
Physicochemical Properties of Ion Pairs of Biological Macromolecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fundamental Concepts on Ion Pairs
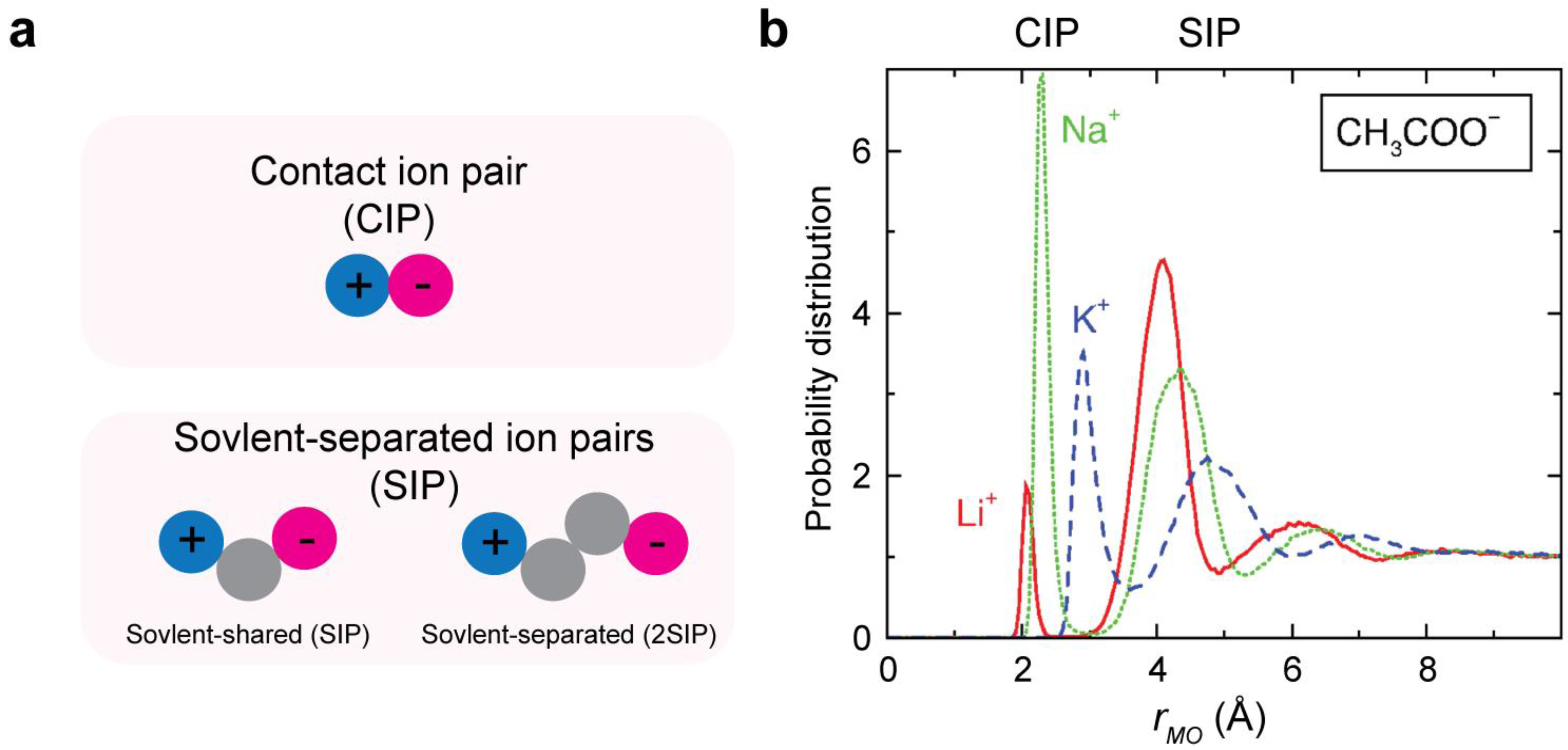
2.1. Contact Ion Pair (CIP) and Solvent-Separated Ion Pair (SIP)
2.2. Electrostriction of Water Molecules by Ions
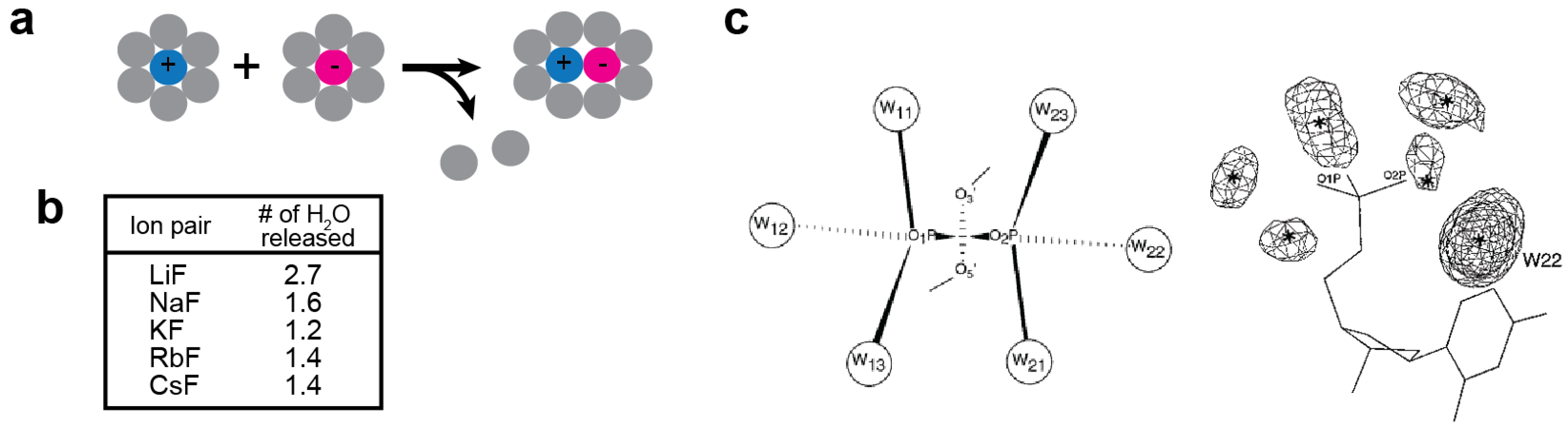
2.3. Kosmotropic and Chaotropic Ions
2.4. Collins’s Law of Matching Water Affinity

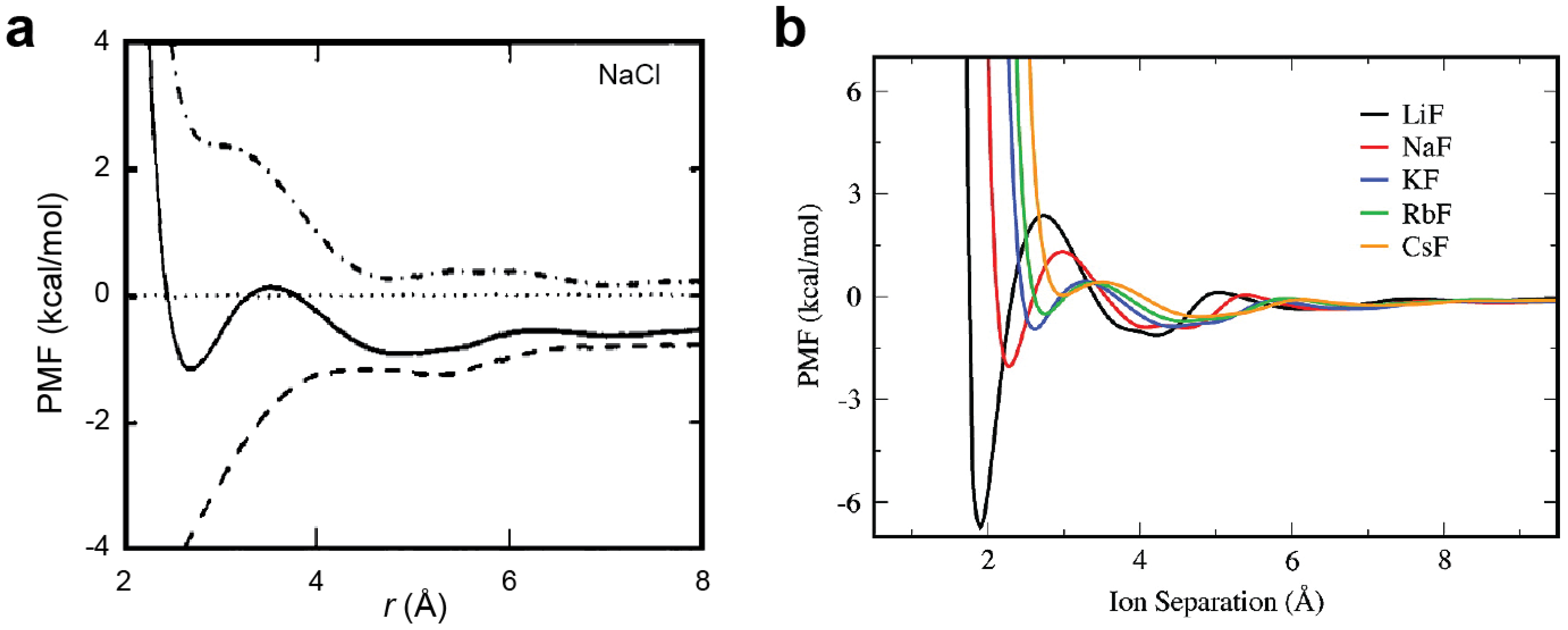
2.5. Potentials of Mean Force (PMFs) for Ion Pairs
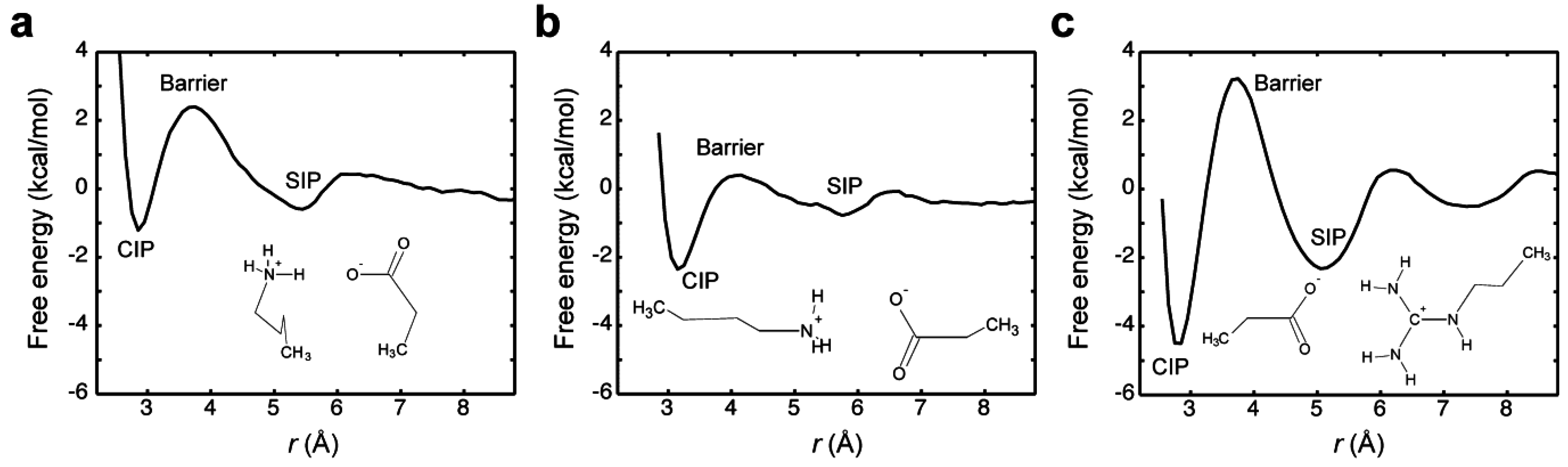
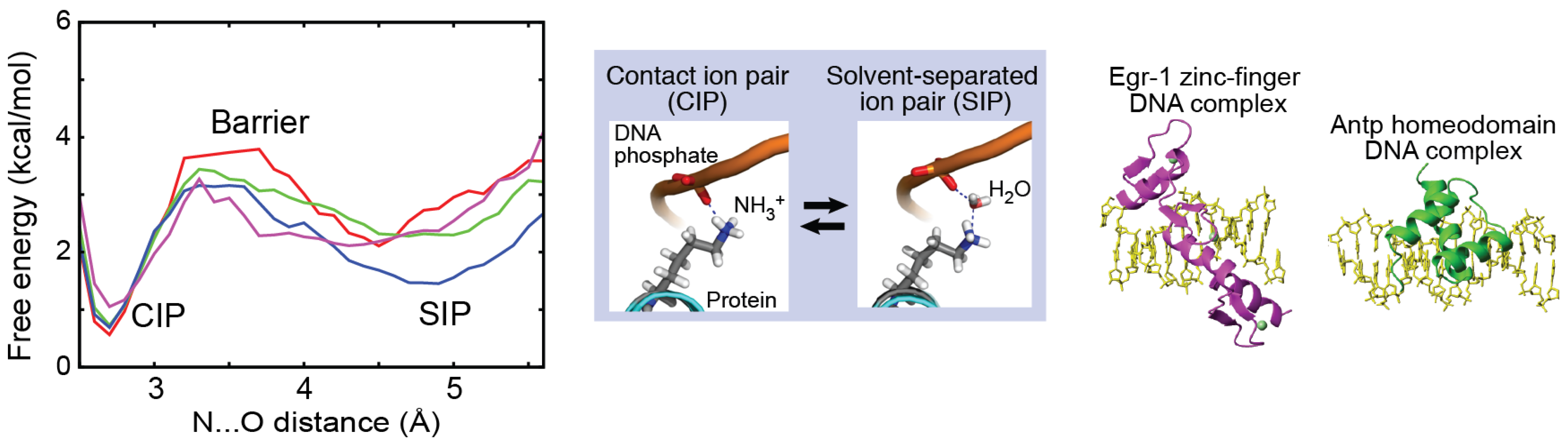
2.6. pKa Shift
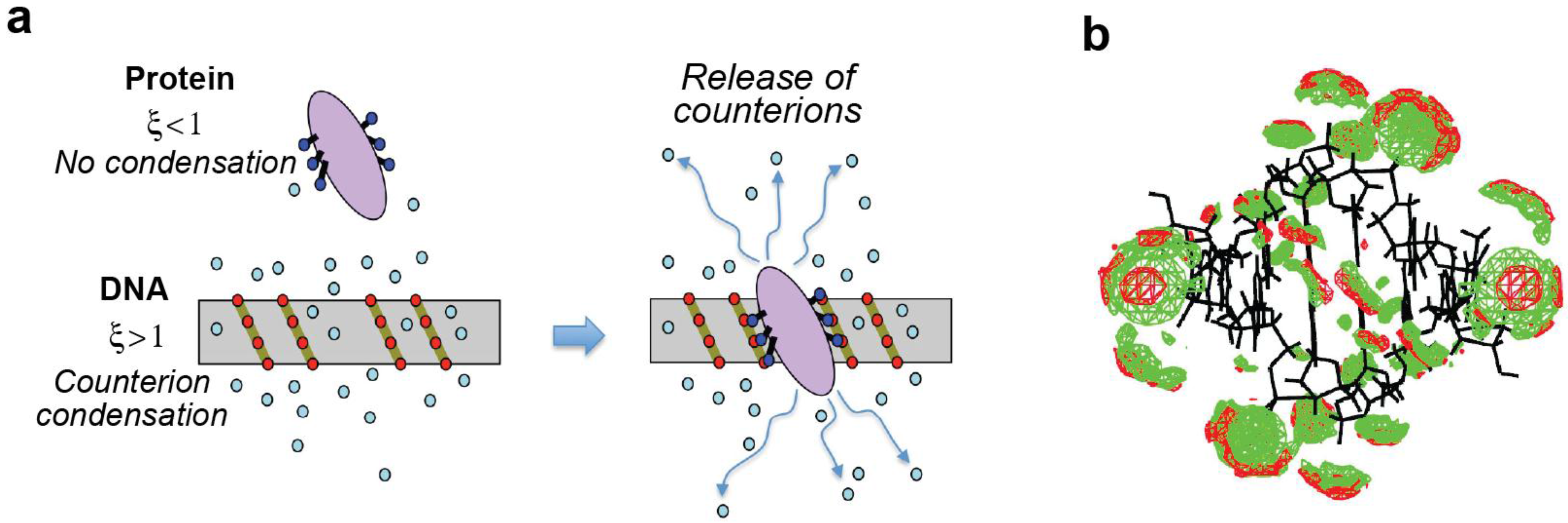
2.7. Polyelectrolyte Effects
3. Studies of Dynamics and Kinetics of Ion Pairs
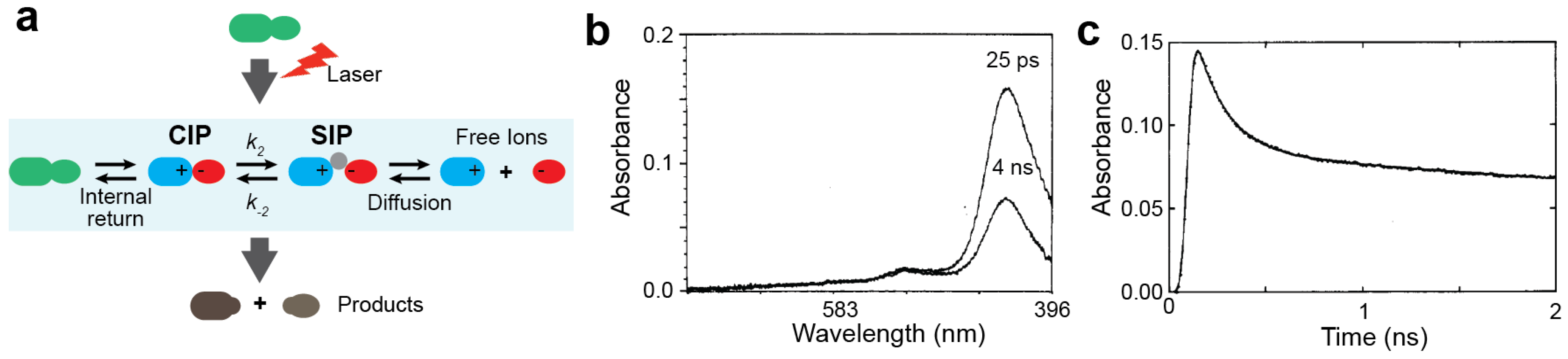
3.1. Experimental Studies on the Ion-pair Dynamics of Small Compounds by Time-resolved Absorption Spectroscopy
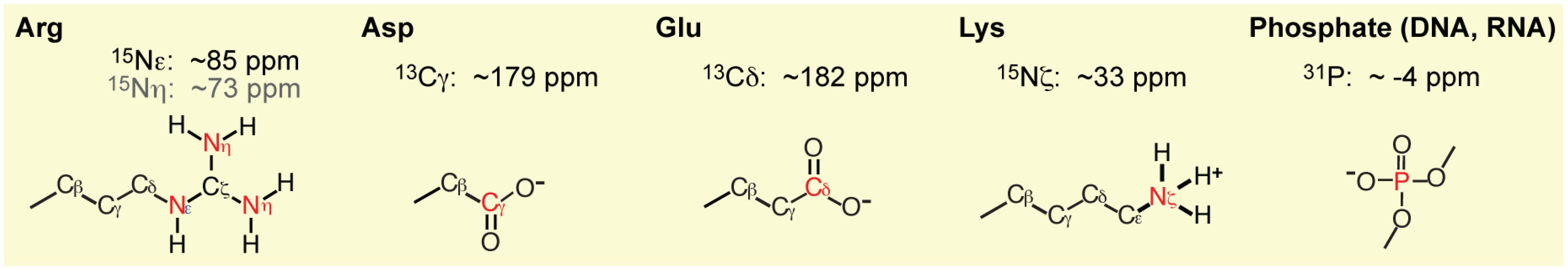
3.2. Experimental Studies on the Macromolecular Ion Pairs by NMR Spectroscopy
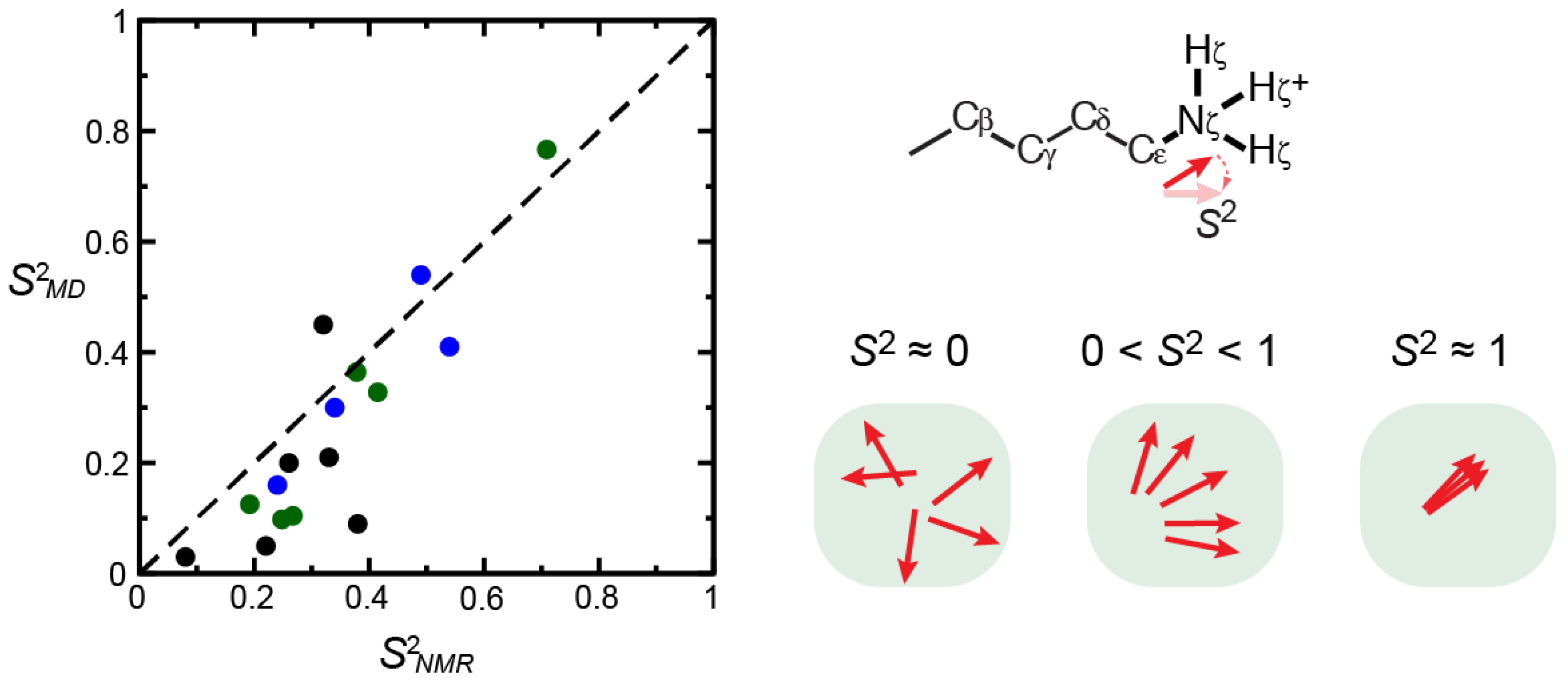

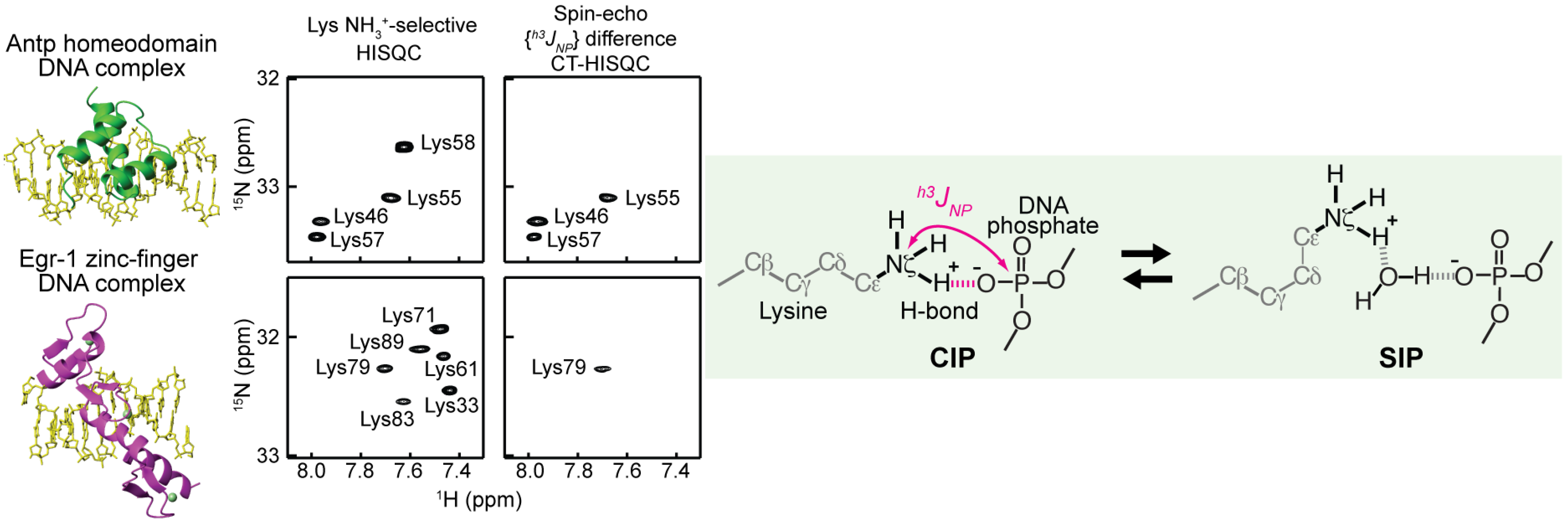
3.3. Computational Studies on Dynamics of Macromolecular Ion Pairs

4. Experimental Studies of the Energetics of Ion Pairs in Biological Macromolecular Systems
4.1. Experimental Analysis of the Energetics of Ion Pairs in Proteins
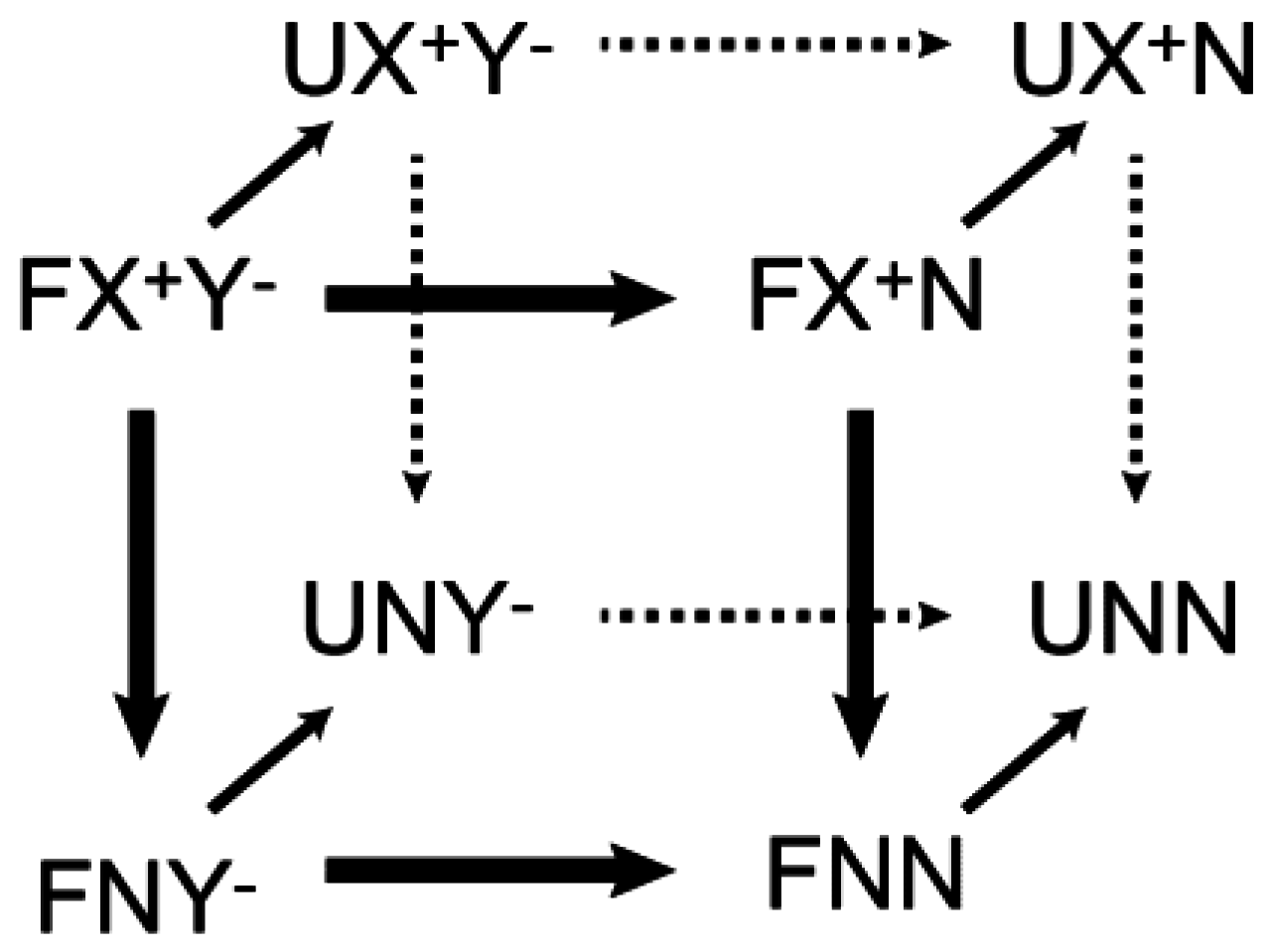
4.2. Entropic Analysis of the Counterion Release upon Ion-Pair Formation between Protein and DNA

5. Future Perspectives
5.1. Potential Roles of CIP–SIP Transitions in Protein Functions
5.2. Controversial Effects of Ion Pairs on Protein Stability
5.3. Necessity of Methodological Development in Ion-Pair Research for Biomolecules
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- McCoy, A.J.; Chandana Epa, V.; Colman, P.M. Electrostatic complementarity at protein/protein interfaces. J. Mol. Biol. 1997, 268, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Nadassy, K.; Wodak, S.J.; Janin, J. Structural features of protein-nucleic acid recognition sites. Biochsemistry 1999, 38, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Daley, D.T.; Luscombe, N.M.; Berman, H.M.; Thornton, J.M. Protein-RNA interactions: A structural analysis. Nucleic Acids Res. 2001, 29, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Pflugl, G.; Kallen, J.; Schirmer, T.; Jansonius, J.N.; Zurini, M.G.; Walkinshaw, M.D. X-ray structure of a decameric cyclophilin-cyclosporin crystal complex. Nature 1993, 361, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhen, J.; Karpowich, N.K.; Goetz, R.M.; Law, C.J.; Reith, M.E.; Wang, D.N. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 2007, 317, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef]
- Collins, K.D. Why continuum electrostatics theories cannot explain biological structure, polyelectrolytes or ionic strength effects in ion-protein interactions. Biophys. Chem. 2012, 167, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Fennell, C.J.; Bizjak, A.; Vlachy, V.; Dill, K.A. Ion pairing in molecular simulations of aqueous alkali halide solutions. J. Phys. Chem. B 2009, 113, 6782–6791. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Schravendijk, P.; Hess, B.; van der Vegt, N.F. Ion pairing in aqueous electrolyte solutions with biologically relevant anions. J. Phys. Chem. B 2011, 115, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Salis, A.; Ninham, B.W. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef] [PubMed]
- Winstein, S.; Klinedinst, P.E.; Clippinger, E. Salt Effects and Ion Paris in Solvolysis and Related Eactions. 21. Acetolysis, Bromide Exchange and Special Salt Effect. J. Am. Chem. Soc. 1961, 83, 4986–4989. [Google Scholar] [CrossRef]
- Record, M.T., Jr.; Lohman, M.L.; de Haseth, P. Ion effects on ligand-nucleic acid interactions. J. Mol. Biol. 1976, 107, 145–158. [Google Scholar] [CrossRef]
- Lü, J.M.; Rosokha, S.V.; Lindeman, S.V.; Neretin, I.S.; Kochi, J.K. “Separated” versus “contact” ion-pair structures in solution from their crystalline states: Dynamic effects on dinitrobenzenide as a mixed-valence anion. J. Am. Chem. Soc. 2005, 127, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Masnovi, J.M.; Kochi, J.K. Direct observation of ion-pair dynamics. J. Am. Chem. Soc. 1985, 107, 7880–7893. [Google Scholar] [CrossRef]
- Yabe, T.; Kochi, J.K. Contact ion-pairs. Picosecond dynamics of solvent separation, internal return, and special salt effect. J. Am. Chem. Soc. 1992, 114, 4491–4500. [Google Scholar] [CrossRef]
- Hess, B.; van der Vegt, N.F. Cation specific binding with protein surface charges. Proc. Natl. Acad. Sci. USA 2009, 106, 13296–13300. [Google Scholar] [CrossRef] [PubMed]
- Laidler, K.J. Reactions between ions. In Chemical Kinetics; Longman: New York, NY, USA, 1987; pp. 191–202. [Google Scholar]
- Marcus, Y. Ionic volumes in solution. Biophys. Chem. 2006, 124, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Marcus, Y.; Hefter, G. Ion pairing. Chem. Rev. 2006, 106, 4585–4621. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Patel, K.; Berman, H.M. Hydration of the phosphate group in double-helical DNA. Biophys. J. 1998, 75, 2422–2434. [Google Scholar] [CrossRef]
- Roe, S.M.; Teeter, M.M. Patterns for prediction of hydration around polar residues in proteins. J. Mol. Biol. 1993, 229, 419–427. [Google Scholar] [CrossRef]
- Thanki, N.; Thornton, J.M.; Goodfellow, J.M. Distributions of water around amino acid residues in proteins. J. Mol. Biol. 1988, 202, 637–657. [Google Scholar] [CrossRef]
- Jones, D.; Dole, M. The viscosity of aqueous solutions of strong electrolytes with special reference to barium chloride. J. Am. Chem. Soc. 1929, 51, 2950–2964. [Google Scholar] [CrossRef]
- Duignan, T.T.; Parsons, D.F.; Ninham, B.W. Collins’s rule, Hofmeister effects and ionic dispersion interactions. Chem. Phys. Lett. 2014, 608, 55–59. [Google Scholar] [CrossRef]
- Hribar, B.; Southall, N.T.; Vlachy, V.; Dill, K.A. How ions affect the structure of water. J. Am. Chem. Soc. 2002, 124, 12302–12311. [Google Scholar] [CrossRef] [PubMed]
- Luksic, M.; Fennell, C.J.; Dill, K.A. Using interpolation for fast and accurate calculation of ion-ion interactions. J. Phys. Chem. B 2014, 118, 8017–8025. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Jagoda-Cwiklik, B.; Woodward, C.E.; Vacha, R.; Jungwirth, P. Dielectric interpretation of specificity of ion pairing in water. J. Phys. Chem. Lett. 2010, 1, 300–303. [Google Scholar] [CrossRef]
- Anderson, K.M.; Esadze, A.; Manoharan, M.; Brüschweiler, R.; Gorenstein, D.G.; Iwahara, J. Direct observation of the ion-pair dynamics at a protein-DNA interface by NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 3613–3619. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Esadze, A.; Zandarashvili, L.; Nguyen, D.; Pettitt, B.M.; Iwahara, J. Dynamic equilibria of short-range electrostatic interactions at molecular interfaces of protein-DNA complexes. J. Phys. Chem. Lett. 2015, 6, 2733–2737. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Nguyen, D.; Anderson, K.M.; White, M.A.; Gorenstein, D.G.; Iwahara, J. Entropic enhancement of protein-DNA affinity by oxygen-to-sulfur substitution in DNA phosphate. Biophys. J. 2015, 109, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, B.M.; Rossky, P.J. Alkali-halides in water: Ion-solvent correlations and ion-ion potentials of mean force at infinite dilution. J. Chem. Phys. 1986, 84, 5836–5844. [Google Scholar] [CrossRef]
- Buckner, J.K.; Jorgensen, W.L. Energetics and hydration of the constituent ion-pairs of tetramethylammonium chloride. J. Am. Chem. Soc. 1989, 111, 2507–2516. [Google Scholar] [CrossRef]
- Dang, L.X.; Pettitt, B.M. Chloride-ion pairs in water. J. Am. Chem. Soc. 1987, 109, 5531–5532. [Google Scholar] [CrossRef]
- Friedman, R.A.; Mezei, M. The Potentials of mean force of sodium-chloride and sodium dimethylphosphate in water: An application of adaptive umbrella sampling. J. Chem. Phys. 1995, 102, 419–426. [Google Scholar] [CrossRef]
- Masunov, A.; Lazaridis, T. Potentials of mean force between ionizable amino acid side chains in water. J. Am. Chem. Soc. 2003, 125, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Resat, H.; Mezei, M.; McCammon, J.A. Use of the grand canonical ensemble in potential of mean force calculations. J. Phys. Chem. 1996, 100, 1426–1433. [Google Scholar] [CrossRef]
- Rozanska, X.; Chipot, C. Modeling ion-ion interaction in proteins: A molecular dynamics free energy calculation of the guanidinium-acetate association. J. Chem. Phys. 2000, 112, 9691–9694. [Google Scholar] [CrossRef]
- Heine, A.; DeSantis, G.; Luz, J.G.; Mitchell, M.; Wong, C.H.; Wilson, I.A. Observation of covalent intermediates in an enzyme mechanism at atomic resolution. Science 2001, 294, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Heine, A.; Luz, J.G.; Wong, C.H.; Wilson, I.A. Analysis of the class I aldolase binding site architecture based on the crystal structure of 2-deoxyribose-5-phosphate aldolase at 0.99 Å resolution. J. Mol. Biol. 2004, 343, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Stanton, C.L.; Houk, K.N. Benchmarking pKa prediction methods for residues in proteins. J. Chem. Theory Comput. 2008, 4, 951–966. [Google Scholar] [CrossRef]
- Jelesarov, I.; Karshikoff, A. Defining the role of salt bridges in protein stability. Methods Mol. Biol. 2009, 490, 227–260. [Google Scholar] [PubMed]
- Pace, C.N.; Grimsley, G.R.; Scholtz, J.M. Protein ionizable groups: PK values and their contribution to protein stability and solubility. J. Biol. Chem. 2009, 284, 13285–13289. [Google Scholar] [CrossRef] [PubMed]
- Barbas, C.F.; Heine, A.; Zhong, G.F.; Hoffmann, T.; Gramatikova, S.; Bjornestedt, R.; List, B.; Anderson, J.; Stura, E.A.; Wilson, I.A.; et al. Immune versus natural selection: Antibody aldolases with enzymic rates but broader scope. Science 1997, 278, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Fitch, C.A.; Karp, D.A.; Lee, K.K.; Stites, W.E.; Lattman, E.E.; Garcia-Moreno, E.B. Experimental pKa values of buried residues: Analysis with continuum methods and role of water penetration. Biophys. J. 2002, 82, 3289–3304. [Google Scholar] [CrossRef]
- Garcia-Moreno, B.; Dwyer, J.J.; Gittis, A.G.; Lattman, E.E.; Spencer, D.S.; Stites, W.E. Experimental measurement of the effective dielectric in the hydrophobic core of a protein. Biophys. Chem. 1997, 64, 211–224. [Google Scholar] [CrossRef]
- Ho, M.C.; Menetret, J.F.; Tsuruta, H.; Allen, K.N. The origin of the electrostatic perturbation in acetoacetate decarboxylase. Nature 2009, 459, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Stites, W.E.; Gittis, A.G.; Lattman, E.E.; Shortle, D. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J. Mol. Biol. 1991, 221, 7–14. [Google Scholar] [CrossRef]
- Takayama, Y.; Castaneda, C.A.; Chimenti, M.; Garcia-Moreno, B.; Iwahara, J. Direct evidence for deprotonation of a lysine side chain buried in the hydrophobic core of a protein. J. Am. Chem. Soc. 2008, 130, 6714–6715. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Gunner, M.R.; Garcia-Moreno, B.E. The pKa Cooperative: A collaborative effort to advance structure-based calculations of pKa values and electrostatic effects in proteins. Proteins 2011, 79, 3249–3259. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte Solutions. 3. An Analysis based on mayer ionic solution theory. J. Chem. Phys. 1969. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte Solutions. I. Colligative properties. J. Chem. Phys. 1969. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte Solutions. 2. Self-diffusion of small ions. J. Chem. Phys. 1969. [Google Scholar] [CrossRef]
- Das, R.; Mills, T.T.; Kwok, L.W.; Maskel, G.S.; Millett, I.S.; Doniach, S.; Finkelstein, K.D.; Herschlag, D.; Pollack, L. Counterion distribution around DNA probed by solution X-ray scattering. Phys. Rev. Lett. 2003. [Google Scholar] [CrossRef]
- Pabit, S.A.; Meisburger, S.P.; Li, L.; Blose, J.M.; Jones, C.D.; Pollack, L. Counting ions around DNA with anomalous small-angle X-ray scattering. J. Am. Chem. Soc. 2010, 132, 16334–16336. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Greenfeld, M.; Travers, K.J.; Chu, V.B.; Lipfert, J.; Doniach, S.; Herschlag, D. Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J. Am. Chem. Soc. 2007, 129, 14981–14988. [Google Scholar] [CrossRef] [PubMed]
- Fenley, M.O.; Russo, C.; Manning, G.S. Theoretical assessment of the oligolysine model for ionic interactions in protein-DNA complexes. J. Phys. Chem. B 2011, 115, 9864–9872. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.S. Molecular theory of polyelectrolyte solutions with applications to electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978, 11, 179–246. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.J.; Lynch, G.C.; Pettitt, B.M. Ion and solvent density distributions around canonical B-DNA from integral equations. J. Phys. Chem. B 2011, 115, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Feig, M.; Pettitt, B.M. Sodium and chlorine ions as part of the DNA solvation shell. Biophys. J. 1999, 77, 1769–1781. [Google Scholar] [CrossRef]
- Pan, F.; Roland, C.; Sagui, C. Ion distributions around left- and right-handed DNA and RNA duplexes: A comparative study. Nucleic Acids Res. 2014, 42, 13981–13996. [Google Scholar] [CrossRef] [PubMed]
- Pasi, M.; Maddocks, J.H.; Lavery, R. Analyzing ion distributions around DNA: Sequence-dependence of potassium ion distributions from microsecond molecular dynamics. Nucleic Acids Res. 2015, 43, 2412–2423. [Google Scholar] [CrossRef]
- Ponomarev, S.Y.; Thayer, K.M.; Beveridge, D.L. Ion motions in molecular dynamics simulations on DNA. Proc. Natl. Acad. Sci. USA 2004, 101, 14771–14775. [Google Scholar] [CrossRef]
- Rueda, M.; Cubero, E.; Laughton, C.A.; Orozco, M. Exploring the counterion atmosphere around DNA: What can be learned from molecular dynamics simulations? Biophys. J. 2004, 87, 800–811. [Google Scholar] [CrossRef]
- Varnai, P.; Zakrzewska, K. DNA and its counterions: A molecular dynamics study. Nucleic Acids Res. 2004, 32, 4269–4280. [Google Scholar] [CrossRef] [PubMed]
- Dragan, A.I.; Li, Z.; Makeyeva, E.N.; Milgotina, E.I.; Liu, Y.; Crane-Robinson, C.; Privalov, P.L. Forces driving the binding of homeodomains to DNA. Biochemistry 2006, 45, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Capp, M.W.; Hohenwalter, M.D.; Baskerville, M.; Record, M.T., Jr. Thermodynamic stoichiometries of participation of water, cations and anions in specific and non-specific binding of lac repressor to DNA. Possible thermodynamic origins of the “glutamate effect” on protein-DNA interactions. J. Mol. Biol. 1992, 228, 252–264. [Google Scholar] [CrossRef]
- Privalov, P.L.; Dragan, A.I.; Crane-Robinson, C. Interpreting protein/DNA interactions: Distinguishing specific from non-specific and electrostatic from non-electrostatic components. Nucleic Acids Res. 2011, 39, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Jelesarov, I.; Read, C.M.; Dragan, A.I.; Crane-Robinson, C. The energetics of HMG box interactions with DNA: Thermodynamics of the DNA binding of the HMG box from mouse sox-5. J. Mol. Biol. 1999, 294, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Record, M.T., Jr.; Zhang, W.; Anderson, C.F. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: A practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 1998, 51, 281–353. [Google Scholar] [PubMed]
- Fenley, M.O.; Harris, R.C.; Jayaram, B.; Boschitsch, A.H. Revisiting the association of cationic groove-binding drugs to DNA using a Poisson-Boltzmann approach. Biophys. J. 2010, 99, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Stigter, D. Evaluation of the counterion condensation theory of polyelectrolytes. Biophys. J. 1995, 69, 380–388. [Google Scholar] [CrossRef]
- Peters, K.S.; Li, B.L. Picosecond dynamics of contact ion-pairs and solvent-separated ion-pairs in the photosolvolysis of diphenylmethyl chloride. J Phys Chem 1994, 98, 401–403. [Google Scholar] [CrossRef]
- Simon, J.D.; Peters, K.S. Picosecond dynamics of ion-pairs—The effect of hydrogen-bonding on ion-pair intermediates. J. Am. Chem. Soc. 1982, 104, 6542–6547. [Google Scholar] [CrossRef]
- Simon, J.D.; Peters, K.S. Direct observation of the special salt effect—Picosecond dynamics of ion-pair exchange. J. Am. Chem. Soc. 1982, 104, 6142–6144. [Google Scholar] [CrossRef]
- Simon, J.D.; Peters, K.S. Na+ and Li+ Effects on the photo-reduction of benzophenone—A picosecond absorption study. J. Am. Chem. Soc. 1983, 105, 4875–4882. [Google Scholar] [CrossRef]
- Simon, J.D.; Peters, K.S. Picosecond studies of organic photoreactions. Acc. Chem. Res. 1984, 17, 277–283. [Google Scholar] [CrossRef]
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. An NMR perspective on enzyme dynamics. Chem. Rev. 2006, 106, 3055–3079. [Google Scholar] [CrossRef] [PubMed]
- Bothe, J.R.; Nikolova, E.N.; Eichhorn, C.D.; Chugh, J.; Hansen, A.L.; Al-Hashimi, H.M. Characterizing RNA dynamics at atomic resolution using solution-state NMR spectroscopy. Nat. Methods 2011, 8, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd. Chemical exchange in biomacromolecules: Past, present, and future. J. Magn. Reson. 2014, 241, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.L.; Kay, L.E. Quantifying millisecond time-scale exchange in proteins by CPMG relaxation dispersion NMR spectroscopy of side-chain carbonyl groups. J. Biomol. NMR 2011, 50, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Paquin, R.; Ferrage, F.; Mulder, F.A.; Akke, M.; Bodenhausen, G. Multiple-timescale dynamics of side-chain carboxyl and carbonyl groups in proteins by 13C nuclear spin relaxation. J. Am. Chem. Soc. 2008, 130, 15805–15807. [Google Scholar] [CrossRef]
- Stafford, K.A.; Ferrage, F.; Cho, J.H.; Palmer, A.G., 3rd. Side chain dynamics of carboxyl and carbonyl groups in the catalytic function of Escherichia coli ribonuclease H. J. Am. Chem. Soc. 2013, 135, 18024–18027. [Google Scholar] [CrossRef]
- Berglund, H.; Baumann, H.; Knapp, S.; Ladenstein, R.; Härd, T. Flexibility of an arginine side chain at a DNA-protein interface. J. Am. Chem. Soc. 1995, 117, 12883–12884. [Google Scholar] [CrossRef]
- Birdsall, B.; Polshakov, V.I.; Feeney, J. NMR studies of ligand carboxylate group interactions with arginine residues in complexes of Lactobacillus casei dihydrofolate reductase with substrates and substrate analogues. Biochemistry 2000, 39, 9819–9825. [Google Scholar] [CrossRef] [PubMed]
- Morgan, W.D.; Birdsall, B.; Nieto, P.M.; Gargaro, A.R.; Feeney, J. 1H/15N HSQC NMR studies of ligand carboxylate group interactions with arginine residues in complexes of brodimoprim analogues and Lactobacillus casei dihydrofolate reductase. Biochemistry 1999, 38, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Nieto, P.M.; Birdsall, B.; Morgan, W.D.; Frenkiel, T.A.; Gargaro, A.R.; Feeney, J. Correlated bond rotations in interactions of arginine residues with ligand carboxylate groups in protein ligand complexes. FEBS Lett. 1997, 405, 16–20. [Google Scholar] [CrossRef]
- Iwahara, J.; Clore, G.M. Sensitivity improvement for correlations involving arginine side-chain Nepsilon/Hepsilon resonances in multi-dimensional NMR experiments using broadband 15N 180 degrees pulses. J. Biomol. NMR 2006, 36, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Zweckstetter, M.; Clore, G.M. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 15062–15067. [Google Scholar] [CrossRef] [PubMed]
- Trbovic, N.; Cho, J.H.; Abel, R.; Friesner, R.A.; Rance, M.; Palmer, A.G., 3rd. Protein side-chain dynamics and residual conformational entropy. J. Am. Chem. Soc. 2009, 131, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Werbeck, N.D.; Kirkpatrick, J.; Hansen, D.F. Probing arginine side-chains and their dynamics with carbon-detected NMR spectroscopy: Application to the 42 kDa human histone deacetylase 8 at high pH. Angew. Chem. Int. Ed. Engl. 2013, 52, 3145–3147. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.A.; Botuyan, M.V.; Kaplan, B.E.; Rossi, J.J.; Chen, Y. Arginine side-chain dynamics in the HIV-1 rev-RRE complex. J. Mol. Biol. 2000, 303, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Pascal, S.M.; Singer, A.U.; Formankay, J.D.; Kay, L.E. NMR pulse schemes for the sequence-specific assignment of arginine guanidino N-15 and H-1 chemical-shifts in proteins. J. Am. Chem. Soc. 1995, 117, 3556–3564. [Google Scholar] [CrossRef]
- Esadze, A.; Li, D.W.; Wang, T.; Brüschweiler, R.; Iwahara, J. Dynamics of lysine side-chain amino groups in a protein studied by heteronuclear 1H-15N NMR spectroscopy. J. Am. Chem. Soc. 2011, 133, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Zandarashvili, L.; Iwahara, J. Effective strategy to assign 1H-15N heteronuclear correlation NMR signals from lysine side-chain NH3+ groups of proteins at low temperature. J. Biomol. NMR 2014, 60, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Jung, Y.S.; Clore, G.M. Heteronuclear NMR spectroscopy for lysine NH3 groups in proteins: Unique effect of water exchange on 15N transverse relaxation. J. Am. Chem. Soc. 2007, 129, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Poon, D.K.; Schubert, M.; Au, J.; Okon, M.; Withers, S.G.; McIntosh, L.P. Unambiguous determination of the ionization state of a glycoside hydrolase active site lysine by 1H-15N heteronuclear correlation spectroscopy. J. Am. Chem. Soc. 2006, 128, 15388–15389. [Google Scholar] [CrossRef] [PubMed]
- Segawa, T.; Kateb, F.; Duma, L.; Bodenhausen, G.; Pelupessy, P. Exchange rate constants of invisible protons in proteins determined by NMR spectroscopy. Chembiochem 2008, 9, 537–542. [Google Scholar] [CrossRef]
- Tomlinson, J.H.; Ullah, S.; Hansen, P.E.; Williamson, M.P. Characterization of salt bridges to lysines in the protein G B1 domain. J. Am. Chem. Soc. 2009, 131, 4674–4684. [Google Scholar] [CrossRef] [PubMed]
- Takayama, Y.; Sahu, D.; Iwahara, J. Observing in-phase single-quantum 15N multiplets for NH2/NH3+ groups with two-dimensional heteronuclear correlation spectroscopy. J. Magn. Reson. 2008, 194, 313–316. [Google Scholar] [CrossRef]
- Williamson, M.P.; Hounslow, A.M.; Ford, J.; Fowler, K.; Hebditch, M.; Hansen, P.E. Detection of salt bridges to lysines in solution in barnase. Chem. Commun. 2013, 49, 9824–9826. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Li, D.W.; Wang, T.; Brüschweiler, R.; Iwahara, J. Signature of mobile hydrogen bonding of lysine side chains from long-range 15N-13C scalar J-couplings and computation. J. Am. Chem. Soc. 2011, 133, 9192–9195. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Esadze, A.; Iwahara, J. NMR studies on the dynamics of hydrogen bonds and ion pairs involving lysine side chains of proteins. Adv. Protein Chem. Struct. Biol. 2013, 93, 37–80. [Google Scholar] [PubMed]
- Zandarashvili, L.; Iwahara, J. Temperature dependence of internal motions of protein side-chain NH3+ groups: Insight into energy barriers for transient breakage of hydrogen bonds. Biochemistry 2015, 54, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Esadze, A.; Vuzman, D.; Kemme, C.A.; Levy, Y.; Iwahara, J. Balancing between affinity and speed in target DNA search by zinc-finger proteins via modulation of dynamic conformational ensemble. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Gorenstein, D.G. Conformation and dynamics of DNA and protein-DNA complexes by 31P NMR. Chem. Rev. 1994, 94, 1315–1338. [Google Scholar] [CrossRef]
- Tian, Y.; Kayatta, M.; Shultis, K.; Gonzalez, A.; Mueller, L.J.; Hatcher, M.E. 31P NMR investigation of backbone dynamics in DNA binding sites. J. Phys. Chem. B 2009, 113, 2596–2603. [Google Scholar] [CrossRef]
- Wu, Z.; Delaglio, F.; Tjandra, N.; Zhurkin, V.B.; Bax, A. Overall structure and sugar dynamics of a DNA dodecamer from homo- and heteronuclear dipolar couplings and 31P chemical shift anisotropy. J. Biomol. NMR 2003, 26, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Szabo, A.; Bax, A.; Kay, L.E.; Driscoll, P.C.; Gronenborn, A.M. Deviations from the simple 2-parameter model-free approach to the interpretation of N-15 nuclear magnetic-relaxation of proteins. J. Am. Chem. Soc. 1990, 112, 4989–4991. [Google Scholar] [CrossRef]
- Kay, L.E.; Torchia, D.A. The effects of dipolar cross-correlation on C-13 Methyl-Carbon T1, T2, and noe measurements in macromolecules. J. Magn. Reson. 1991, 95, 536–547. [Google Scholar]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in Macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in Macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Case, D.A.; Scheurer, C.; Brüschweiler, R. Static and dynamic effects on vicinal scalar J couplings in proteins and peptides: A MD/DFT analysis. J. Am. Chem. Soc. 2000, 122, 10390–10397. [Google Scholar] [CrossRef]
- Chou, J.J.; Case, D.A.; Bax, A. Insights into the mobility of methyl-bearing side chains in proteins from 3JCC and 3JCN couplings. J. Am. Chem. Soc. 2003, 125, 8959–8966. [Google Scholar] [CrossRef]
- Perez, C.; Löhr, F.; Rüterjans, H.; Schmidt, J.M. Self-consistent Karplus parametrization of 3J couplings depending on the polypeptide side-chain torsion chi1. J. Am. Chem. Soc. 2001, 123, 7081–7093. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Cordier, F.; Grzesiek, S. Direct observation of hydrogen bonds in proteins by interresidue 3hJNC scalar couplings. J. Am. Chem. Soc. 1999, 121, 1601–1602. [Google Scholar] [CrossRef]
- Cornilescu, G.; Hu, J.S.; Bax, A. Identification of the hydrogen bonding network in a protein by scalar couplings. J. Am. Chem. Soc. 1999, 121, 2949–2950. [Google Scholar] [CrossRef]
- Dingley, A.J.; Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Pervushin, K.; Ono, A.; Fernandez, C.; Szyperski, T.; Kainosho, M.; Wüthrich, K. NMR scalar couplings across Watson-Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 14147–14151. [Google Scholar] [CrossRef] [PubMed]
- Grzesiek, S.; Cordier, F.; Dingley, A.J. Scalar couplings across hydrogen bonds. Nuclear Magn. Reson. Biol. Macromol. A 2001, 338, 111–133. [Google Scholar]
- Grzesiek, S.; Cordier, F.; Jaravine, V.; Barfield, M. Insights into biomolecular hydrogen bonds from hydrogen bond scalar couplings. Prog. NMR Spectrosc. 2004, 45, 275–300. [Google Scholar] [CrossRef]
- Liu, A.; Hu, W.; Majumdar, A.; Rosen, M.K.; Patel, D.J. NMR detection of side chain-side chain hydrogen bonding interactions in 13C/15N-labeled proteins. J. Biomol. NMR 2000, 17, 305–310. [Google Scholar] [CrossRef]
- Anderson, K.M.; Nguyen, D.; Esadze, A.; Zandrashvili, L.; Gorenstein, D.G.; Iwahara, J. A chemical approach for site-specific identification of NMR signals from protein side-chain NH3+ groups forming intermolecular ion pairs in protein-nucleic acid complexes. J. Biomol. NMR 2015, 62, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Bjelic, S.; Wieninger, S.; Jelesarov, M.; Karshikoff, A. Electrostatic contribution to the thermodynamic and kinetic stability of the homotrimeric coiled coil Lpp-56: A computational study. Proteins Struct Funct. Bioinform. 2008, 70, 810–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caflisch, A.; Karplus, M. Acid and thermal-denaturation of barnase investigated by molecular-dynamics simulations. J. Mol. Biol. 1995, 252, 672–708. [Google Scholar] [CrossRef] [PubMed]
- Gruia, A.D.; Fischer, S.; Smith, J.C. Kinetics of breaking a salt-bridge critical in protein unfolding. Chem. Phys. Lett. 2004, 385, 337–340. [Google Scholar] [CrossRef]
- Huang, X.Q.; Zhou, H.X. Similarity and difference in the unfolding of thermophilic and mesophilic cold shock proteins studied by molecular dynamics. Biophys. J. 2006, 91, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Koumanov, A.; Karshikoff, A.; Friis, E.P.; Borchert, T.V. Conformational averaging in pK calculations: Improvement and limitations in prediction of ionization properties of proteins. J. Phys. Chem. B 2001, 105, 9339–9344. [Google Scholar] [CrossRef]
- Sheldahl, C.; Harvey, S.C. Molecular dynamics on a model for nascent high-density lipoprotein: Role of salt bridges. Biophys. J. 1999, 76, 1190–1198. [Google Scholar] [CrossRef]
- De Bakker, P.I.; Hunenberger, P.H.; McCammon, J.A. Molecular dynamics simulations of the hyperthermophilic protein Sac7d from Sulfolobus acidocaldarius: Contribution of salt bridges to thermostability. J. Mol. Biol. 1999, 285, 1811–1830. [Google Scholar] [CrossRef] [PubMed]
- Bashford, D.; Gerwert, K. Electrostatic calculations of the pKa Values of Ionizable Groups in Bacteriorhodopsin. J. Mol. Biol. 1992, 224, 473–486. [Google Scholar] [CrossRef]
- Gorfe, A.A.; Ferrara, P.; Caflisch, A.; Marti, D.N.; Bosshard, H.R.; Jelesarov, I. Calculation of protein ionization equilibria with conformational sampling: PKa of a model leucine zipper, GCN4 and barnase. Proteins Struct. Funct. Genet. 2002, 46, 41–60. [Google Scholar] [CrossRef]
- Van Vlijmen, H.W.T.; Schaefer, M.; Karplus, M. Improving the accuracy of protein pKa calculations: Conformational averaging versus the average structure. Proteins Struct. Funct. Genet. 1998, 33, 145–158. [Google Scholar] [CrossRef]
- Yang, A.S.; Honig, B. On the pH-dependence of protein stability. J. Mol. Biol. 1993, 231, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Honig, B. Structural origins of ph and ionic-strength effects on protein stability—Acid denaturation of sperm whale apomyoglobin. J. Mol. Biol. 1994, 237, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Bosshard, H.R.; Marti, D.N.; Jelesarov, I. Protein stabilization by salt bridges: Concepts, experimental approaches and clarification of some misunderstandings. J. Mol. Recognit. 2004, 17, 1–16. [Google Scholar] [CrossRef]
- Fersht, A.R. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman and Company: New York, USA, 1998; pp. 128–131. [Google Scholar]
- Horovitz, A. Double-mutant cycles: A powerful tool for analyzing protein structure and function. Fold. Des. 1996, 1, R121–R126. [Google Scholar] [CrossRef]
- Serrano, L.; Horovitz, A.; Avron, B.; Bycroft, M.; Fersht, A.R. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry 1990, 29, 9343–9352. [Google Scholar] [CrossRef] [PubMed]
- Blasie, C.A.; Berg, J.M. Electrostatic interactions across a beta-sheet. Biochemistry 1997, 36, 6218–6222. [Google Scholar] [CrossRef] [PubMed]
- Lassila, K.S.; Datta, D.; Mayo, S.L. Evaluation of the energetic contribution of an ionic network to beta-sheet stability. Protein Sci. 2002, 11, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Merkel, J.S.; Sturtevant, J.M.; Regan, L. Sidechain interactions in parallel beta sheets: The energetics of cross-strand pairings. Structure 1999, 7, 1333–1343. [Google Scholar] [CrossRef]
- Horovitz, A.; Serrano, L.; Avron, B.; Bycroft, M.; Fersht, A.R. Strength and co-operativity of contributions of surface salt bridges to protein stability. J. Mol. Biol. 1990, 216, 1031–1044. [Google Scholar] [CrossRef]
- Makhatadze, G.I.; Loladze, V.V.; Ermolenko, D.N.; Chen, X.; Thomas, S.T. Contribution of surface salt bridges to protein stability: Guidelines for protein engineering. J. Mol. Biol. 2003, 327, 1135–1148. [Google Scholar] [CrossRef]
- Smith, C.K.; Regan, L. Guidelines for protein design: The energetics of beta sheet side chain interactions. Science 1995, 270, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Record, M.T.; Anderson, C.F.; Lohman, T.M. Thermodynamic analysis of ion effects on binding and conformational equilibria of proteins and nucleic-acids—Roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 1978, 11, 103–178. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Iwahara, J. Stopped-flow fluorescence kinetic study of protein sliding and intersegment transfer in the target DNA search process. J. Mol. Biol. 2014, 426, 230–244. [Google Scholar] [CrossRef]
- Spolar, R.S.; Record, M.T., Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science 1994, 263, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Esadze, A.; Kemme, C.A.; Kolomeisky, A.B.; Iwahara, J. Positive and negative impacts of nonspecific sites during target location by a sequence-specific DNA-binding protein: Origin of the optimal search at physiological ionic strength. Nucleic Acids Res. 2014, 42, 7039–7046. [Google Scholar] [CrossRef] [PubMed]
- Winstein, S.; Klinedinst, P.E.; Robinson, G.C. Salt effects and ion pairs in solvolysis and related Reactions. 17. Induced common ion rate depression and mechanism of special salt effect. J. Am. Chem. Soc. 1961, 83, 885–895. [Google Scholar] [CrossRef]
- Karshikoff, A.; Ladenstein, R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: A “traffic rule” for hot roads. Trends Biochem. Sci. 2001, 26, 550–556. [Google Scholar] [CrossRef]
- Vogt, G.; Woell, S.; Argos, P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Sauer, R.T. Tolerance of Arc repressor to multiple-alanine substitutions. Proc. Natl. Acad. Sci. USA 1999, 96, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Kim, P.S. Folding of bovine pancreatic trypsin inhibitor (BPTI) variants in which almost half the residues are alanine. J. Mol. Biol. 2000, 298, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K.; Garcia-Moreno, B.; Shortle, D. Contributions of the ionizable amino acids to the stability of staphylococcal nuclease. Biochemistry 1996, 35, 6443–6449. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. Close-range electrostatic interactions in proteins. Chembiochem 2002, 3, 604–617. [Google Scholar] [CrossRef]
- Cho, J.H.; Raleigh, D.P. Mutational analysis demonstrates that specific electrostatic interactions can play a key role in the denatured state ensemble of proteins. J. Mol. Biol. 2005, 353, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Raleigh, D.P. Electrostatic interactions in the denatured state and in the transition state for protein folding: Effects of denatured state interactions on the analysis of transition state structure. J. Mol. Biol. 2006, 359, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Siu, C.K.; Fox-Beyer, B.S.; Beyer, M.K.; Bondybey, V.E. Ab initio molecular dynamics studies of ionic dissolution and precipitation of sodium chloride and silver chloride in water clusters, NaCl(H2O)n and AgCl(H2O)n, n = 6, 10, and 14. Chemistry 2006, 12, 6382–6392. [Google Scholar] [CrossRef] [PubMed]
- Timko, J.; Bucher, D.; Kuyucak, S. Dissociation of NaCl in water from ab initio molecular dynamics simulations. J. Chem. Phys. 2010. [Google Scholar] [CrossRef] [PubMed]
- Wiedemair, M.J.; Weiss, A.K.; Rode, B.M. Ab initio quantum mechanical simulations confirm the formation of all postulated species in ionic dissociation. Phys. Chem. Chem. Phys. 2014, 16, 7368–7376. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, K.A.; Lin, Y.S.; Das, R.; Pande, V.S. Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Hummer, G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 2009, 113, 9004–9015. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Brüschweiler, R. NMR-based protein potentials. Angew. Chem. Int. Ed. Engl. 2010, 49, 6778–6780. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, L.; Okur, A.; Simmerling, C. Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys. J. 2009, 97, 853–856. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwahara, J.; Esadze, A.; Zandarashvili, L. Physicochemical Properties of Ion Pairs of Biological Macromolecules. Biomolecules 2015, 5, 2435-2463. https://doi.org/10.3390/biom5042435
Iwahara J, Esadze A, Zandarashvili L. Physicochemical Properties of Ion Pairs of Biological Macromolecules. Biomolecules. 2015; 5(4):2435-2463. https://doi.org/10.3390/biom5042435
Chicago/Turabian StyleIwahara, Junji, Alexandre Esadze, and Levani Zandarashvili. 2015. "Physicochemical Properties of Ion Pairs of Biological Macromolecules" Biomolecules 5, no. 4: 2435-2463. https://doi.org/10.3390/biom5042435