbrey, C.; Bechinger, B. Investigations of the synergistic enhancement of antimicrobial activity in mixtures of magainin 2 and PGLa. Biophys. Chem. 2016, 210, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Horn, D.; Reisser, S.; Zerweck, J.; Wadhwani, P.; Ulrich, A.S. 2H-NMR and MD Simulations Reveal Membrane-Bound Conformation of Magainin 2 and Its Synergy with PGLa. Biophys. J. 2016, 111, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Imura, Y.; Yamamoto, M.; Kobayashi, S.; Yano, Y.; Matsuzaki, K. Interaction of a magainin-PGLa hybrid peptide with membranes: Insight into the mechanism of synergism. Biochemistry 2007, 46, 14284–14290. [Google Scholar] [CrossRef] [PubMed]
- Zerweck, J.; Strandberg, E.; Kukharenko, O.; Reichert, J.; Burck, J.; Wadhwani, P.; Ulrich, A.S. Molecular mechanism of synergy between the antimicrobial peptides PGLa and magainin 2. Sci. Rep. 2017, 7, 13153. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Horn, D.; Pritz, G.; Berditsch, M.; Bürck, J.; Wadhwani, P.; Ulrich, A.S. Influence of hydrophobic residues on the activity of the antimicrobial peptide magainin 2 and its synergy with PGLa. J. Pept. Sci. 2015, 21, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Benz, R.; Hancock, R.E. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 1999, 38, 8102–8111. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Lee, H. Synergistic effects of magainin 2 and PGLa on their heterodimer formation, aggregation, and insertion into the bilayer. RSC Adv. 2015, 5, 2047–2055. [Google Scholar] [CrossRef]
- Zerweck, J.; Strandberg, E.; Burck, J.; Reichert, J.; Wadhwani, P.; Kukharenko, O.; Ulrich, A.S. Homo- and heteromeric interaction strengths of the synergistic antimicrobial peptides PGLa and magainin 2 in membranes. Eur. Biophys. J. 2016, 45, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Beyermann, M.; Seelig, J. Membrane binding and pore formation of the antibacterial peptide PGLa: Thermodynamic and mechanistic aspects. Biochemistry 2000, 39, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Apostolov, O.; Beyermann, M.; Seelig, J. Thermodynamics of the alpha-helix-coil transition of amphipathic peptides in a membrane environment: Implications for the peptide-membrane binding equilibrium. J. Mol. Biol. 1999, 294, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Wieprecht, T.; Beyermann, M.; Seelig, J. Thermodynamics of the coil-alpha-helix transition of amphipathic peptides in a membrane environment: The role of vesicle curvature. Biophys. Chem. 2002, 96, 191–201. [Google Scholar] [CrossRef]
- Vermeer, L.S.; Marquette, A.; Schoup, M.; Fenard, D.; Galy, A.; Bechinger, B. Simultaneous Analysis of Secondary Structure and Light Scattering from Circular Dichroism Titrations: Application to Vectofusin-1. Sci. Rep. 2016, 6, 39450. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.R.; Andresen, T.L. Thermodynamic profiling of peptide membrane interactions by isothermal titration calorimetry: A search for pores and micelles. Biophys. J. 2011, 101, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Kodama, H.; Kondo, M.; Wakamatsu, K.; Takeda, A.; Tachi, T.; Matsuzaki, K. Effects of peptide dimerization on pore formation: Antiparallel disulfide-dimerized magainin 2 analogue. Biopolymers 2001, 58, 437–446. [Google Scholar] [CrossRef]
- Dempsey, C.E.; Ueno, S.; Avison, M.B. Enhanced membrane permeabilization and antibacterial activity of a disulfide-dimerized magainin analogue. Biochemistry 2003, 42, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, E.N.; Santos-Filho, N.A.; Ramos, M.A.; Bauab, T.M.; Camargo, I.L.; Cilli, E.M. C-terminal Lysine-Linked Magainin 2 with Increased Activity Against Multidrug-Resistant Bacteria. Protein Pept. Lett. 2016, 23, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Verly, R.M.; Resende, J.M.; Junior, E.F.C.; de Magalhães, M.T.C.; Guimarães, C.F.C.R.; Munhoz, V.H.O.; Bemquerer, M.P.; Almeida, F.C.L.; Santoro, M.M.; Piló-Veloso, D.; et al. Structure and membrane interactions of the homodimeric antibiotic peptide homotarsinin. Sci. Rep. 2017, 7, 40854. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Kim, Y.; Chirlian, L.E.; Gesell, J.; Neumann, J.M.; Montal, M.; Tomich, J.; Zasloff, M.; Opella, S.J. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR 1991, 1, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Vogt, T.C.B.; Bechinger, B. The interactions of histidine-containing amphipathic helical peptide antibiotics with lipid bilayers: The effects of charges and pH. J. Biol. Chem. 1999, 274, 29115–29121. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef]
Figure 1.
Schematic models illustrating how antimicrobial peptides work and interact with membranes (A–D), and how two peptides can interact synergistically in a membrane environment (E–G). (A) Peptides such as magainin partition into the membrane interface and cause disordering of the lipid packing. (B) Bilayer openings form stochastically when the peptide concentration increases locally, or when the membrane disrupts at high peptide-to-lipid ratios [72]. Along the openings, the peptides can insert and cross in in-planar or at tilted alignments. (C) In molecular dynamics calculations schematic, amphipathic helices have been simulated to form double belts [73], an arrangement which also agrees with the in-planar alignment of the peptide helices observed by solid-state nuclear magnetic resonance NMR spectroscopy [38]. (D) Fluorescence quenching experiments suggest mesophase structures formed by in-plane oriented helices [74]. (E) The membrane disruptive properties of one peptide (yellow) help the insertion of another one (blue), which by itself is less likely to partition into membranes of high negative curvature [75]. (F) Peptide-peptide contacts result in the agglutination of liposomes (Figure 3) [76], and could be responsible for synergistic enhancement of activities. (G) A more densely packed mesophase arrangement forms in the presence of two peptides with complementary charge distribution such as magainin 2 and PGLa [77]. Notably, multiple mechanisms, such as a combination of E, F, and G may apply. Panels A, B, E, and F show side views, panels C, D, and G show top views of the lipid bilayer.
Figure 1.
Schematic models illustrating how antimicrobial peptides work and interact with membranes (A–D), and how two peptides can interact synergistically in a membrane environment (E–G). (A) Peptides such as magainin partition into the membrane interface and cause disordering of the lipid packing. (B) Bilayer openings form stochastically when the peptide concentration increases locally, or when the membrane disrupts at high peptide-to-lipid ratios [72]. Along the openings, the peptides can insert and cross in in-planar or at tilted alignments. (C) In molecular dynamics calculations schematic, amphipathic helices have been simulated to form double belts [73], an arrangement which also agrees with the in-planar alignment of the peptide helices observed by solid-state nuclear magnetic resonance NMR spectroscopy [38]. (D) Fluorescence quenching experiments suggest mesophase structures formed by in-plane oriented helices [74]. (E) The membrane disruptive properties of one peptide (yellow) help the insertion of another one (blue), which by itself is less likely to partition into membranes of high negative curvature [75]. (F) Peptide-peptide contacts result in the agglutination of liposomes (Figure 3) [76], and could be responsible for synergistic enhancement of activities. (G) A more densely packed mesophase arrangement forms in the presence of two peptides with complementary charge distribution such as magainin 2 and PGLa [77]. Notably, multiple mechanisms, such as a combination of E, F, and G may apply. Panels A, B, E, and F show side views, panels C, D, and G show top views of the lipid bilayer.
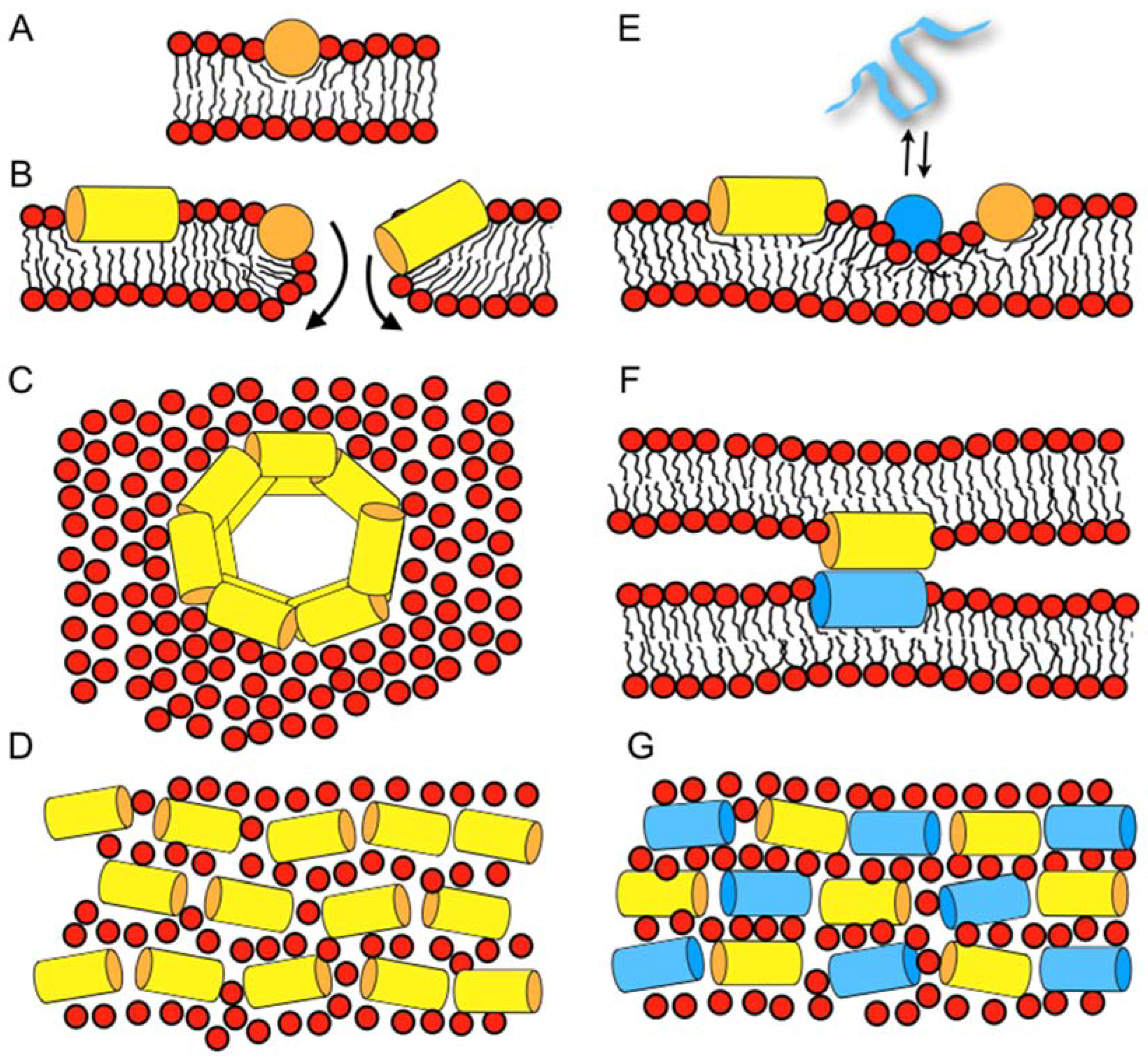
Figure 2.
Isothermal Titration Calorimetry measurements of large unilamellar vesicles (LUVs) made of palmitoyl-oleoyl-phosphatidylethanolamine/ palmitoyl-oleoyl-phosphatidylglycerol POPE/POPG (3/1) at 440 µM total lipid concentration, into which solutions of 85 µM magainin 2 (A), 140 µM PGLa (B), and the equimolar mixture of PGLa and magainin 2 at 105 µM total peptide concentration (C) have been injected successively. Buffer: 10 mM Tris-HCl, pH 7, 100 mM NaCl. The inserts show close-ups of the regions t > 2215 s. The fittings of the data with a single binding site model are displayed as solid lines in panels D–F. The open circles in panel F show the difference of the heat of reactions when comparing the mixture of peptides with the sums obtained from the magainin 2 and PGLa titrations (i.e., half the intensities of the fitted curves shown in panels D and E, dashed line). The values of the enthalpies (ΔH), entropies (ΔS), binding constant (K), and apparent stoichiometry (N) are reported in the corresponding graphs.
Figure 2.
Isothermal Titration Calorimetry measurements of large unilamellar vesicles (LUVs) made of palmitoyl-oleoyl-phosphatidylethanolamine/ palmitoyl-oleoyl-phosphatidylglycerol POPE/POPG (3/1) at 440 µM total lipid concentration, into which solutions of 85 µM magainin 2 (A), 140 µM PGLa (B), and the equimolar mixture of PGLa and magainin 2 at 105 µM total peptide concentration (C) have been injected successively. Buffer: 10 mM Tris-HCl, pH 7, 100 mM NaCl. The inserts show close-ups of the regions t > 2215 s. The fittings of the data with a single binding site model are displayed as solid lines in panels D–F. The open circles in panel F show the difference of the heat of reactions when comparing the mixture of peptides with the sums obtained from the magainin 2 and PGLa titrations (i.e., half the intensities of the fitted curves shown in panels D and E, dashed line). The values of the enthalpies (ΔH), entropies (ΔS), binding constant (K), and apparent stoichiometry (N) are reported in the corresponding graphs.
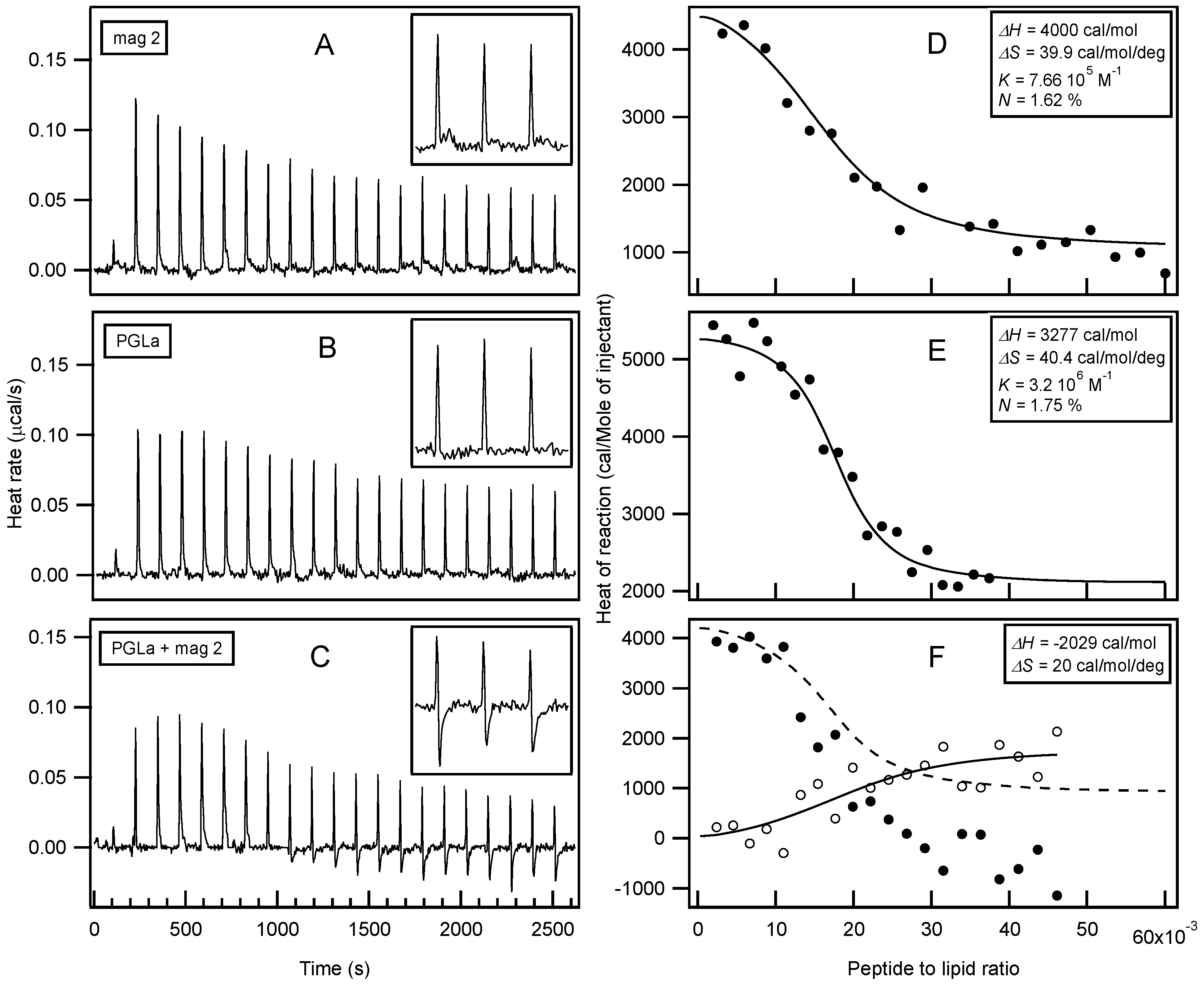
Figure 3.
Dynamic light s cattering (DLS) size measurement of LUVs made of 440 µM POPE/POPG (3/1) and incubated with magainin 2 (long dashed line), PGLa (short dashed line) or an equimolar mixture of PGLa and magainin 2 (continuous line) in 10 mM Tris-HCl buffer pH 7. The vesicles were made by mechanical extrusion through a 100 nm pore filter and the peptide to lipid ratio was kept constant at P/L = 3.6% for all tree measurements (this corresponds to the same condition for peptide injection at t ≈ 2030 s in Figure 2C). The averaged hydrodynamic radius < RH > is indicated in the upper right of the graphs.
Figure 3.
Dynamic light s cattering (DLS) size measurement of LUVs made of 440 µM POPE/POPG (3/1) and incubated with magainin 2 (long dashed line), PGLa (short dashed line) or an equimolar mixture of PGLa and magainin 2 (continuous line) in 10 mM Tris-HCl buffer pH 7. The vesicles were made by mechanical extrusion through a 100 nm pore filter and the peptide to lipid ratio was kept constant at P/L = 3.6% for all tree measurements (this corresponds to the same condition for peptide injection at t ≈ 2030 s in Figure 2C). The averaged hydrodynamic radius < RH > is indicated in the upper right of the graphs.
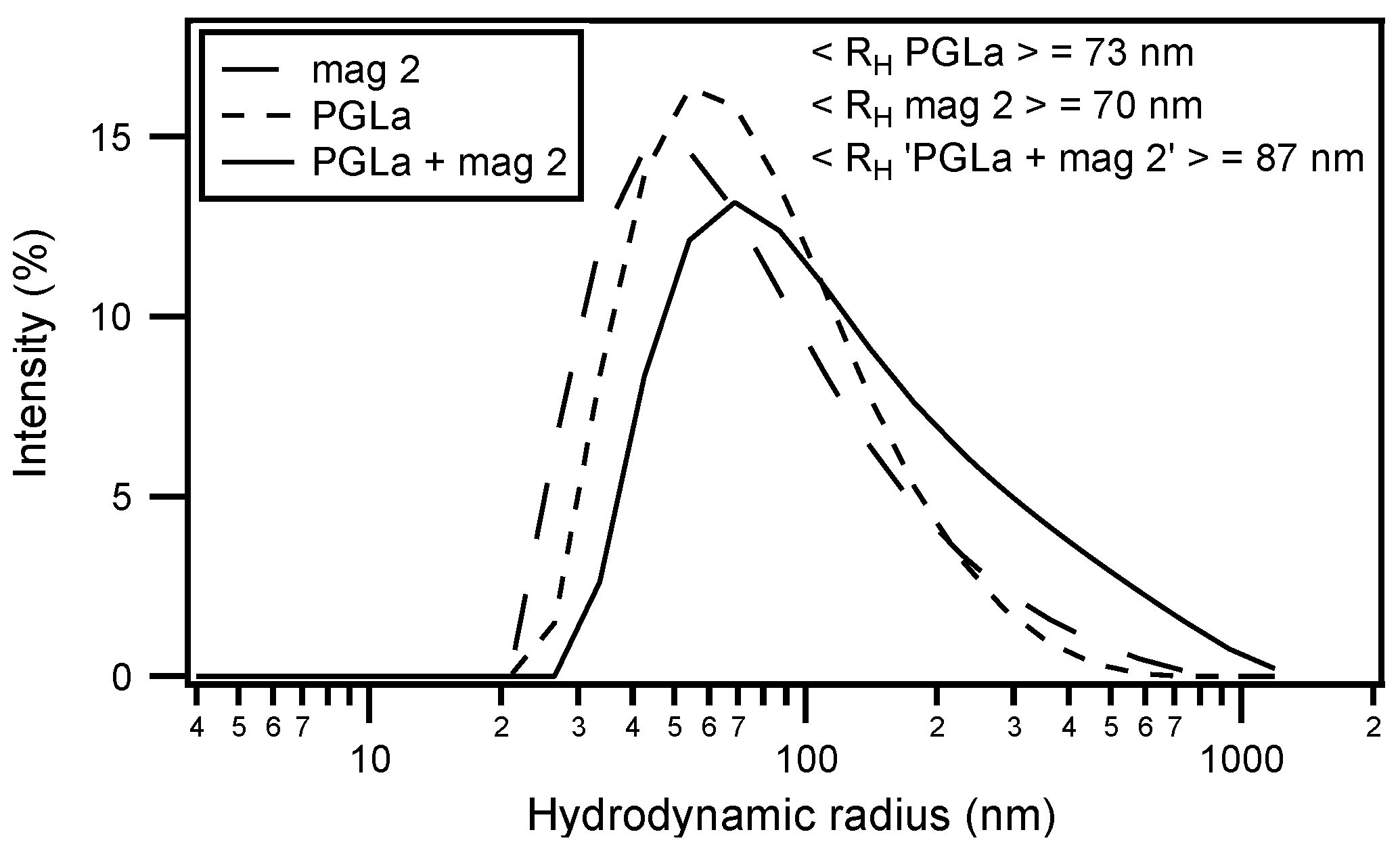

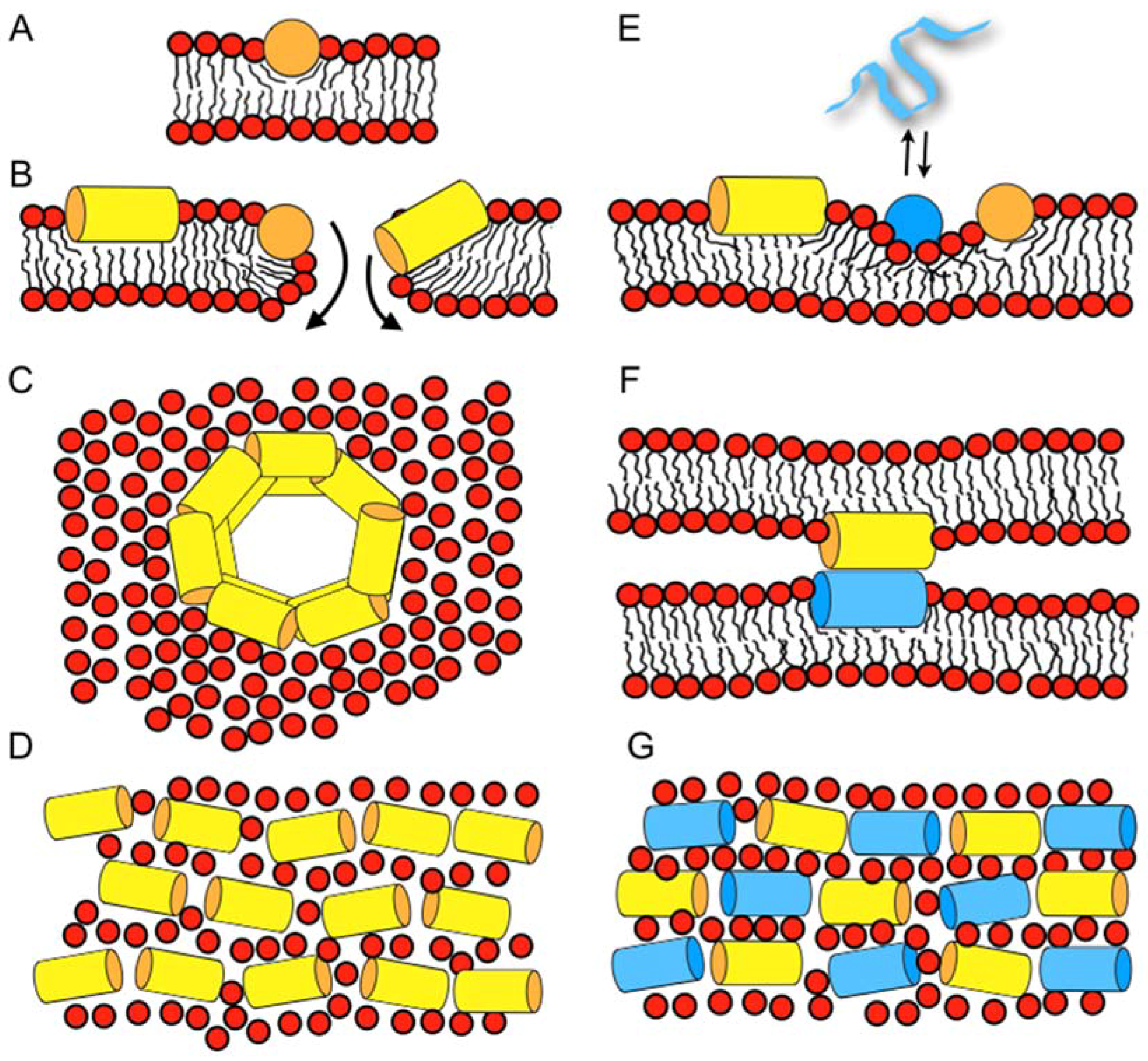{kind=link}
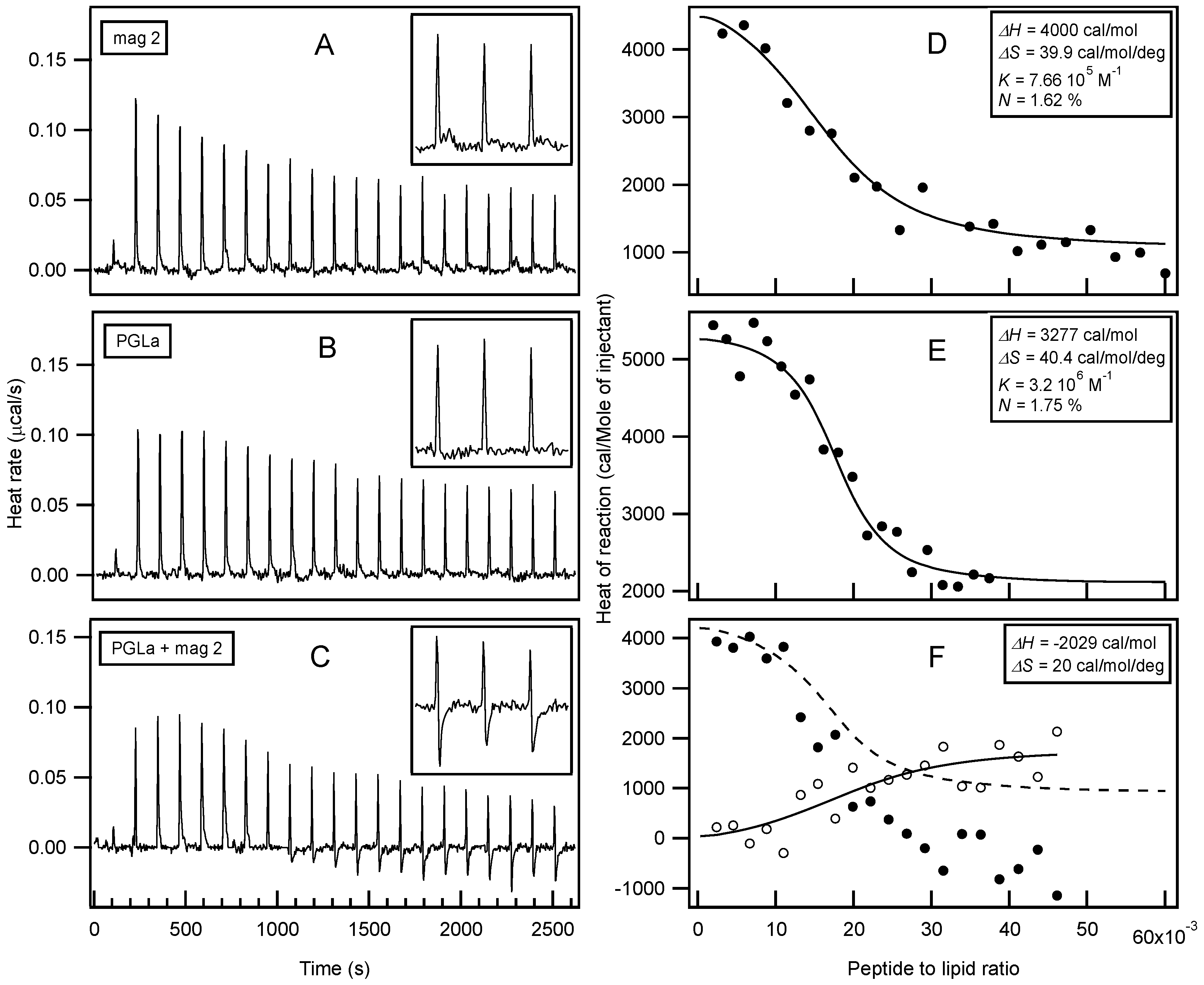{kind=link}
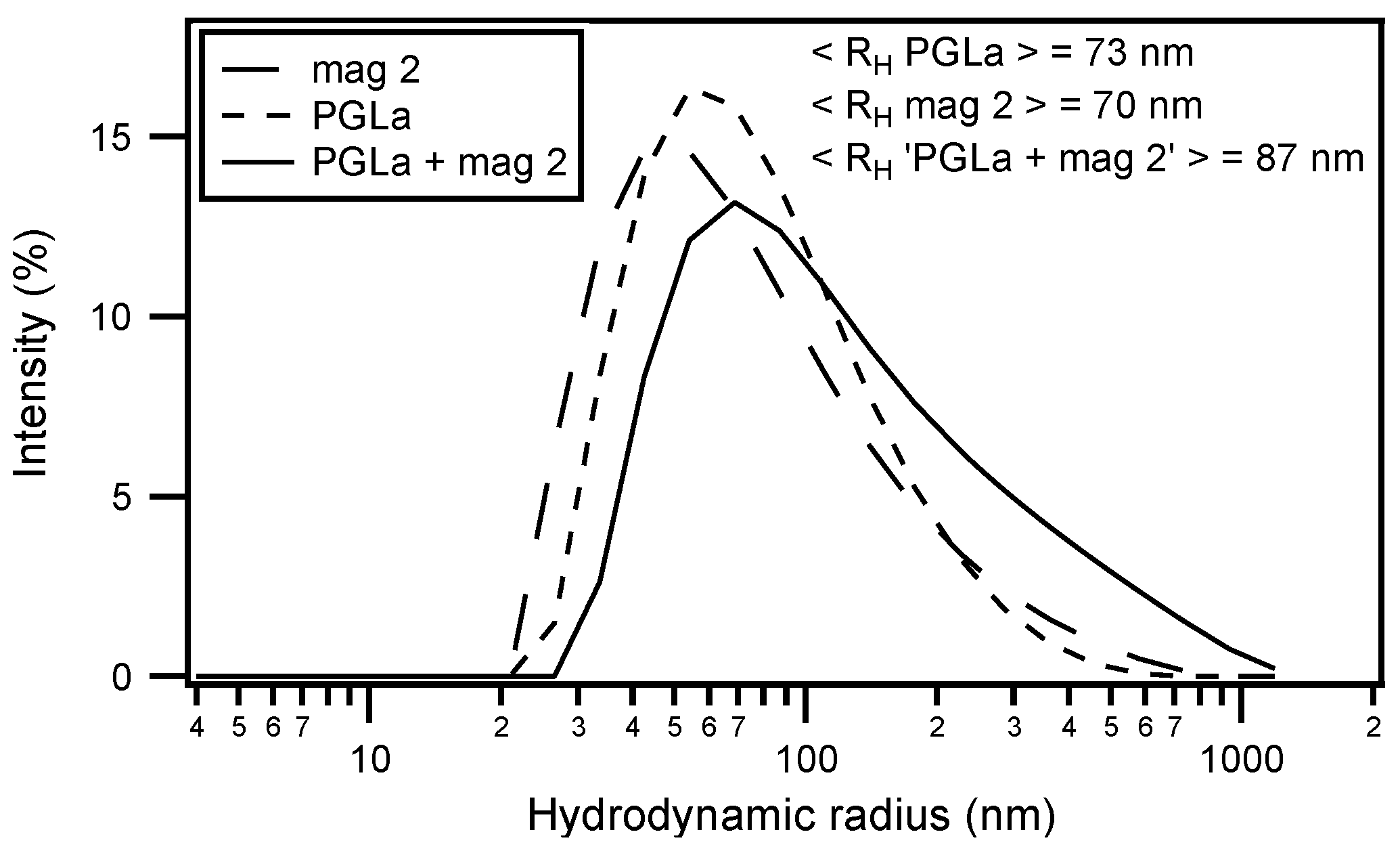{kind=link}
Table 1.
Sequences of peptides discussed in this paper. The one-letter code is used for peptides made from conventional amino acids only. The alamethicin sequence is given by the three-letter code with the following non-standard residues: Aib: α-aminoisobutyric acid, Phl: L-phenylalaninol, Ac- for acetyl- and -NH2 for the carboxamide terminus, respectively.
Table 1.
Sequences of peptides discussed in this paper. The one-letter code is used for peptides made from conventional amino acids only. The alamethicin sequence is given by the three-letter code with the following non-standard residues: Aib: α-aminoisobutyric acid, Phl: L-phenylalaninol, Ac- for acetyl- and -NH2 for the carboxamide terminus, respectively.
| magainin 2 | GIGKF LHSAK KFGKA FVGEI MNS |
| PGLa | GMASK AGAIA GKIAK VALKA L-NH2 |
| cecropin A | KWKLF KKIEK VGQNI RDGII KAGPA VAVVG QATQI AK-NH2 |
| LL37 | LLGDF FRKSK EKIGK EFKRI VQRIK DFLRN LVPRT ES |
| melittin | GIGAV LKVLT TGLPA LISWI KRKRQ Q-NH2 |
| LAH4 | KKALL ALALH HLAHL ALHLA LALKK A-NH2 |
| alamethicin (F50/7) | Ac-Aib-Pro-Aib-Ala-Aib-Aib-Gln-Aib-Val-Aib-Gly-Leu-Aib-Pro-Val-Aib-Aib-Gln-Gln-Phl |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. https://doi.org/10.3390/biom8020018
AMA Style
Marquette A, Bechinger B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules. 2018; 8(2):18. https://doi.org/10.3390/biom8020018
Chicago/Turabian StyleMarquette, Arnaud, and Burkhard Bechinger. 2018. "Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism" Biomolecules 8, no. 2: 18. https://doi.org/10.3390/biom8020018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.