1. Introduction
Protein tyrosine phosphatase interacting protein 51 (PTPIP51) was identified in various protein complexes fulfilling opposite cellular functions, namely proliferation, differentiation and apoptosis [
1,
2]. Meanwhile, a growing number of PTPIP51 interaction partners have been described. Recent studies specified the identified interaction partners including kinases and phosphatases, linker proteins such as 14-3-3, cell receptors (epidermal growth factor receptor (EGFR) and insulin receptor (IR)) [
3,
4,
5,
6], transcription factors (nuclear factor κB [NFκB]) [
7], transport associated proteins (neurophysin) [
8], mitotic associated proteins (CGI-99 and Nuf2) [
3], diacylglycerol kinase α (DGKα), a modulator of the second messenger diacylglycerol [
9], and regulators of Ca
2+ homeostasis (vesicle associated protein B (VAPB)) [
10,
11,
12].
Many tumors display an altered PTPIP51 protein expression and an anomalous subcellular distribution [
13,
14] as has been observed in the cells of basal cell carcinoma and squamous cell carcinoma [
15]. Additionally, in malignant blasts of acute myeloid leukemia (AML) samples PTPIP51 is expressed in contrast to cells of healthy bone marrow specimen with no PTPIP51 protein expression [
16,
17]. In AML blasts the interaction of PTPIP51 with the mitogen activated protein kinase (MAPK) pathway is inhibited due to its high Tyr176 phosphorylation level [
16].
The MAPK pathway modulation is arbitrated by the scaffold protein family 14-3-3 [
18]. When bound to 14-3-3 proteins, then PTPIP51 is empowered to modulate the MAPK pathway [
18]. To avoid an over-activation of the MAPK pathway, the formation of PTPIP51 protein complexes with 14-3-3 and Raf-1 is tightly regulated by distinct kinases and phosphatases [
4,
9,
16]. When PTPIP51 is phosphorylated at its Tyr176 residue, the ternary complex of PTPIP51, 14-3-3 and Raf-1 dissociates into a PTPIP51/14-3-3 complex and Raf-1 [
9]. The Tyr176 phosphorylation of PTPIP51 is mediated by the EGFR and c-Src and antagonized by protein tyrosine phosphatase 1B (PTP1B) [
4]. Besides its regulatory function, the Tyr176 phosphorylation of PTPIP51 also alters subcellular localization [
4]. When Tyr176 is phosphorylated, PTPIP51 localizes to the mitotic spindle apparatus interacting with the microtubular system [
3]. CGI-99 and Nuf-2 were identified as interaction partners of PTPIP51 forming the mitotic protein complex of PTPIP51 [
3]. Of note, this mitotic PTPIP51 protein complex was also traced in interphase cells [
3]. Besides its involvement in the MAPK mediated cell survival and in the mitotic process, PTPIP51 is also linked to apoptosis [
19]. In addition, PTPIP51 impacts the contact of mitochondria to the endoplasmic reticulum (ER) by interacting with VAPB and regulates normal calcium homeostasis [
10,
11].
Yet, up until now the exact regulation of the interactions and relocation of PTPIP51 is not fully understood. Recently, small molecule compounds have gained the spotlight, especially in cancer research and for therapeutical applications [
20]. LDC-3 is a newly synthesized small molecule that targets the cytoplasmic dynein when used in nanomolar concentrations [
21]. Yet, application of the small molecule in µmolar concentration resulted in an interaction with PTPIP51 [
21], an off-target, and thereby modulates its function. Studying the effects of LDC-3 on the interactome has the potential to give deep and detailed insights into the formation and regulation of PTPIP51 protein complexes.
This new compound has enabled us to initiate such a study using the PTPIP51 expressing HaCaT cell line. This non-tumorigenic spontaneously immortalized keratinocyte cell line was extensively investigated for the functional regulation and interaction patterns of PTPIP51. Thus, this cell line is optimally suited to a more detailed protein complex investigation using the small molecule compound LDC-3. HaCaT cells were exposed to LDC-3 in a dose and time dependent manner. The protein–protein interactions were evaluated using the Duolink proximity ligation assay (DPLA), which provides real time in situ interaction data suitable for quantification. By combing LDC-3 with the in situ interaction-profiling assay—DPLA—this study gives critical new insights in the regulation of the endogenous interactome of PTPIP51 in normal non-tumorigenic cells. Analysis of these protein networks by LDC-3 provides a drugable alteration in the PTPIP51 interactions, thereby, interacting and activating the MAPK pathway. This information might be useful for the generation of a therapeutic strategy targeting the PTPIP51 interactome in dysregulated, disease-relevant pathways.
3. Discussion
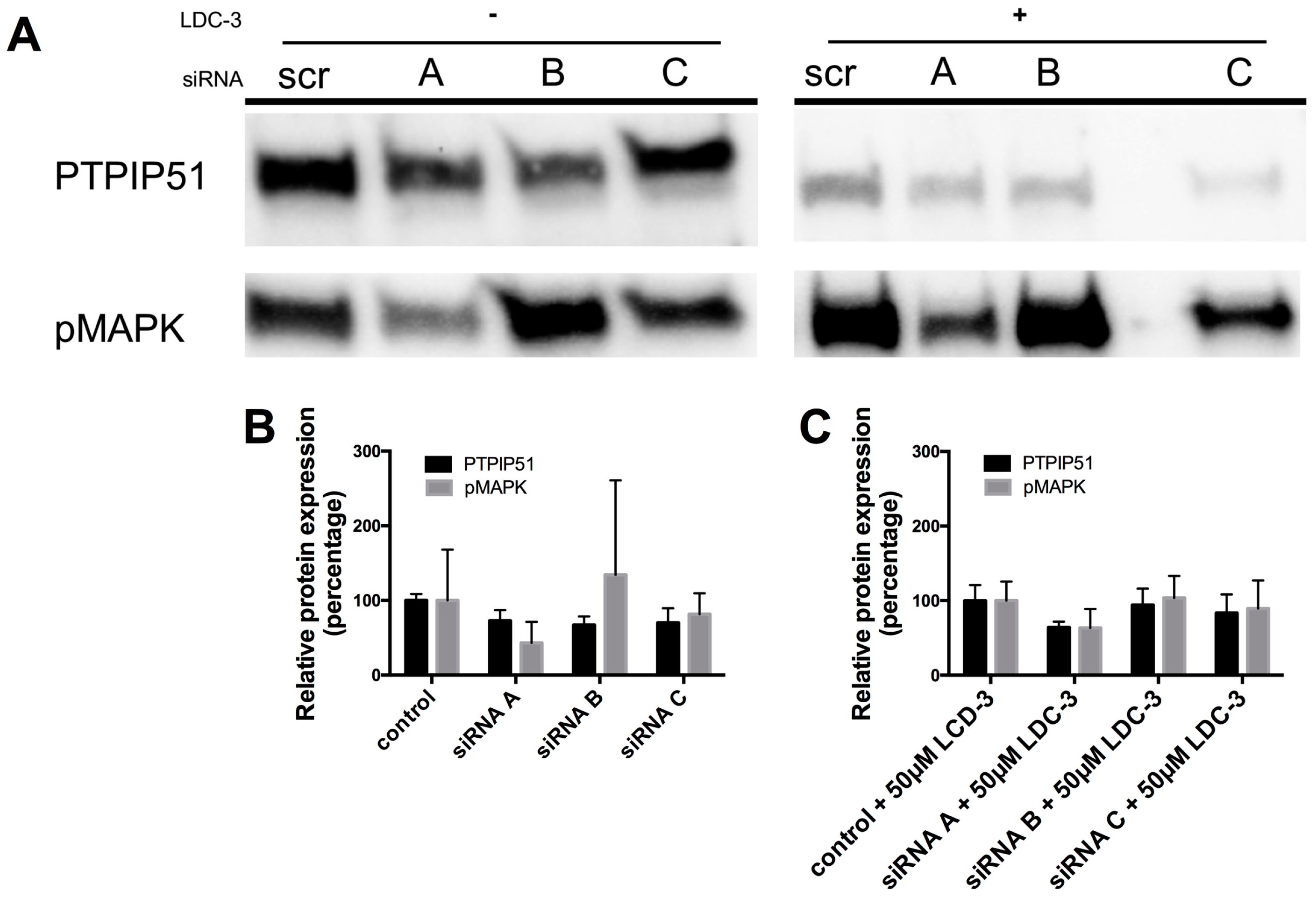
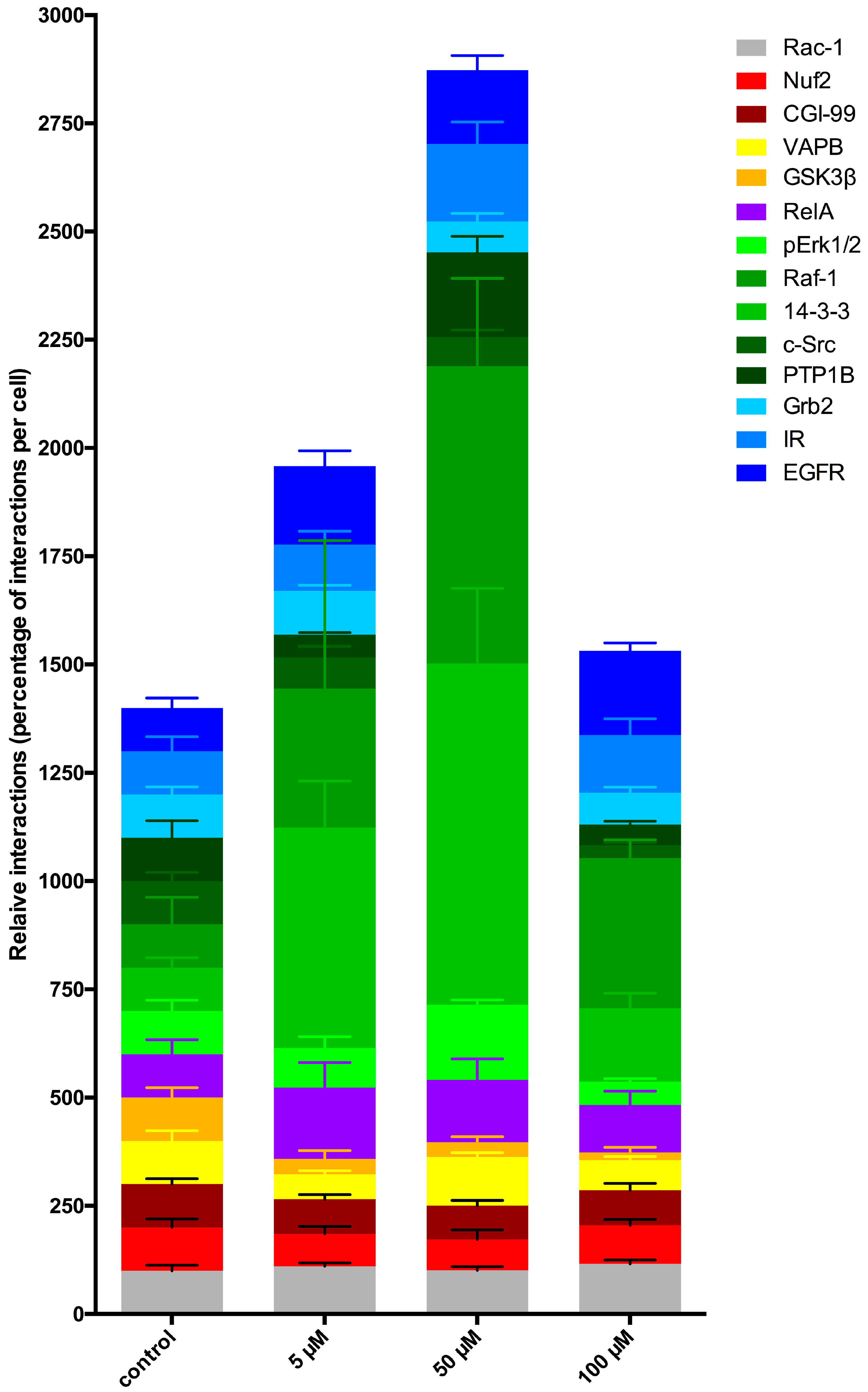
To date, PTPIP51 is a relatively well described protein regarding its multiple participations within cellular signaling pathways. In the current study, we give an overview of physiological protein complex clustering in HaCat cells. A number of these signaling pathways are inflicted in pathogenic processes. Such interaction profiles build the bases for targeted therapy of deteriorated signal transduction. Yet, up until now it was impossible to directly target PTPIP51 protein function and, therefore, the associated interactome. In a high-throughput screen for small molecules interfering with the Hedgehog signaling pathway, the aminothiazole LDC-3 was identified to bind to PTPIP51 [
21,
24]. The specificity of the LDC-3 binding to PTPIP51 was corroborated in a yeast-three-hybrid assay [
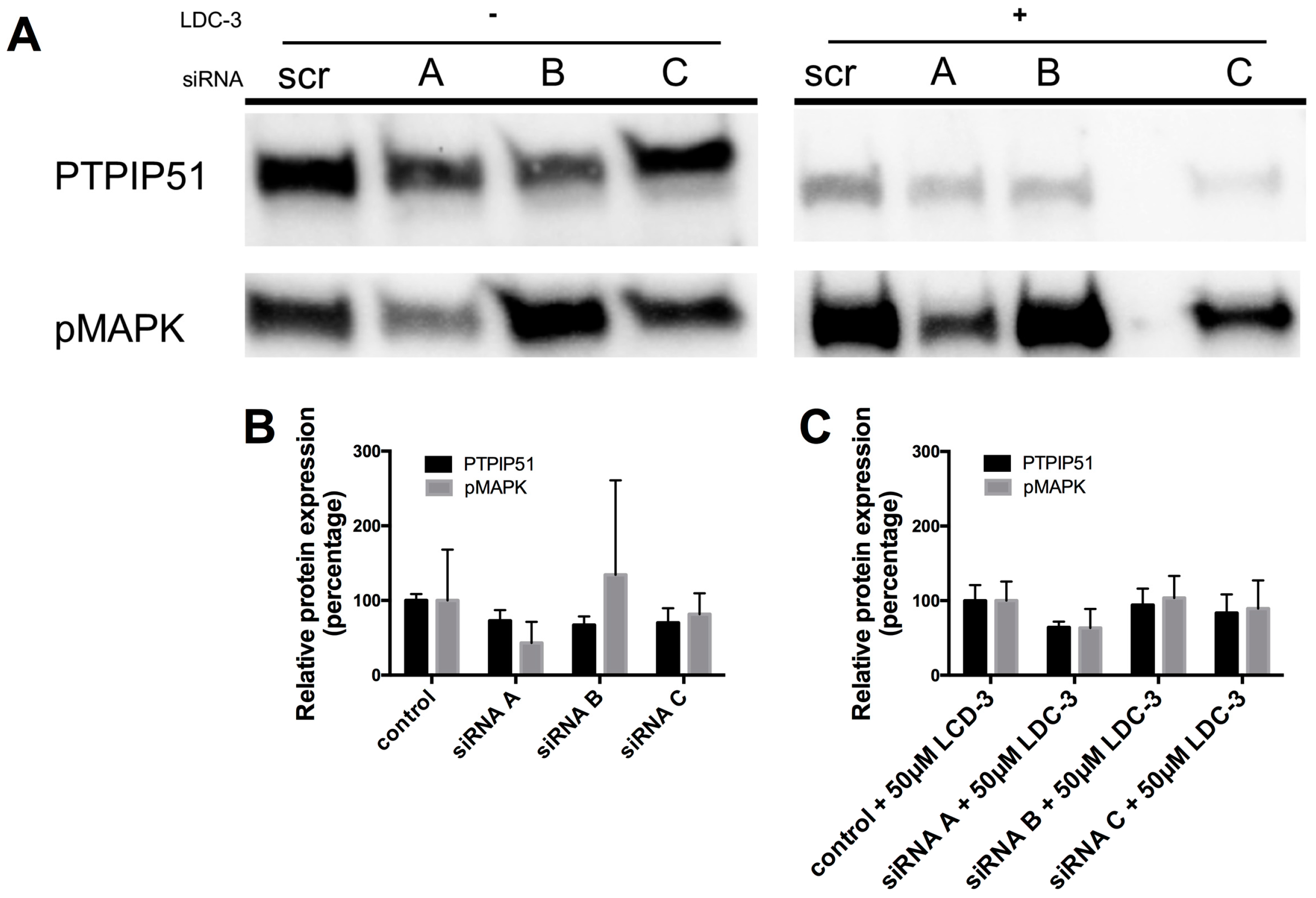
21]. Furthermore, in own functional experiments to better understanding the efficacy and molecular mechanism of LDC-3 effects, we employed a knock down approach. These knock-down experiments of PTPIP51 abolished the effect of LDC-3 on the MAPK pathway. Therefore, we claim that LDC-3 is able to modulate PTPIP51 protein interactions.
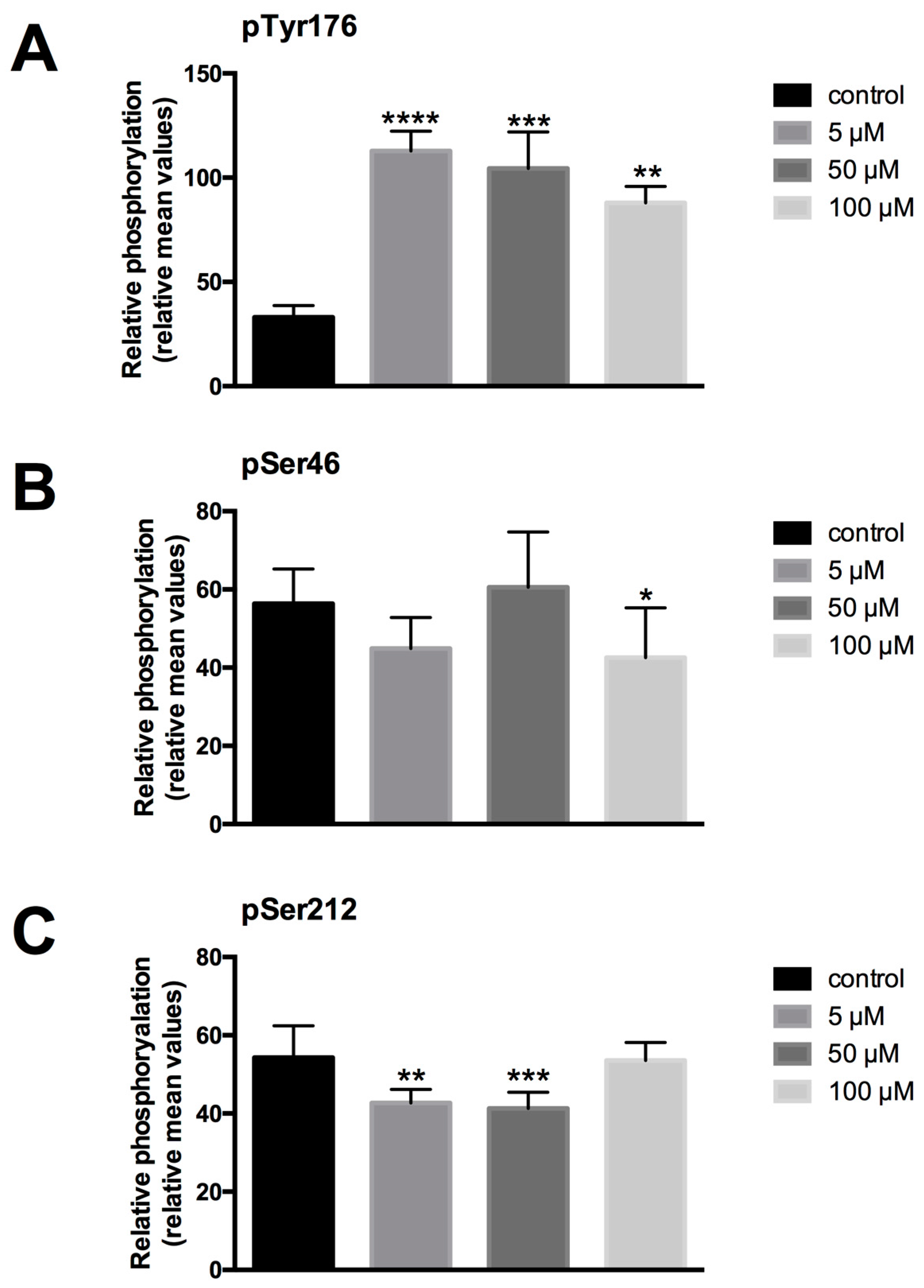
Physiological regulation of PTPIP51 protein–protein interactions is mediated by the tyrosine and serine phosphorylation status of PTPIP51 [
4,
9]. If phosphorylated at its Tyr176 residue PTPIP51 interaction with Raf-1 through 14-3-3 is inhibited and, therefore, PTPIP51 is incapacitated to stimulate the MAPK pathway [
4,
9]. This inhibition is mediated by receptor tyrosine kinases, e.g., the EGFR [
4]. Ser212 phosphorylation antagonizes the aforementioned effect by augmenting the affinity of PTPIP51 to Raf-1 [
9].
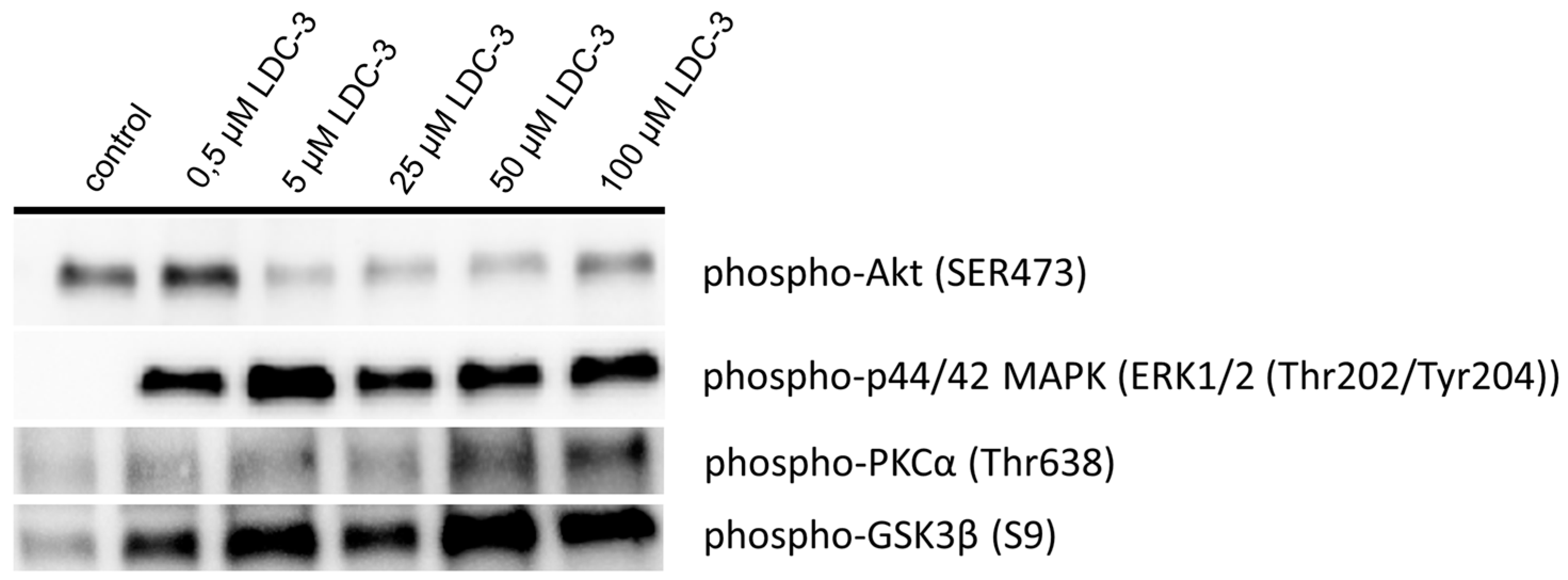
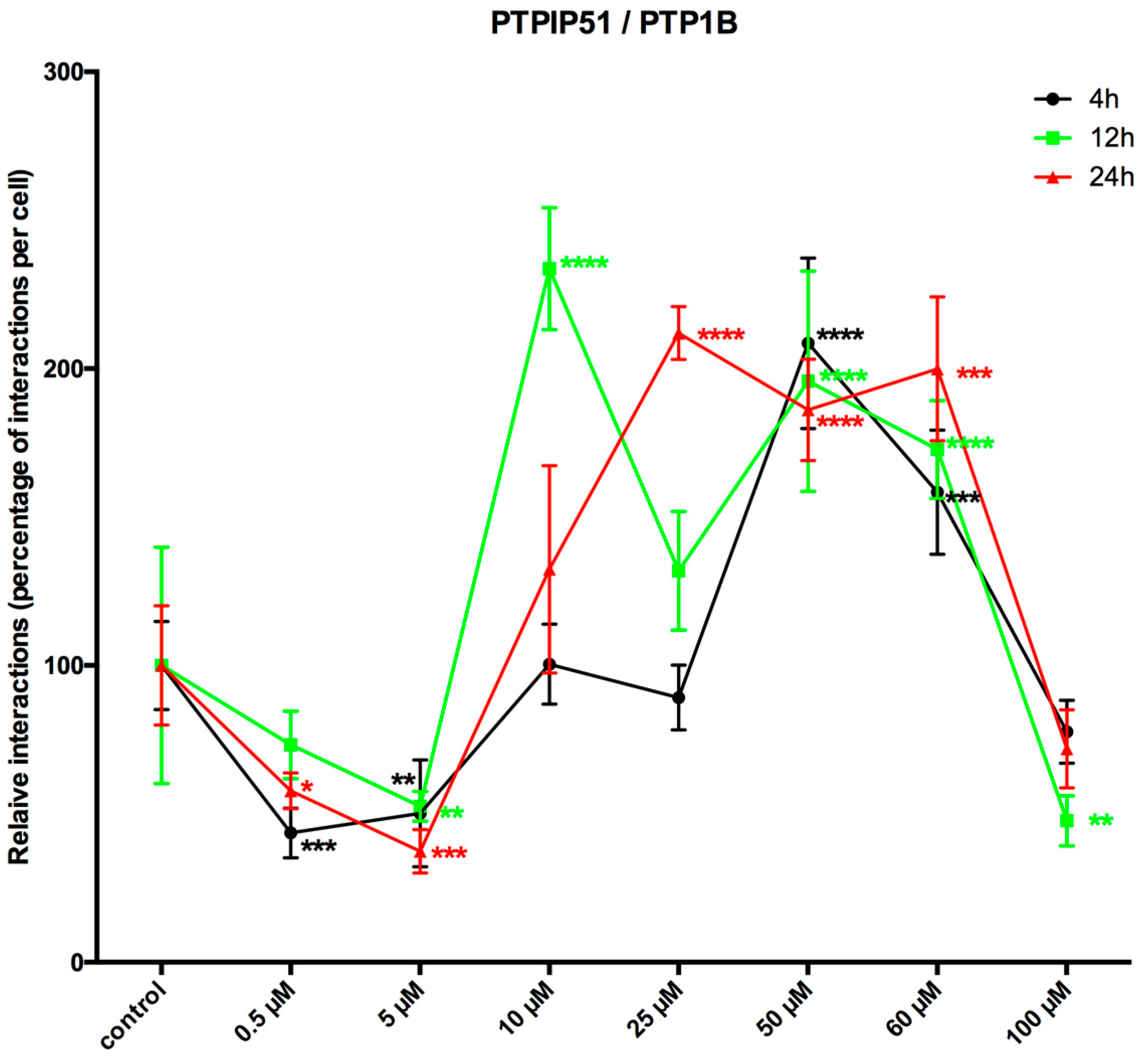
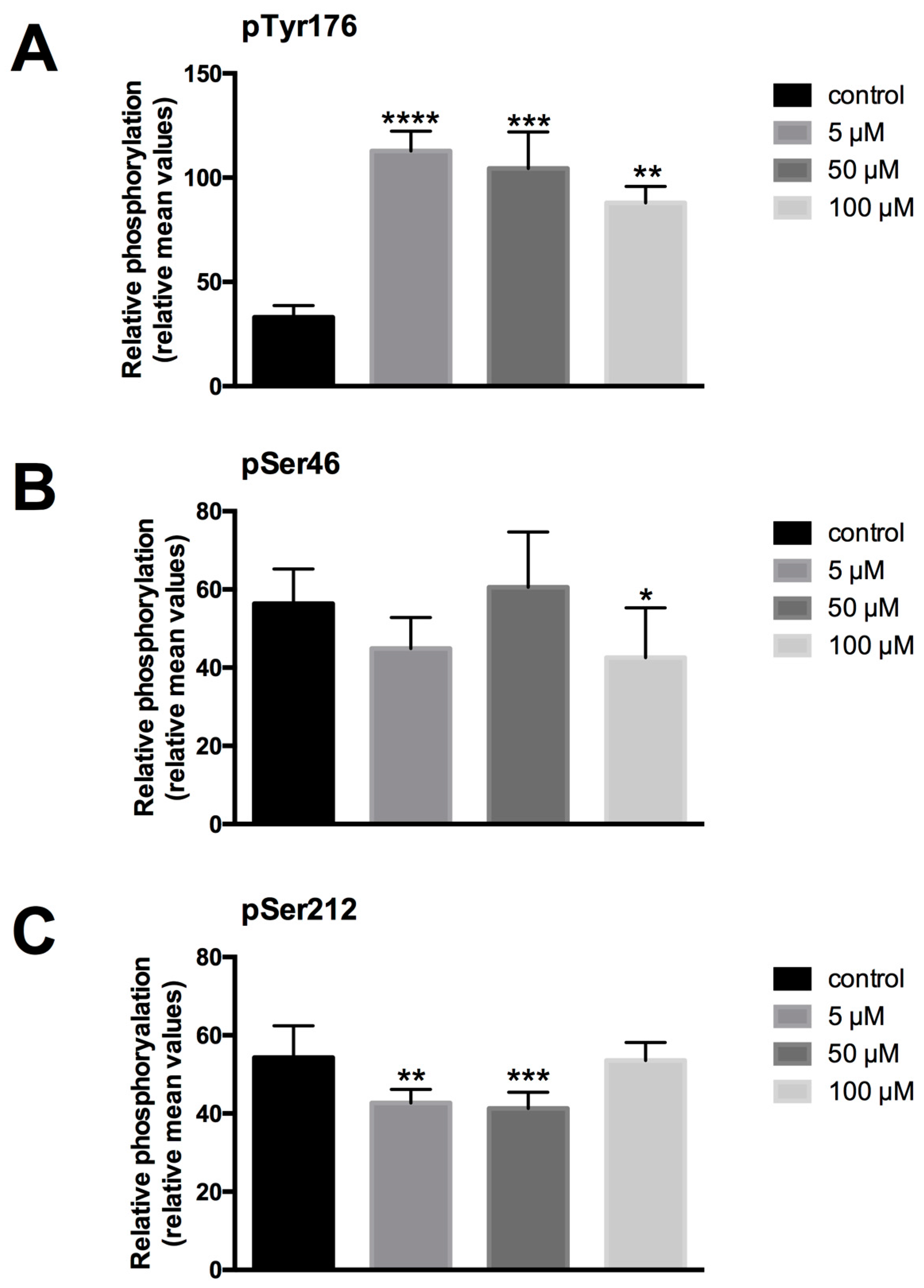
LDC-3 treatment of HaCaT cells strongly enhanced the MAPK binding affinity of PTPIP51 in a dose dependent manner regardless of its regulatory phosphorylation status. PTPIP51 showed a high phosphorylation level at Tyr176 in HaCaT cells, paralleled by the enhanced interaction of PTPIP51/EGFR and PTPIP51/IR, respectively. The detected decrease of the Tyr176 phosphorylation by immunoblotting compared to the semi-quantitative analyses may due to the incomplete registration of the various isoforms of PTPIP51. Facing the high Tyr176 phosphorylation status, the PTPIP51/PTP1B protein complex is augmented. This regulatory circuit is disabled by LDC-3. Therefore, LDC-3 forces PTPIP51 into the MAPK protein cluster and stabilizes the interactions of PTPIP51 with 14-3-3 and Raf-1, respectively. Consequently, the MAPK pathway activity is augmented as verified by immunoblotting experiments. Interestingly, sustained p42/p44-MAPK (Erk1/2) activation can lead to anti-proliferative effects with subsequent induction of apoptosis [
25]. LDC-3 also prohibited the mitotic function of PTPIP51 by reducing CGI-99 and Nuf2 interaction, although PTPIP51 shows high Tyr176 phosphorylation levels normally seen during mitosis [
3].
The MAPK pathway regulates cellular proliferation, differentiation, apoptosis and migration. These processes play a central role in human diseases, not only in tumorigenesis, but also in neurodegeneration, making it a main target for therapeutic intervention [
26,
27].
Recent studies linked the interaction of PTPIP51/VAPB to the GSK3β activation status in a neurodegenerative disease, namely ALS [
28]. If GSK3β is activated it perturbs the PTPIP51/VAPB interaction and therefore the attachment of mitochondria to the mitochondria associated membrane (MAM) of the ER. This mechanism leads to an abnormal calcium homeostasis and might be involved in the induction of apoptosis [
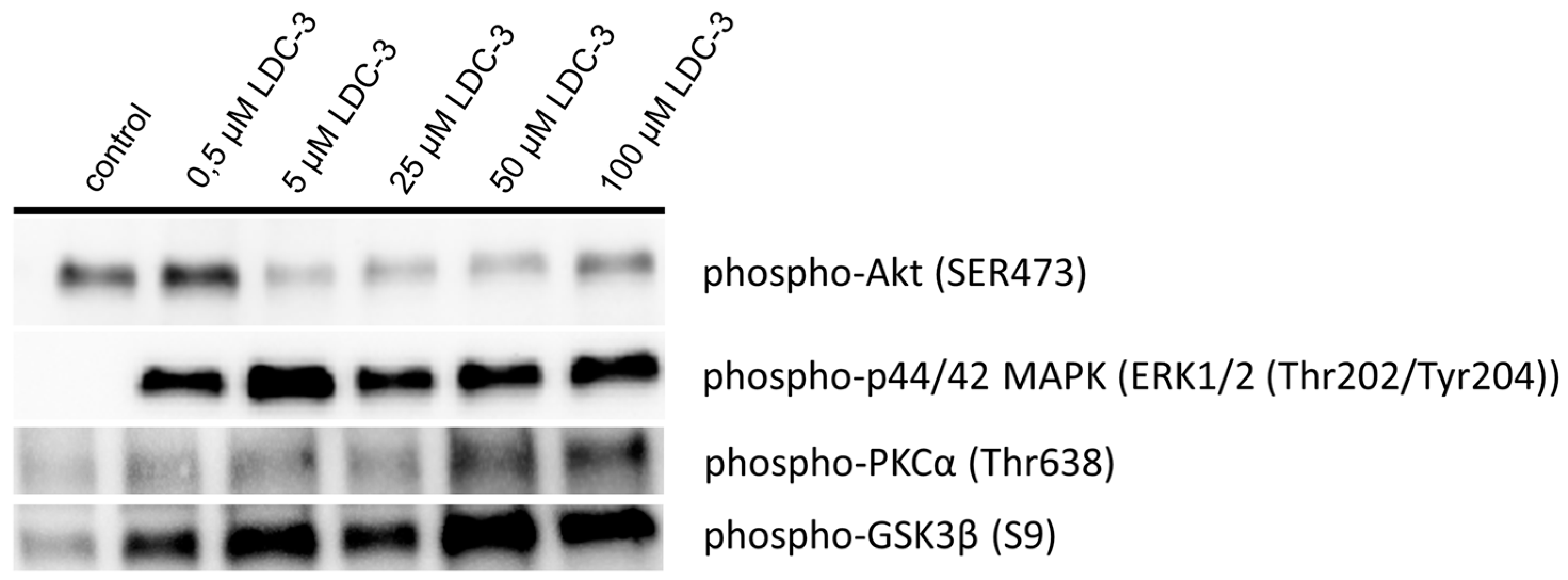
28]. In the current study, we verified a direct interaction of PTPIP51 with GSK3β. LDC-3 also perturbs the mitochondria/ER binding by preventing PTPIP51/VAPB complex formation. The reduced PTPIP51/GSK3β interactions presumably maintain cell survival by the protection of the residual PTPIP51/VAPB complexes followed by the inactivation of GSK3β. LDC-3 treatment also reduced the mitochondrial metabolic rate as an additional expression for the perturbed interaction of PTPIP51 with VAPB. Interestingly, Akt can deactivate GSK3β by phosphorylating its Ser9 residue [
29]. Yet, the activation status of Akt is depleted under the influence of LDC-3. This effect can be observed for concentrations of 5 µM and higher. The lowest concentration of 0.5 µM LDC-3 has no effect on the Akt activation status, whereas p42/p44-MAPK (Erk1/2) is activated. We assumed that the MAPK signaling cascade is the preferentially modulated pathway by the LDC-3 targeted PTPIP51 in µmolar concentrations. Yet, the exact mechanism remains to be solved. PKCα was further analyzed in regard to its activation status. PKCα directly targets the inhibitory Ser9 residue by phosphorylating it [
30]. PKC can deactivate Akt in murine keratinocytes [
31] and was probably activated by the altered calcium release due to the disturbed interaction of PTPIP51/VAPB interaction. This may be followed by a reduction of the MAM area. Notably, PKCα itself interacts with PTPIP51 [
4]. In conclusion, we assume that the PTPIP51/VAPB complex formation is monitored by the activity of PKCα and GSK3β.
A probable mechanism explaining the described altered interaction of PTPIP51 with its endogenous interactome might be an allosteric switch within the PTPIP51 protein structure. PTPIP51 exerts its function by specific binding sites [
2,
32]. These binding sites can either be exposed or concealed by LDC-3, thus facilitating or impairing interactions.
Protein complexes and their underlying protein–protein interactions play critical roles in normal cellular function and malignant transformation [
33]. For two reasons, detailed insights into variations and alteration in protein complexes are strongly needed: (1) If cellular protein complexes are not sufficiently characterized, normal cellular function and probably malignant transformations will not be fully understood. (2) Lately, small molecule drugs affecting protein–protein interactions were developed for anti-cancer therapy [
34]. In this context, it is very critical to identify and analyze altered protein complex formation to determine the mode of action as well as the effectiveness of the new drug. In further studies, it will be crucial to examine how the strong stabilization of the PTPIP51/14-3-3/Raf1 protein complex by LDC-3 impacts a malignantly transformed cellular system with deregulated signal transduction in regard to the MAPK pathway. A study analyzing the effects of LDC-3 on breast cancer cells has already been performed proving a possible therapeutical application of LDC-3 [
35].
Moreover, LDC-3 or a pharmacological derivative of it might also be a therapeutic option for the treatment of ALS normalizing the calcium homeostasis.
The intervention by LDC-3 in the PTPIP51 interactome and the associated cross-talks offers an enormous potential to interfere in deranged signal transduction pathways and may, therefore, give many options for therapeutic applications.
4. Materials and Methods
4.1. Compounds
LDC-3, LDC-4 and LDC-9 were synthesized by the Lead Discovery Center, GmbH, Dortmund, Germany; the chemical structures and broad specificity experiments have been submitted for publication. Stock solutions, dissolved in DMSO, were stored at −80 °C. For treatment, the substances were diluted in culture media and applied in the indicated concentrations.
4.2. Cell Culture
All experiments of this study were performed with HaCaT cells kindly provided by Dr. Teschemacher (Department of Pharmacology, Justus-Liebig-University, Giessen, Germany) with the permission of Dr. Fusenig (DKFZ, Heidelberg, Germany, MTA number L-4598). Cells were kept at 37 °C in a humidified 5% CO2 atmosphere and were cultured in Roswell Park Memorial Institute 1640 (RPMI1640) medium (PAA, Paching, Austria) supplemented with 10% fetal calf serum (FCS), 1% penicillin and 1% streptomycin (ThermoFisher Scientific, Langenselbold, Germany). For experiments cells were grown on culture slides coated with FCS until near confluency. Subsequently, the medium was removed and the cells were treated with LDC-3 containing RMPI1640. The control cells were solely incubated in RPMI1640 medium. The reactions were terminated by withdrawing the medium, adding ice cold phosphate buffered saline (PBS, pH 7.4) and fixation of the cells with 4% paraformaldehyde. Cells were subsequently fixed with methanol and immunocytochemistry was applied.
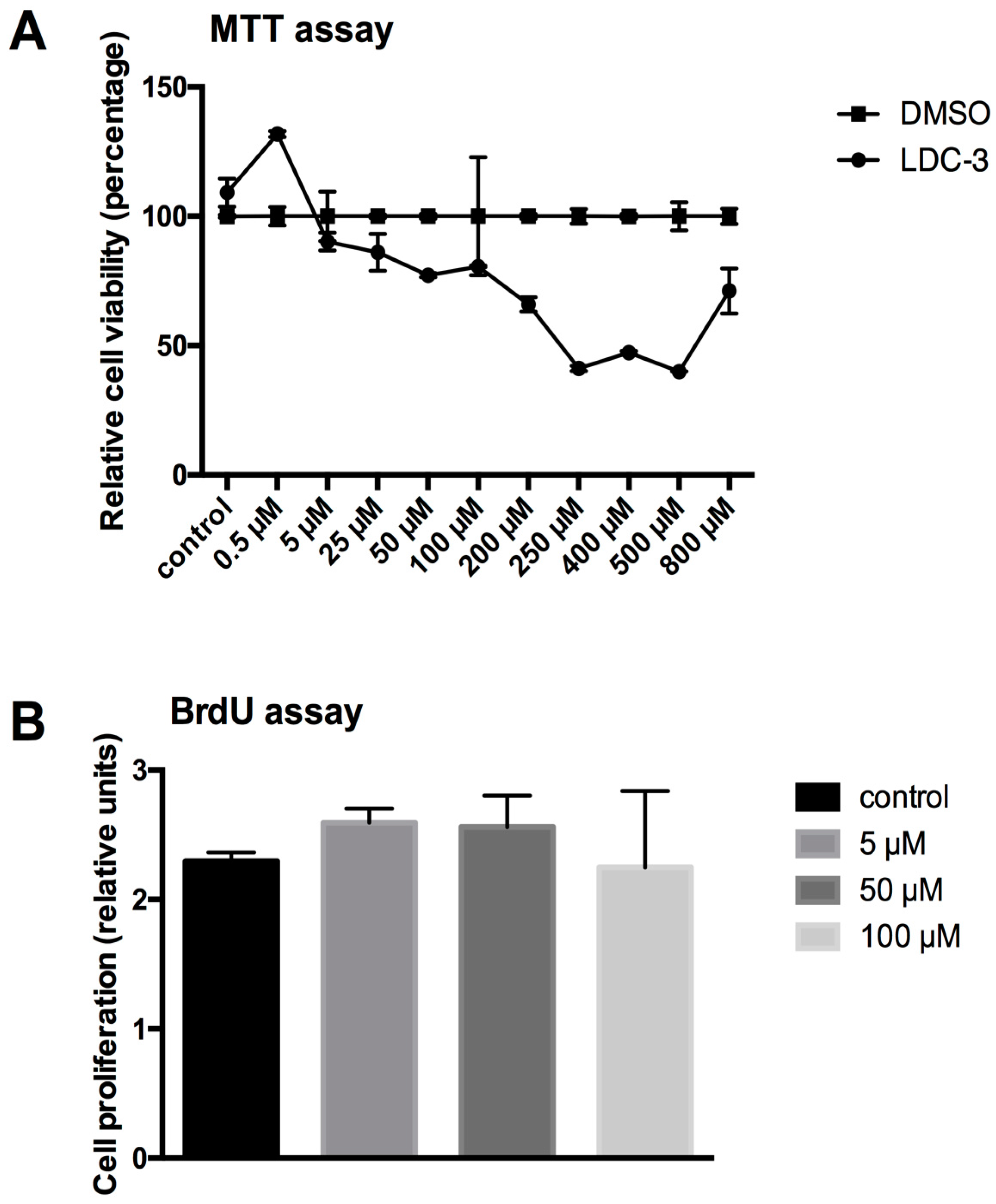
4.3. Determination of Mitochondrial Metabolic Rate by the MTT Assay
Mitochondrial metabolic rate was tested using an MTT assay (Roche Diagnostics, Mannheim, Germany). The assay was performed according to the manufacture’s manual. In short: HaCaT cells (10,000 cells/well) were cultured in flat-bottomed 96-well tissue culture plates either with medium (controls) or with LDC-3 supplemented medium. After preincubation of 24 h the MTT labeling solution was added and incubated for further 6 h. The reaction was stopped by adding the solubilizing solution overnight. The assay was analyzed by reading absorbance at 560 nm.
4.4. BrdU Proliferation Assay
The BrdU proliferation assay was performed according to the manufacture’s manual (Cell signaling technology, Frankfurt, Germany). In short: HaCaT cells (10,000 cells/well) were cultured in flat-bottomed 96-well tissue culture plates either with medium (controls) or with LDC-3 supplemented medium. After preincubation of 24 h without or with the LDC effectors BrdU was added and incubated for further 24 h. The reaction was stopped by fixation and lysis of the cells followed by the addition of a horseradish peroxidase (HRP)-linked antibody and tetramethylbenzidine (TMB) substrate. Proliferation was analyzed by reading absorbance at 450 nm.
4.5. Immunoblotting
Samples of HaCat cell lysates (
n = 3) were separated on Mini-PROTEAN TGX Stain-Free Precast Gels (Bio-Rad, München, Germany). Transfer on an Immobilon P membrane (Millipore, Billerica, MA, USA) was performed according to manufacturer’s instructions using the Bio-Rad Trans-Blot Turbo Transfer System (Bio-Rad) with the settings for mixed molecular weight proteins. The membrane was blocked with 1× Rotiblock for 1 h at room temperature. Incubation with polyclonal rabbit anti-PTPIP51, anti-pMAPK or anti-CDK-1 was done overnight at 4 °C. HRP-conjugated anti-rabbit immunoglobulins were applied for 1 h at room temperature diluted in 1× Rotiblock. The reaction was visualized with the enhanced chemiluminescence (ECL) prime substrate. For documentation the Bio-Rad ChemiDoc Touch Imaging System (Bio-Rad) was used. Calibration was performed with a molecular weight marker suitable for chemiluminescence (Life technologies GmbH, Darmstadt, Germany). The blots were equalized to the obtained stain-free blot for comparison using the Bio-Rad Image Lab (Bio-Rad). Therefore, no loading control is required. The stain-free blots and the quantification data for the shown experiments are given in the
supplementary information.
4.6. siRNA Experiment
siRNA constructs were obtained from Origene (Rockville, MD, USA). Cells were grown in flat-bottomed 24-well tissue culture plates for 24 h with a starting cell number of 100,000 cells per well before transfection. The provided siRNA constructs were initially dissolved in the supplied siRNA dilution buffer to a final concentration of 20 µM. For RNA interference (RNAi) experiments the constructs were further diluted using Opti-Mem and the adequate amount of siTrans 1.0 transfection reagent according to the manufacturer’s protocol resulting in a final working dilution of 10 nM.
HaCat cells (n = 3) were incubated for 24 h with one of the siRNA contructs and the scramble control. A subset of the cells was left untreated for another 24 h, whereas the other subgroup was treated with 50 µM of LDC-3 for 24 h. The reaction was terminated by the administration of the NuPAGE LDS Sample Buffer (Thermo Fischer Scientific, Waltham, MA, USA).
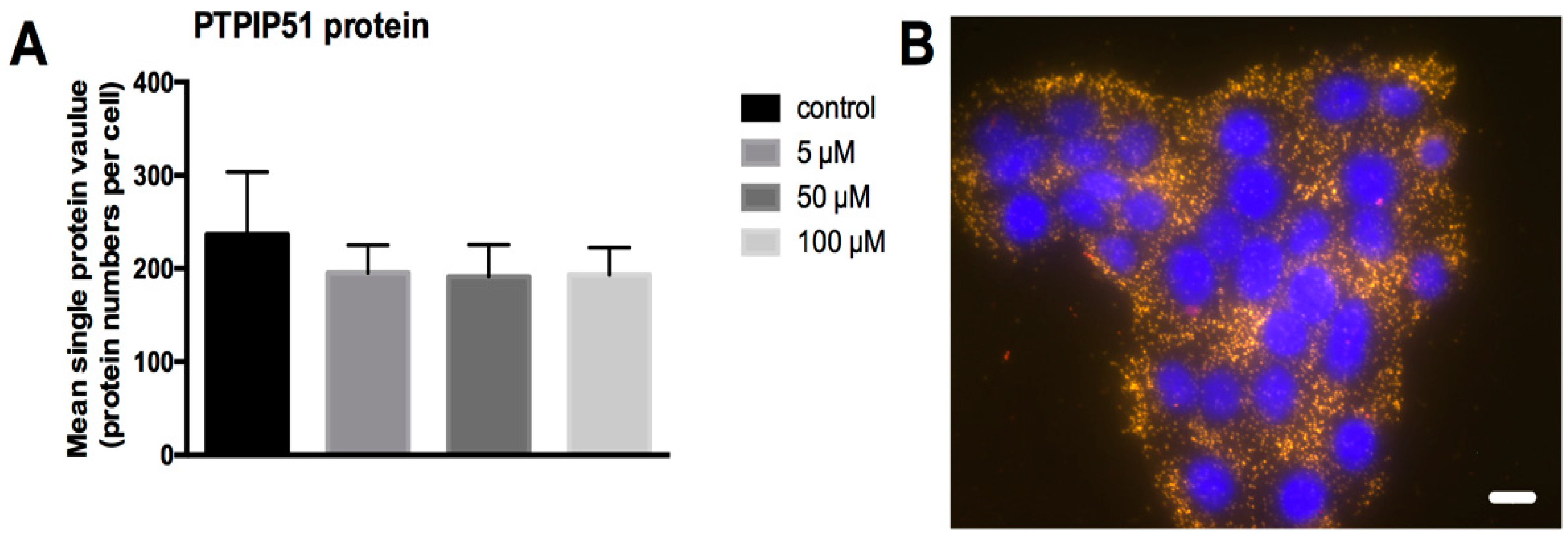
4.7. Quantification of PTPIP51 Protein
To evaluate the influence of LDC-3 on the amount of PTPIP51 protein per single cell the DPLA test was applied, using two PLA probes (DPLA rb plus/minus) directed against the primary rabbit antibody. Both PLA probes bind to the same antibody and are ligated. The signal is several hundred-fold enhanced by rolling circle amplification. Each interaction is presented by a dot and corresponds to one PTPIP51 molecule.
4.8. PTPIP51 Antibody
The PTPIP51 antibody (P51ab) was produced as described previously [
8].
4.9. Immunohistochemistry
Immunohistochemistry was performed as previously described by Koch et al. [
8]. Prior to immunostaining nonspecific binding sites were blocked with 0.1 M phosphate buffered saline (pH 7.4) containing 5% bovine serum albumin and 5% normal goat serum for 1 h. Samples were incubated overnight at room temperature with primary antibodies (
Supplementary Materials Table S1) diluted in PBS, followed by washing in PBS (three times for 10 min) and subsequent incubation for 1 h at room temperature with the corresponding secondary antibodies (
Supplementary Materials Table S1). The slides were washed in PBS, coverslipped in carbonate buffered glycerol at pH 8.6 and evaluated either by epifluorescence microscopy or by sequential confocal laser scanning microscopy. (Carl Zeiss, Jena, Germany). PTPIP51 (aa131–470) and peptide specific PTPIP51 antibodies (
Table S1) were visualized either by an Alexa Fluor 555 coupled anti-rabbit secondary antibody (
Table S1) or a Cy3 coupled anti-guinea pig antibody (
Table S1). Primary mouse antibodies used for double staining were visualized by using an Alexa Fluor 488 coupled anti-mouse secondary antibody (
Table S1). Nuclei were displayed by DAPI.
4.10. Semiquantification of Protein Expression
For the semiquantive analysis of the expressed protein, images were taken under standard conditions (time of exposition). The stored images were analyzed by the ImageJ program, measuring the immunfluorescence intensities of immunostained HaCaT cells by encircling the stained cells. The intensities of 800–1000 cells/group were measured and averaged. The resulting mean values of ten images for Tyr176, Ser46 and Ser212 phosphorylated PTPIP51 were referred to as relative units and plotted in a diagram. The experiments were repeated three times (n = 3).
4.11. Epifluorescence Microscopy
The Axioplan 2 fluorescence microscope equipped with Plan-Apochromat objectives (Carl Zeiss Jena, Germany) was used for photo documentation. For visualization of the secondary antibody Alexa Fluor 555 an excitation filter with a spectrum of 530–560 nm and an emission filter with a spectrum 572–647 nm were used. Alexa Fluor 488 was visualized by an excitation filter with a range of 460–500 nm and an emission filter with a range of 512–542 nm.
4.12. Peptide Specific Phospho-Tyr176 PTPIP51 Antibody
For analysis of the tyrosine phosphorylation state of PTPIP51, an antibody (BioLux, Stuttgart, Germany) to the Tyr176 phosphorylated sequence DAESEGGYTTANAE was used (PTPIP51-PTyr176). The identity and the purity of the synthetized antigenic peptide was approved by electrospray ionization mass spectrometry (ESI-MS) and ultraviolet (UV)-analysis. Guinea pigs were immunized with the keyhole limpet hemocyanin (KLH)-conjugated peptide. The specificity of the antibody was tested by enzyme-linked immunosorbent assay (ELISA) and Western blot. To verify the use of the peptide specific phospho-antibody for immunostaining, preabsorption experiments were performed [
3].
4.13. Peptide Specific Phospho-Serine 46 and 212 PTPIP51 Antibody
For analysis of the serine phosphorylation state of PTPIP51, an antibody (Genosphere Biotechnologies, Paris, France) to the serine 46 phosphorylated sequence CQRHGRSQ[pS]LPNS and an antibody (Genosphere Biotechnologies) to the Ser212 phosphorylated sequence CETVKMGRKD[pS]LDLE were used.
4.14. Confocal Laser Scanning Microscopy
Confocal images of HaCaT cells were obtained with a Zeiss confocal laser scanning microscope (CLSM, LSM 800, Carl Zeiss). Confocal images of Cy3 fluorescence were acquired using Plan-Apochromat 40×/1.1 water objective. The laser voltage was set to 550 V for all analyses. The pinhole diameter was set to 1 Airy unit. Acquisition of confocal images was done using the Zeiss Zen 2.1 software. Subsequently the acquired images were processed by Arivis vision4D 2.12 (Arivis, München, Germany) for producing 3D reconstructions.
4.15. Duolink Proximity Ligation Assay (DPLA)
In situ interactions were detected by the proximity ligation assay kit Duolink (PLA probe anti-rabbit minus, PLA probe anti-mouse plus (Sigma-Aldrich, St. Louis, MO, USA); Dection Kit Orange, (Sigma-Aldrich)). The DPLA probe anti-rabbit minus binds to the PTPIP51 antibody, whereas the PLA probe anti-mouse plus binds to the antibody against the probable interaction partner (
Supplementary Materials Table S1), respectively. The Duolink proximity ligation assay secondary anti-bodies generate only a signal when the two DPLA probes have bound, which only takes place if both proteins are closer than 40 nm, indicating their interaction [
36]. Paraformaldehyde-fixed HaCaT cells were pre-incubated with blocking agent for 1 h. After washing in PBS for 10 min, primary PTPIP51 antibody (1:1000) was applied to the samples. Primary antibodies to the interacting partner (
Supplementary Materials Table S1) were used for proving the interaction by co-incubation with the PTPIP51 antibody. The specificity of the antibodies is documentated in the
supplementary Table S1 by giving the appropriate scientific reference. Incubation was done overnight in a pre-heated humidity chamber. Slides were washed three times in PBS for 10 min. Duolink PLA probes detecting rabbit or mouse antibodies were diluted in the blocking agent in a concentration of 1:5 and applied to the slides followed by incubation for 1 h in a pre-heated humidity chamber at 37°C. Unbound DPLA probes were removed by washing two times in PBS for 5 min. The samples were incubated with the ligation solution consisting of Duolink II Ligation stock (1:5) and Duolink Ligase (1:40) diluted in high purity water for 30 min at 37 °C. After ligation the Duolink Amplification and Detection stock, diluted 1:5 by the addition of polymerase (1:80), was applied to the slides for 100 min. Afterwards the slides were incubated with DAPI for the identification of nuclei. After the final washing steps the slides were dried and coverslips were applied. Quantification was done with the DuoLink Image Tool v1.0.1.2 (OlinkBioscience, Uppsala, Sweden). The signal threshold was adjusted to 100 and the pixel size for spot detection to 5 pixels for each picture.
4.16. Statistical Analysis
The quantified DPLA spots were calculated per cell (number of dots/cell) for each picture. The results were standardized to the number of interactions/cell. Approximately 2000 cells were analyzed of four independent experiments for all protein–protein interactions and PTPIP51 protein quantification (n = 4). The values were subsequently analyzed by GraphPad Prism 6 (GraphPad Software) using the Dunnett’s multiple comparisons test. Results were considered as significant with p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}