
Endoplasmic Reticulum Stress and Ethanol Neurotoxicity

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ER Stress and CNS Pathology
3. ER Stress and Ethanol-Induced Organ Damage
4. ER Stress and Ethanol-Induced Brain Damage
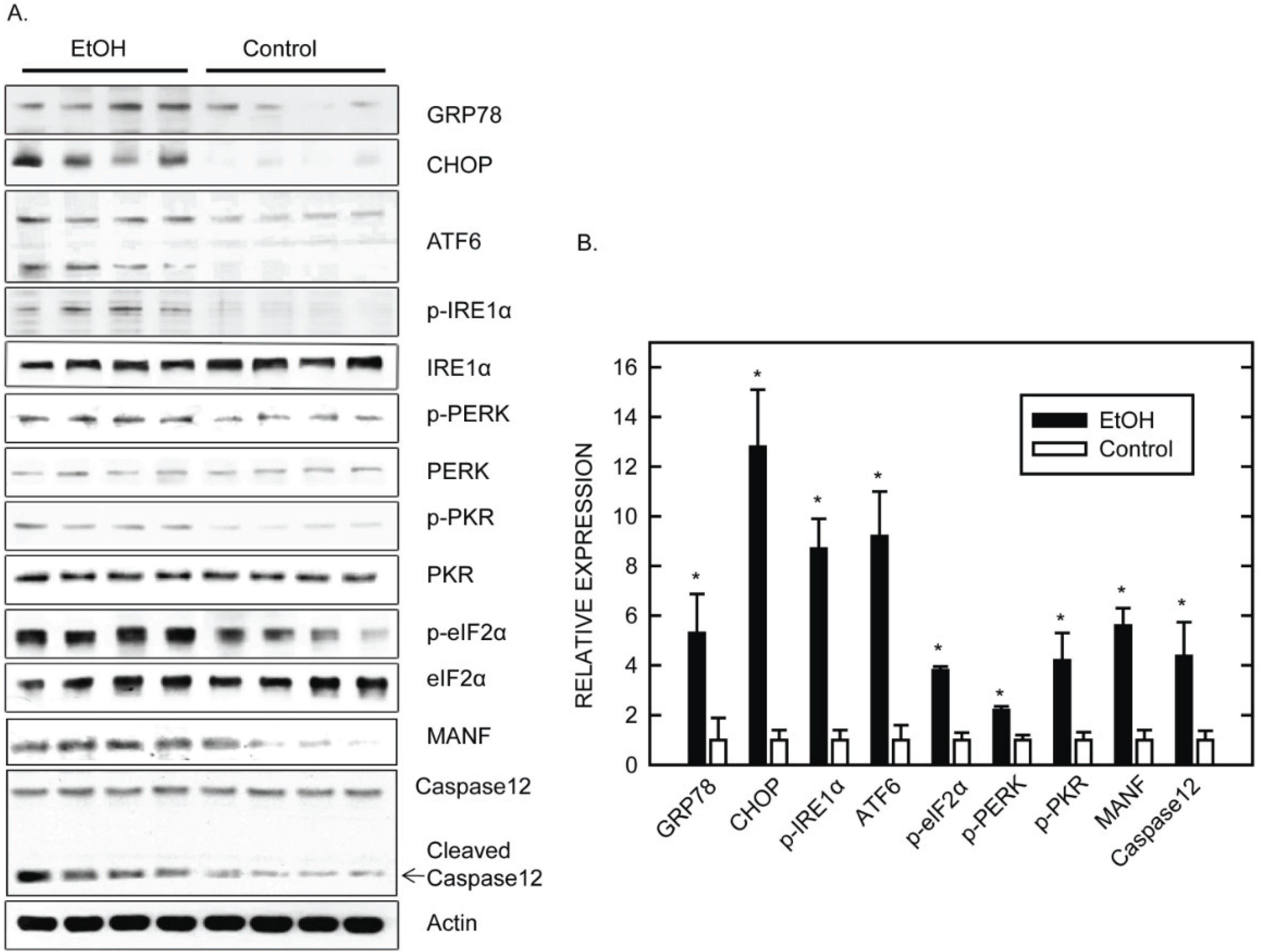
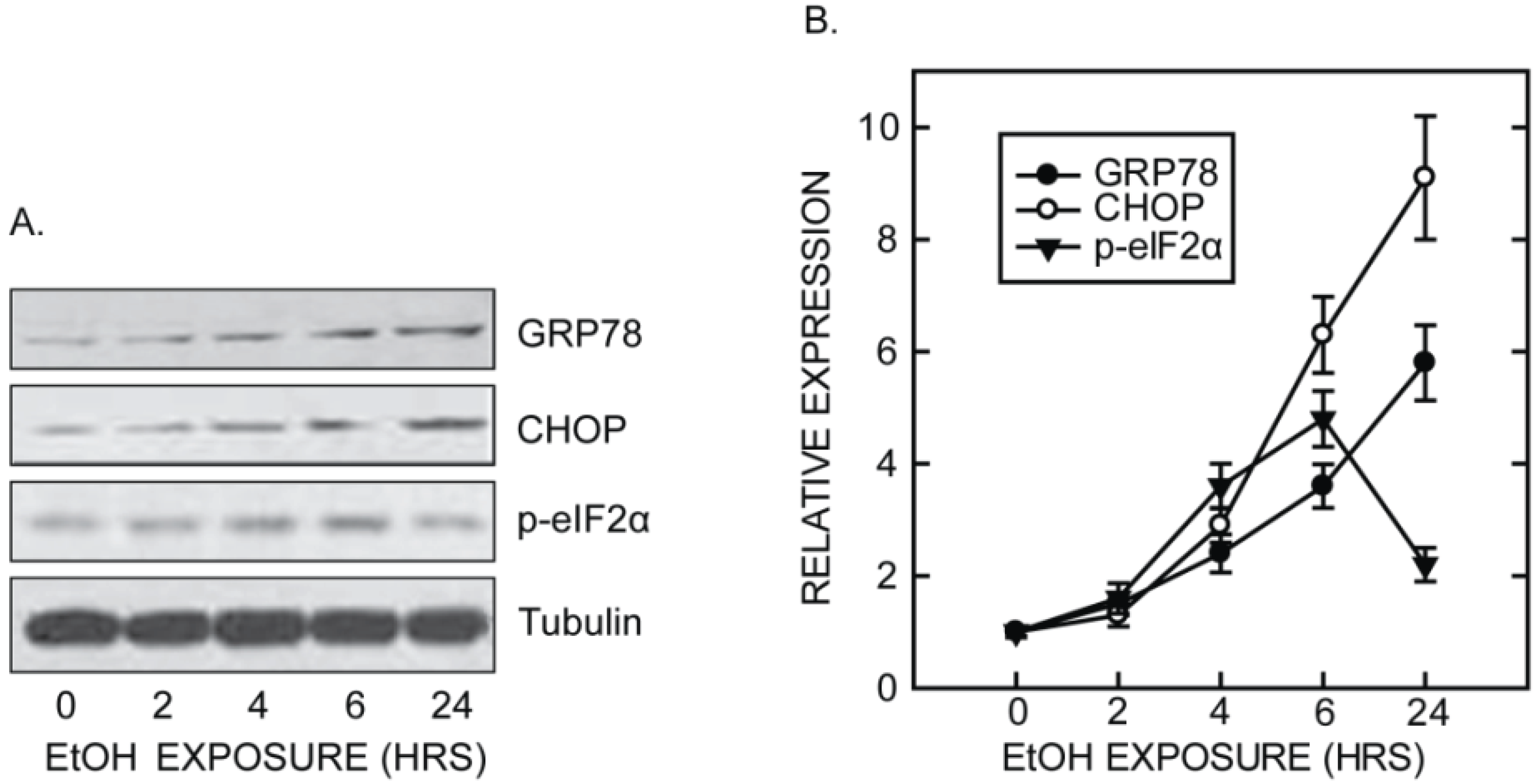
5. Mechanisms Underlying Ethanol-Induced ER Stress
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- National Institute on Alcohol Abuse and Alcoholism, Alcohol Facts and Statistics. Available online: http://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics (accessed on 1 December 2013).
- Substance Abuse and Mental Health Services Administration. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-48, HHS Publication No. (SMA) 14-4863; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2014. [Google Scholar]
- Bouchery, E.E.; Harwood, H.J.; Sacks, J.J.; Simon, C.J.; Brewer, R.D. Economic costs of excessive alcohol consumption in the US, 2006. Am. J. Prev. Med. 2011, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Alimov, A.; Wang, H.; Liu, M.; Frank, J.A.; Xu, M.; Ou, X.; Luo, J. Expression of autophagy and UPR genes in the developing brain during ethanol-sensitive and resistant periods. Metab. Brain Dis. 2013, 28, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Wang, X.; Liu, Y.; Fan, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Li, M.; Fang, S.; et al. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol. Clin. Exp. Res. 2011, 35, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Dlugos, C.A. ATF6 and caspase 12 expression in purkinje neurons in acute slices from adult, ethanol-fed rats. Brain Res. 2014, 1577, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.; Howe, C.; Battaglia, F.C. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment; National Academy Press: Washington, DC, USA, 1996. [Google Scholar]
- Ji, C. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem. Res. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, J.; Qi, Y.; Dai, L.; Zhang, M.; Frank, J.A.; Handshoe, J.W.; Cui, J.; Xu, W.; Chen, G. Deficient PKR in RAX/PKR association ameliorates ethanol-induced neurotoxicity in the developing cerebellum. Cerebellum 2015, 14, 386–397. [Google Scholar] [CrossRef]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes Nutr. 2010, 5, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Hiran, K.R.; Mukherjee, S.; Vasudevan, D.M. Oxidative stress is the primary event: Effects of ethanol consumption in brain. Indian J. Clin. Biochem. 2007, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Zima, T.; Fialova, L.; Mestek, O.; Janebova, M.; Crkovska, J.; Malbohan, I.; Stipek, S.; Mikulikova, L.; Popov, P. Oxidative stress, metabolism of ethanol and alcohol-related diseases. J. Biomed. Sci. 2001, 8, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Pastorino, J.G. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002, 27, 63–68. [Google Scholar] [CrossRef]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 2003, 27, 277–284. [Google Scholar]
- Qi, Y.; Zhang, M.; Li, H.; Frank, J.A.; Dai, L.; Liu, H.; Chen, G. Microrna-29b regulates ethanol-induced neuronal apoptosis in the developing cerebellum through SP1/RAX/PKR cascade. J. Biol. Chem. 2014, 289, 10201–10210. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Zhang, X.; Szabo, E.; Michalak, M.; Opas, M. Endoplasmic reticulum stress during the embryonic development of the central nervous system in the mouse. Int. J. Dev. Neurosci. 2007, 25, 455–463. [Google Scholar] [CrossRef]
- Michalak, M.; Gye, M.C. Endoplasmic reticulum stress in periimplantation embryos. Clin. Exp. Reprod. Med. 2015, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Placido, A.I.; Pereira, C.M.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. Modulation of endoplasmic reticulum stress: An opportunity to prevent neurodegeneration? CNS Neurol. Disord. Drug Targets 2015, 14, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; PSP Genetics Study Group; Lee, V.M.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and alzheimer’s disease. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Veerhuis, R.; van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cedazo-Minguez, A.; Hallbeck, M.; Jerhammar, F.; Marcusson, J.; Terman, A. Intracellular distribution of amyloid beta peptide and its relationship to the lysosomal system. Transl. Neurodegener. 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell 2012, 11, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Guo, Q.; Furukawa, K.; Pedersen, W.A. Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer’s disease. J. Neurochem. 1998, 70, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.S.; Preisler, J.; Baum, L.; Lee, D.H.; Ng, H.K.; Hugon, J.; So, K.F.; Chang, R.C. Low molecular weight Aβ induces collapse of endoplasmic reticulum. Mol. Cell. Neurosci. 2009, 41, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yang, X.; Lau, J.C.; Hung, C.H.; Wuwongse, S.; Zhang, Q.; Wang, J.; Baum, L.; So, K.F.; Chang, R.C. Endoplasmic reticulum stress induces tau pathology and forms a vicious cycle: Implication in Alzheimer’s disease pathogenesis. J. Alzheimer’s Dis. 2012, 28, 839–854. [Google Scholar]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gan, M.; Ebrahim, A.S.; Lin, W.L.; Melrose, H.L.; Yen, S.H. ER stress response plays an important role in aggregation of alpha-synuclein. Mol. Neurodegener. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Yan, S.; Shi, Y. Single-prolonged stress induces endoplasmic reticulum-dependent apoptosis in the hippocampus in a rat model of post-traumatic stress disorder. PLoS ONE 2013, 8, e69340. [Google Scholar] [CrossRef] [PubMed]
- Ni Fhlathartaigh, M.; McMahon, J.; Reynolds, R.; Connolly, D.; Higgins, E.; Counihan, T.; Fitzgerald, U. Calreticulin and other components of endoplasmic reticulum stress in rat and human inflammatory demyelination. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, H.; Zheng, G.; Shi, Z.N. Region-specific vulnerability to endoplasmic reticulum stress-induced neuronal death in rat brain after status epilepticus. J. Biosci. 2013, 38, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, H.; Gu, W.; Liu, Y.; Zhang, M. Ginsenoside Rb1 protects hippocampal neurons from high glucose-induced neurotoxicity by inhibiting GSK3β-mediated CHOP induction. Mol. Med. Rep. 2014, 9, 1434–1438. [Google Scholar] [PubMed]
- Begum, G.; Yan, H.Q.; Li, L.; Singh, A.; Dixon, C.E.; Sun, D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J. Neurosci. 2014, 34, 3743–3755. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Zhang, X.Y.; Han, R.; Zhang, T.T.; Chen, C.; Qin, Z.H.; Sheng, R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol. Sin. 2013, 34, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, Y.; Zhou, K.; Freeze, H.H.; Zhang, Y.W.; Xu, H. Insufficient ER-stress response causes selective mouse cerebellar granule cell degeneration resembling that seen in congenital disorders of glycosylation. Mol. Brain 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Vitte, J.; Traver, S.; de Paula, A.M.; Lesage, S.; Rovelli, G.; Corti, O.; Duyckaerts, C.; Brice, A. Leucine-rich repeat kinase 2 is associated with the endoplasmic reticulum in dopaminergic neurons and accumulates in the core of Lewy bodies in Parkinson disease. J. Neuropathol. Exp. Neurol. 2010, 69, 959–972. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, P.; Smith, M.A.; Kramp, K.; Huang, Y.; Hisamoto, N.; Matsumoto, K.; Hatzoglou, M.; Jin, H.; Feng, Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. Elegans. PLoS ONE 2011, 6, e22354. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. New insights into the pathogenesis of alcohol-induced ER stress and liver diseases. Int. J. Hepatol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.W.; Ma, Y.X.; Wang, X.N.; Wang, C.F.; Lu, J.; Cao, W.; Wu, X.D. Ethanol promotes saturated fatty acid-induced hepatoxicity through endoplasmic reticulum (ER) stress response. Chin. J. Nat. Med. 2015, 13, 250–256. [Google Scholar] [CrossRef]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of chop in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Pandol, S.J.; Gorelick, F.S.; Gerloff, A.; Lugea, A. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig. Dis. 2010, 28, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Ren, J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: Role of insulin signaling and ER stress. J. Mol. Cell. Cardiol. 2008, 44, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.; Lee, J.H.; Nyomba, B.L. Ethanol causes endoplasmic reticulum stress and impairment of insulin secretion in pancreatic beta-cells. Alcohol 2012, 46, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011, 140, 987–997. [Google Scholar] [CrossRef]
- Sutherland, G.T.; Sheedy, D.; Kril, J.J. Using autopsy brain tissue to study alcohol-related brain damage in the genomic age. Alcohol. Clin. Exp. Res. 2014, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elofson, J.; Gongvatana, W.; Carey, K.B. Alcohol use and cerebral white matter compromise in adolescence. Addict. Behav. 2013, 38, 2295–2305. [Google Scholar] [CrossRef] [PubMed]
- Brolese, G.; Lunardi, P.; de Souza, D.F.; Lopes, F.M.; Leite, M.C.; Goncalves, C.A. Pre- and postnatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal glutamate uptake in adolescent offspring. PLoS ONE 2015, 10, e0127845. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.C.; Pereira, M.C.; Santana, L.N.; Fernandes, R.M.; Teixeira, F.B.; Oliveira, G.B.; Fernandes, L.M.; Fontes-Junior, E.A.; Prediger, R.D.; Crespo-Lopez, M.E.; et al. Chronic ethanol exposure during adolescence through early adulthood in female rats induces emotional and memory deficits associated with morphological and molecular alterations in hippocampus. J. Psychopharmacol. 2015, 29, 712–724. [Google Scholar] [CrossRef]
- Chen, G.; Ma, C.; Bower, K.A.; Shi, X.; Ke, Z.; Luo, J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: Involvement of oxidative stress. J. Neurosci. Res. 2008, 86, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing c57bl/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Yang, W.; Shen, Y.; Chen, Y.; Chen, L.; Wang, L.; Wang, H.; Xu, S.; Fang, S.; Fu, Y.; Yu, Y.; et al. Mesencephalic astrocyte-derived neurotrophic factor prevents neuron loss via inhibiting ischemia-induced apoptosis. J. Neurol. Sci. 2014, 344, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Sun, A.; Wang, Y.; Cha, D.; Wang, H.; Wang, F.; Feng, L.; Fang, S.; Shen, Y. Upregulation of mesencephalic astrocyte-derived neurotrophic factor in glial cells is associated with ischemia-induced glial activation. J. Neuroinflammation 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Y.; Cheng, L.; Liu, B.; Zhang, W.; Guo, Y.J.; Nie, L. Mesencephalic astrocyte-derived neurotrophic factor inhibits oxygen-glucose deprivation-induced cell damage and inflammation by suppressing endoplasmic reticulum stress in rat primary astrocytes. J. Mol. Neurosci. 2013, 51, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Huang, S.; Gaertig, M.A.; Li, X.J.; Li, S. Age-dependent decrease in chaperone activity impairs MANF expression, leading to Purkinje cell degeneration in inducible SCA17 mice. Neuron 2014, 81, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Goral, J.; Meyer, A. Ethanol down-regulates microglia inflammatory responses augmented by endoplasmic reticulum stress. FASEB J. 2013. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Ke, Z.J.; Comer, A.L.; Xu, M.; Frank, J.A.; Zhang, Z.; Shi, X.; Luo, J. Tunicamycin-induced unfolded protein response in the developing mouse brain. Toxicol. Appl. Pharmacol. 2015, 283, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Autophagy and ethanol neurotoxicity. Autophagy 2014, 10, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ke, Z.; Xu, M.; Liao, M.; Wang, X.; Qi, Y.; Zhang, T.; Frank, J.A.; Bower, K.A.; Shi, X.; et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012, 8, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; d’Hellencourt, C.L.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Fron. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Davies, K.J.; Maiorino, M.; Parasassi, T.; Sevanian, A. Atherosclerosis: Another protein misfolding disease? Trends Mol. Med. 2002, 8, 370–374. [Google Scholar] [CrossRef]
- Mills, K.R.; Ward, K.; Martin, F.; Peters, T.J. Peripheral neuropathy and myopathy in chronic alcoholism. Alcohol Alcohol. 1986, 21, 357–362. [Google Scholar] [PubMed]
- Nishitani, Y.; Matsumoto, H. Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Lett. 2006, 580, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Iwahashi, K.; Furukawa, A.; Ameno, K.; Kinoshita, H.; Ijiri, I.; Sekine, Y.; Suzuki, K.; Iwata, Y.; Minabe, Y.; et al. Acetaldehyde adducts in the brain of alcoholics. Arch. Toxicol. 2003, 77, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Iwahashi, K.; Itoh, M.; Ameno, K.; Ijiri, I.; Takeuchi, Y.; Suwaki, H. Immunohistochemical study on acetaldehyde adducts in alcohol-fed mice. Alcohol. Clin. Exp. Res. 2000, 24, 93S–96S. [Google Scholar] [PubMed]
- Worrall, S.; Niemela, O.; Parkkila, S.; Peters, T.J.; Preedy, V.R. Protein adducts in type I and type II fibre predominant muscles of the ethanol-fed rat: Preferential localisation in the sarcolemmal and subsarcolemmal region. Eur. J. Clin. Invest. 2001, 31, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.H.; Li, Y.Q.; Feng, S.L.; Li, B.X.; Pan, Z.W.; Xu, C.Q.; Li, T.T.; Yang, B.F. Calcium-sensing receptor activation contributed to apoptosis stimulates TRPC6 channel in rat neonatal ventricular myocytes. Biochem. Biophys. Res. Commun. 2010, 394, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; de Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Walker, D.W.; Heaton, M.B. Nerve growth factor and chronic ethanol treatment alter calcium homeostasis in developing rat septal neurons. Brain Res. Dev. Brain Res. 2003, 143, 57–71. [Google Scholar] [CrossRef]
- Kouzoukas, D.E.; Li, G.; Takapoo, M.; Moninger, T.; Bhalla, R.C.; Pantazis, N.J. Intracellular calcium plays a critical role in the alcohol-mediated death of cerebellar granule neurons. J. Neurochem. 2013, 124, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int. J. Clin. Exp. Med. 2010, 3, 33–40. [Google Scholar] [PubMed]
- Lieber, C.S.; Teschke, R.; Hasumura, Y.; Decarli, L.M. Differences in hepatic and metabolic changes after acute and chronic alcohol consumption. Fed. Proc. 1975, 34, 2060–2074. [Google Scholar] [PubMed]
- Iseri, O.A.; Lieber, C.S.; Gottlieb, L.S. The ultrastructure of fatty liver induced by prolonged ethanol ingestion. Am. J. Pathol. 1966, 48, 535–555. [Google Scholar] [PubMed]
- Lane, B.P.; Lieber, C.S. Ultrastructural alterations in human hepatocytes following ingestion of ethanol with adequate diets. Am. J. Pathol. 1966, 49, 593–603. [Google Scholar] [PubMed]
- Koch, O.R.; de Conti, L.L.R.; Bolanos, L.P.; Stoppani, A.O. Ultrastructural and biochemical aspects of liver mitochondria during recovery from ethanol-induced alterations. Experimental evidence of mitochondrial division. Am. J. Pathol. 1978, 90, 325–344. [Google Scholar] [PubMed]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. NY Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.W.; Morgan, M.Y.; Laulicht, M.; Hoffbrand, A.V.; Sherlock, S. Hepatic iron stores and markers of iron overload in alcoholics and patients with idiopathic hemochromatosis. Dig. Dis. Sci. 1982, 27, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Irving, M.G.; Halliday, J.W.; Powell, L.W. Association between alcoholism and increased hepatic iron stores. Alcohol. Clin. Exp. Res. 1988, 12, 7–13. [Google Scholar] [CrossRef] [PubMed]
- De Feo, T.M.; Fargion, S.; Duca, L.; Cesana, B.M.; Boncinelli, L.; Lozza, P.; Cappellini, M.D.; Fiorelli, G. Non-transferrin-bound iron in alcohol abusers. Alcohol. Clin. Exp. Res. 2001, 25, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Taroni, F.; Cortopassi, G.A. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid. Redox Signal. 2006, 8, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loreal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Luo, J. Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules 2015, 5, 2538-2553. https://doi.org/10.3390/biom5042538
Yang F, Luo J. Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules. 2015; 5(4):2538-2553. https://doi.org/10.3390/biom5042538
Chicago/Turabian StyleYang, Fanmuyi, and Jia Luo. 2015. "Endoplasmic Reticulum Stress and Ethanol Neurotoxicity" Biomolecules 5, no. 4: 2538-2553. https://doi.org/10.3390/biom5042538
APA StyleYang, F., & Luo, J. (2015). Endoplasmic Reticulum Stress and Ethanol Neurotoxicity. Biomolecules, 5(4), 2538-2553. https://doi.org/10.3390/biom5042538