
Rho GTPases: Novel Players in the Regulation of the DNA Damage Response?

{kind=link}
{kind=link}
Abstract
:1. Regulation and Biological Function of Rho GTPases
2. Rho GTPases in the Regulation of Genotoxic Stress Responses
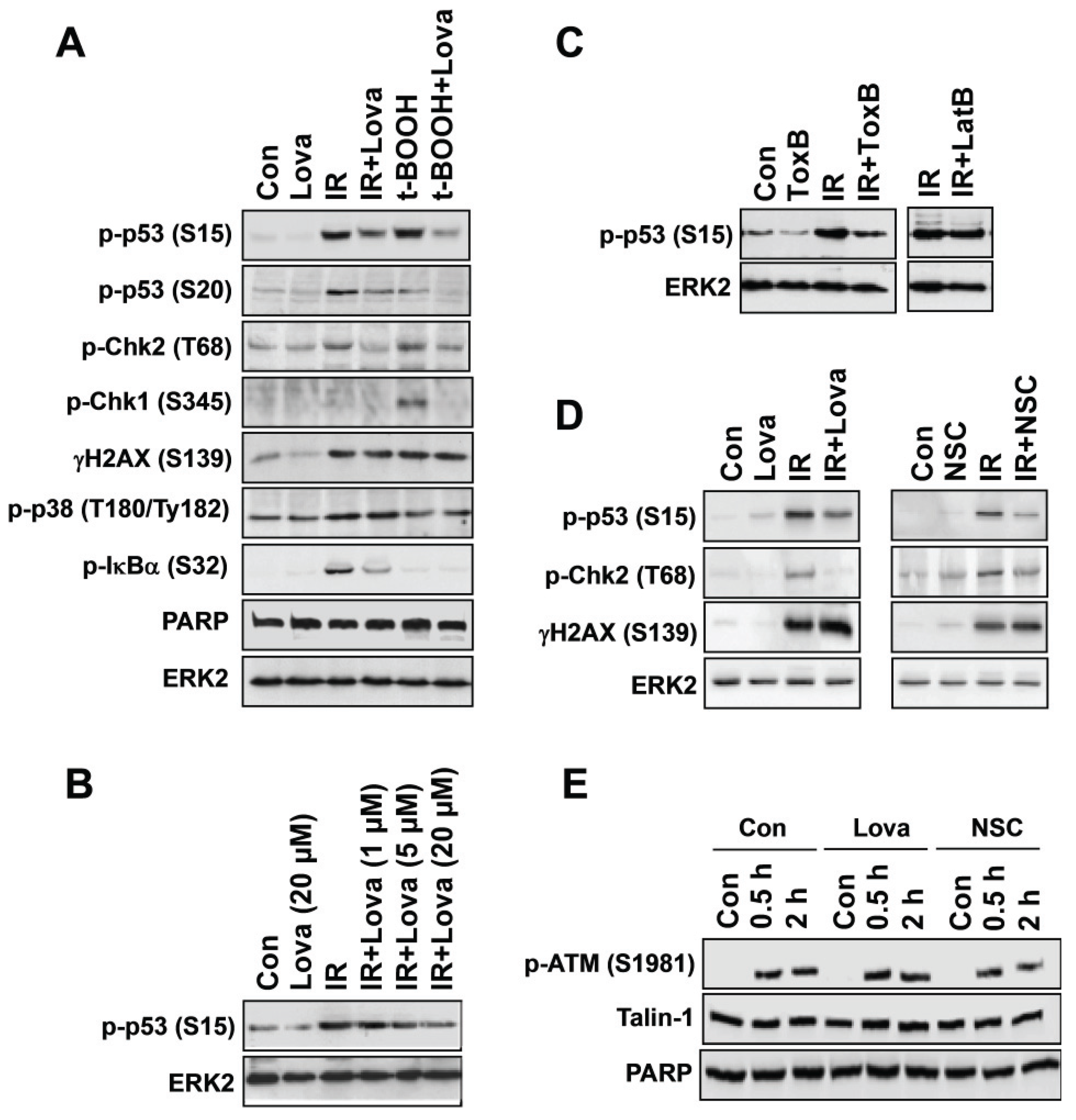
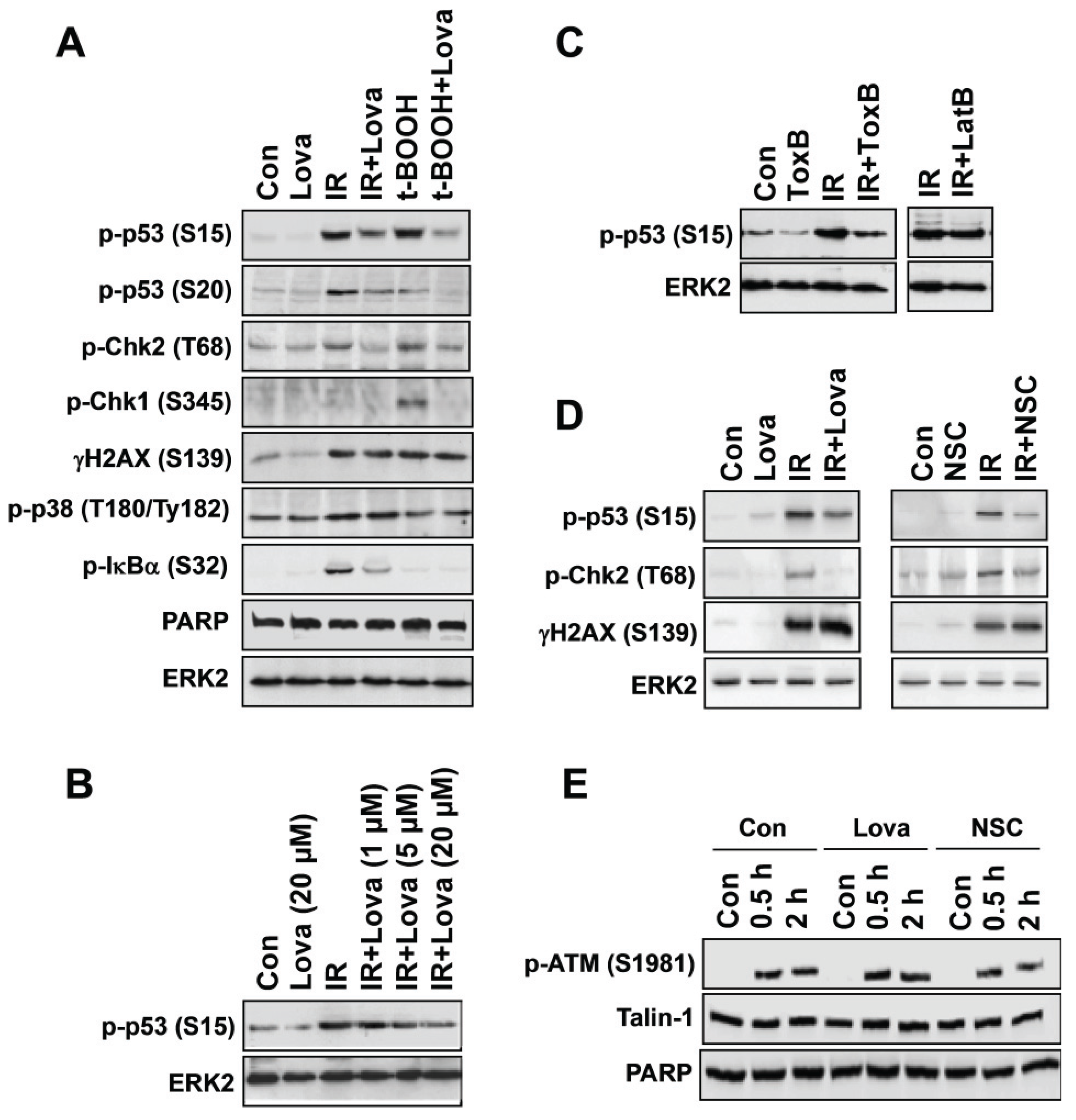
3. Pharmacological and Genetic Targeting of Rac1-signaling Impacts Activation of DDR Mechanisms
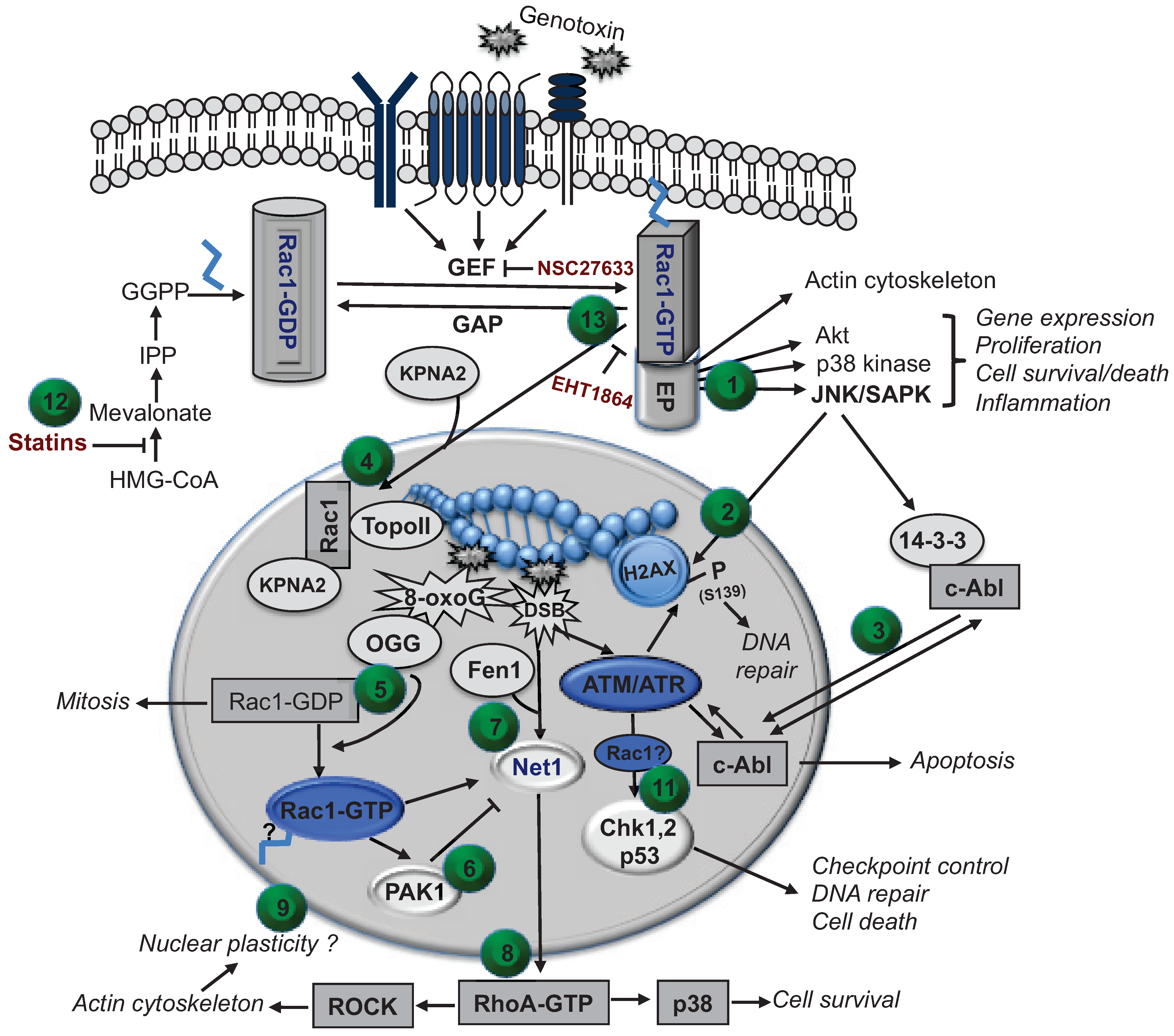
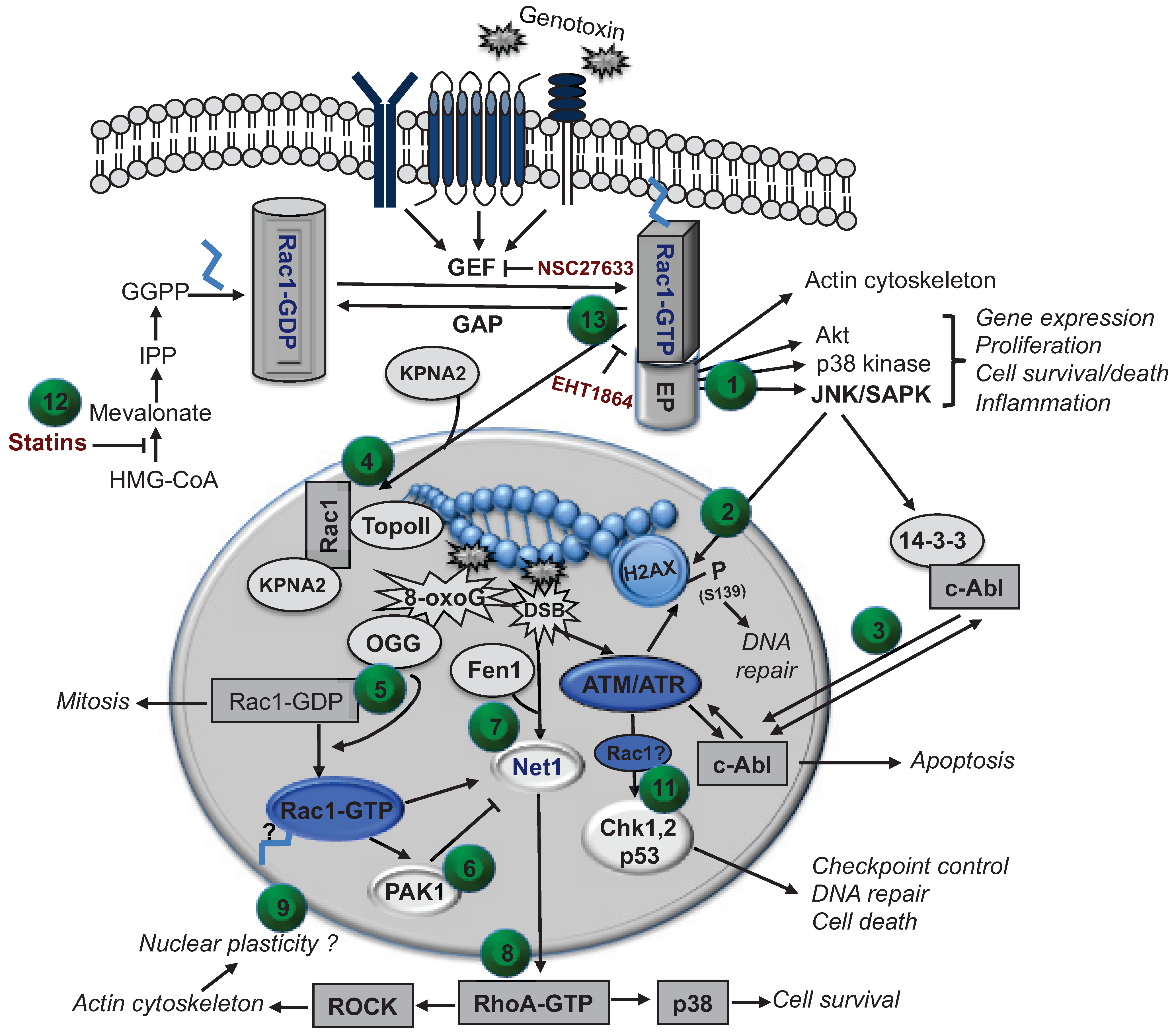
4. Putative Role of Nuclear Rho GTPases in the Regulation of the DNA Damage Response (DDR)
5. Translational Aspects of Rac1 Targeting Strategies in Anticancer Therapy
6. Outlook and Conclusions—Further Validation of Rac1 as Promising Target to Modulate DDR and Repair
Acknowledgments
Conflicts of Interest
References
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chernoff, J.; Zheng, Y. Interaction of Rac1 with GTPase-activating proteins and putative effectors. A comparison with Cdc42 and RhoA. J. Biol. Chem. 1998, 273, 8776–8782. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M. Rho signalling at a glance. J. Cell Sci. 2004, 117, 5457–5458. [Google Scholar] [CrossRef] [PubMed]
- Iden, S.; Collard, J.G. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 2008, 9, 846–859. [Google Scholar] [CrossRef]
- Kjoller, L.; Hall, A. Signaling to Rho GTPases. Exp. Cell Res. 1999, 253, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Van Aelst, L.; D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 1997, 11, 2295–2322. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.; Marshall, C.J.; Hall, A.; Tilbrook, P.A. Post-translational modifications of p21Rho proteins. J. Biol. Chem. 1992, 267, 20033–20038. [Google Scholar] [PubMed]
- Adamson, P.; Paterson, H.F.; Hall, A. Intracellular localization of the p21Rho proteins. J. Cell Biol. 1992, 119, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.R.; Nassar, N.; Cerione, R.A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator Rhogdi. Cell 2000, 100, 345–356. [Google Scholar] [CrossRef]
- Olofsson, B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell Signal 1999, 11, 545–554. [Google Scholar] [CrossRef]
- Busch, C.; Aktories, K. Microbial toxins and the glycosylation of Rho family GTPases. Curr. Opin. Struct. Biol. 2000, 10, 528–535. [Google Scholar] [CrossRef]
- Aktories, K.; Schmidt, G.; Just, I. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 2000, 381, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Ras-related GTPases and the cytoskeleton. Mol. Cell Biol. 1992, 3, 475–479. [Google Scholar] [CrossRef]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Claret, F.X.; Abo, A.; Karin, M. Selective activation of the JNK signaling cascade and c-jun transcriptional activity by the small GTPases Rac and Cdc42hs. Cell 1995, 81, 1147–1157. [Google Scholar] [CrossRef]
- Canman, C.E.; Kastan, M.B. Three paths to stress relief. Nature 1996, 384, 213–214. [Google Scholar] [CrossRef]
- Verheij, M.; Bose, R.; Lin, X.H.; Yao, B.; Jarvis, W.D.; Grant, S.; Birrer, M.J.; Szabo, E.; Zon, L.I.; Kyriakis, J.M.; et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 1996, 380, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 map kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, J.; Depatie, C.; Ohmichi, M.; Mercola, D. The activation of c-jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J. Biol. Chem. 2003, 278, 20582–20592. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sanchez-Perez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-κB by Rho, Cdc42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.A.; Govek, E.E.; Bottner, B.; van Aelst, L. Rho GTPases: Signaling, migration, and invasion. Exp. Cell Res. 2000, 261, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. Rho GTPases in transformation and metastasis. Adv. Cancer Res. 2002, 84, 57–80. [Google Scholar] [PubMed]
- Ridley, A.J. Rho proteins and cancer. Breast Cancer Res. Treat. 2004, 84, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Braga, V.M.; Machesky, L.M.; Hall, A.; Hotchin, N.A. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J. Cell Biol. 1997, 137, 1421–1431. [Google Scholar] [CrossRef]
- Ridley, A.J.; Hall, A. The small GTP-binding protein Rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Olson, M.F.; Ashworth, A.; Hall, A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 1995, 269, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Aznar, S.; Lacal, J.C. Rho signals to cell growth and apoptosis. Cancer Lett. 2001, 165, 1–10. [Google Scholar] [CrossRef]
- Knebel, A.; Rahmsdorf, H.J.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996, 15, 5314–5325. [Google Scholar] [PubMed]
- Sachsenmaier, C.; Radler-Pohl, A.; Zinck, R.; Nordheim, A.; Herrlich, P.; Rahmsdorf, H.J. Involvement of growth factor receptors in the mammalian uvc response. Cell 1994, 78, 963–972. [Google Scholar] [CrossRef]
- Herrlich, P.; Karin, M.; Weiss, C. Supreme enlightenment: Damage recognition and signaling in the mammalian UV response. Mol. Cell 2008, 29, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Knebel, A.; Tenev, T.; Neininger, A.; Gaestel, M.; Herrlich, P.; Bohmer, F.D. Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction. J. Biol. Chem. 1999, 274, 26378–26386. [Google Scholar] [PubMed]
- Ichijo, H. From receptors to stress-activated map kinases. Oncogene 1999, 18, 6087–6093. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Baskaran, R.; Lea-Chou, E.T.; Wood, L.D.; Chen, Y.; Karin, M.; Wang, J.Y. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 1996, 384, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y. Regulation of cell death by the Abl tyrosine kinase. Oncogene 2000, 19, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Ren, R.; Pandey, P.; Shafman, T.D.; Feller, S.M.; Weichselbaum, R.R.; Kufe, D.W. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995, 376, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Baskaran, R.; Aebi, S.; Fink, D.; Nebel, S.; Cenni, B.; Wang, J.Y.; Howell, S.B.; Christen, R.D. Differential induction of c-Jun NH2-terminal kinase and c-Abl kinase in DNA mismatch repair-proficient and -deficient cells exposed to cisplatin. Cancer Res. 1997, 57, 3253–3257. [Google Scholar] [PubMed]
- Shaulian, E.; Karin, M. Ap-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Kaina, B. Late activation of stress kinases (SAPK/JNK) by genotoxins requires the DNA repair proteins DNA-PKcs and CSB. Mol. Biol. Cell 2006, 17, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Baskaran, R.; Nebel, S.; Fink, D.; Howell, S.B.; Wang, J.Y.; Christen, R.D. Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. Br. J. Cancer 1999, 79, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Damrot, J.; Helbig, L.; Roos, W.P.; Barrantes, S.Q.; Kaina, B.; Fritz, G. DNA replication arrest in response to genotoxic stress provokes early activation of stress-activated protein kinases (SAPK/JNK). J. Mol. Biol. 2009, 385, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- De Feraudy, S.; Revet, I.; Bezrookove, V.; Feeney, L.; Cleaver, J.E. A minority of foci or pan-nuclear apoptotic staining of γH2AX in the s phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Sluss, H.K.; Davis, R.J. H2AX is a target of the JNK signaling pathway that is required for apoptotic DNA fragmentation. Mol. Cell 2006, 23, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Kopper, F.; Bierwirth, C.; Schon, M.; Kunze, M.; Elvers, I.; Kranz, D.; Saini, P.; Menon, M.B.; Walter, D.; Sorensen, C.S.; et al. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc. Natl. Acad. Sci. USA 2013, 110, 16856–16861. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.; Wang, X.; Linding, R.; Ong, S.E.; Weaver, D.; Carr, S.A.; et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated rna stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. P53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Gnad, R.; Aktories, K.; Kaina, B.; Fritz, G. Inhibition of protein isoprenylation impairs Rho-regulated early cellular response to genotoxic stress. Mol. Pharmacol. 2000, 58, 1389–1397. [Google Scholar] [PubMed]
- Rashid, M.; Tawara, S.; Fukumoto, Y.; Seto, M.; Yano, K.; Shimokawa, H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-coA reductase inhibitors. Circ. J. 2009, 73, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Pleiotropic effects of statins—Basic research and clinical perspectives. Circ. J. 2010, 74, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U. Effects of statins on endothelium and signaling mechanisms. Stroke 2004, 35, 2708–2711. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Kaina, B. Rho GTPases: Promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 2006, 6, 1–14. [Google Scholar] [PubMed]
- Fritz, G. Targeting the mevalonate pathway for improved anticancer therapy. Curr. Cancer Drug Targets 2009, 9, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Damrot, J.; Nubel, T.; Epe, B.; Roos, W.P.; Kaina, B.; Fritz, G. Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide. Br. J. Pharmacol. 2006, 149, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Huelsenbeck, J.; Fritz, G. Mevalonate pathway inhibitors affect anticancer drug-induced cell death and DNA damage response of human sarcoma cells. Cancer Lett. 2011, 304, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Helbig, L.; Damrot, J.; Hulsenbeck, J.; Koberle, B.; Brozovic, A.; Osmak, M.; Fiket, Z.; Kaina, B.; Fritz, G. Late activation of stress-activated protein kinases/c-jun N-terminal kinases triggered by cisplatin-induced DNA damage in repair-defective cells. J. Biol. Chem. 2011, 286, 12991–13001. [Google Scholar] [CrossRef] [PubMed]
- Devary, Y.; Gottlieb, R.A.; Smeal, T.; Karin, M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell 1992, 71, 1081–1091. [Google Scholar] [CrossRef]
- Onesto, C.; Shutes, A.; Picard, V.; Schweighoffer, F.; Der, C.J. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. Methods Enzymol. 2008, 439, 111–129. [Google Scholar] [PubMed]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, S.C.; Schorr, A.; Roos, W.P.; Huelsenbeck, J.; Henninger, C.; Kaina, B.; Fritz, G. Rac1 protein signaling is required for DNA damage response stimulated by topoisomerase II poisons. J. Biol. Chem. 2012, 287, 38590–38599. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.; Henninger, C.; Schad, A.; Lackner, K.J.; Kaina, B.; Fritz, G. Inhibition of Rac1 signaling by lovastatin protects against anthracycline-induced cardiac toxicity. Cell Death Dis. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wartlick, F.; Bopp, A.; Henninger, C.; Fritz, G. DNA damage response (DDR) induced by topoisomerase ii poisons requires nuclear function of the small GTPase Rac. Biochim. Biophys. Acta 2013, 1833, 3093–3103. [Google Scholar] [CrossRef] [PubMed]
- Riad, A.; Bien, S.; Westermann, D.; Becher, P.M.; Loya, K.; Landmesser, U.; Kroemer, H.K.; Schultheiss, H.P.; Tschope, C. Pretreatment with statin attenuates the cardiotoxicity of doxorubicin in mice. Cancer Res. 2009, 69, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Shiojima, I.; Ikeda, H.; Komuro, I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J. Mol. Cell Cardiol. 2009, 47, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc. Res. 2013, 97, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Henninger, C.; Huelsenbeck, S.; Wenzel, P.; Brand, M.; Huelsenbeck, J.; Schad, A.; Fritz, G. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol. Res. 2015, 91, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Bopp, A.; Wartlick, F.; Henninger, C.; Kaina, B.; Fritz, G. Rac1 modulates acute and subacute genotoxin-induced hepatic stress responses, fibrosis and liver aging. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bopp, A.; Wartlick, F.; Henninger, C.; Schwarz, M.; Kaina, B.; Fritz, G. Rac1 promotes diethylnitrosamine (den)-induced formation of liver tumors. Carcinogenesis 2015, 36, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Paajarvi, G.; Roudier, E.; Crisby, M.; Hogberg, J.; Stenius, U. HMG-coA reductase inhibitors, statins, induce phosphorylation of Mdm2 and attenuate the p53 response to DNA damage. FASEB J. 2005, 19, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, M.; Gorenne, I.; Mercer, J.; Figg, N.; Littlewood, T.; Bennett, M. Statins use a novel nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ. Res. 2008, 103, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.G.; Costanzo, A.; Yang, H.Q.; Melino, G.; Kaelin, W.G., Jr.; Levrero, M.; Wang, J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809. [Google Scholar]
- Yuan, Z.M.; Huang, Y.; Ishiko, T.; Kharbanda, S.; Weichselbaum, R.; Kufe, D. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 1437–1440. [Google Scholar] [CrossRef]
- Yuan, S.S.; Chang, H.L.; Lee, E.Y. Ionizing radiation-induced Rad51 nuclear focus formation is cell cycle-regulated and defective in both ATM−/− and c-Abl−/− cells. Mutat. Res. 2003, 525, 85–92. [Google Scholar] [CrossRef]
- Chen, G.; Yuan Shyng Shiou, F.; Liu, W.; Xu, Y.; Trujillo, K.; Song, B.; Cong, F.; Goff Stephen, P.; Wu, Y.; Arlinghaus, R.; et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 1999, 274, 12748–12752. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Yuan Zhi, M.; Weichselbaum, R.; Kufe, D. Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene 1998, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zeng, L.; Wang, J.; Chau, J.F.; Lai, K.P.; Jia, D.; Poonepalli, A.; Hande, M.P.; Liu, H.; He, G.; et al. A positive role for c-Abl in ATM and ATR activation in DNA damage response. Cell Death Differ. 2011, 18, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamaguchi, T.; Natsume, T.; Kufe, D.; Miki, Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell Biol. 2005, 7, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Zandy, N.L.; Playford, M.; Pendergast, A.M. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc. Natl. Acad. Sci. USA 2007, 104, 17686–17691. [Google Scholar] [CrossRef] [PubMed]
- Bassermann, F.; Jahn, T.; Miething, C.; Seipel, P.; Bai, R.Y.; Coutinho, S.; Tybulewicz, V.L.; Peschel, C.; Duyster, J. Association of Bcr-Abl with the proto-oncogene vav is implicated in activation of the Rac-1 pathway. J. Biol. Chem. 2002, 277, 12437–12445. [Google Scholar] [CrossRef] [PubMed]
- Sandrock, K.; Bielek, H.; Schradi, K.; Schmidt, G.; Klugbauer, N. The nuclear import of the small GTPase Rac1 is mediated by the direct interaction with karyopherin α2. Traffic 2010, 11, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, D.; Abidi, W.; Guardavaccaro, D.; Zhou, M.; Ahearn, I.; Pagano, M.; Philips, M.R. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 2008, 181, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Greer, P.M.; Cao, P.T.; Kolb, R.H.; Cowan, K.H. Rac1 GTPase plays an important role in γ-irradiation induced G2/M checkpoint activation. Breast Cancer Res. 2012. [Google Scholar] [CrossRef]
- Dion, V.; Shimada, K.; Gasser, S.M. Actin-related proteins in the nucleus: Life beyond chromatin remodelers. Curr. Opin. Cell Biol. 2010, 22, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hinde, E.; Yokomori, K.; Gaus, K.; Hahn, K.M.; Gratton, E. Fluctuation-based imaging of nuclear Rac1 activation by protein oligomerisation. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Hajas, G.; Bacsi, A.; Aguilera-Aguirre, L.; Hegde, M.L.; Tapas, K.H.; Sur, S.; Radak, Z.; Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase-1 links DNA repair to cellular signaling via the activation of the small GTPase Rac1. Free Radic. Biol. Med. 2013, 61, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motwani, M.; Li, D.Q.; Horvath, A.; Kumar, R. Identification of novel gene targets and functions of p21-activated kinase 1 during DNA damage by gene expression profiling. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.; Tanaka, M.; Otsuki, Y.; Fujiyama, T.; Kataoka, H.; Arai, H.; Hanai, H.; Sugimura, H. Involvement of α-PAK-interacting exchange factor in the PAK1-c-jun NH2-terminal kinase 1 activation and apoptosis induced by benzo[a]pyrene. Mol. Cell Biol 2001, 21, 6796–6807. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Carr, H.S.; Richter-Dahlfors, A.; Masucci, M.G.; Thelestam, M.; Frost, J.A.; Frisan, T. A bacterial cytotoxin identifies the RhoA exchange factor net1 as a key effector in the response to DNA damage. PLoS ONE 2008. [Google Scholar] [CrossRef]
- Guerra, L.; Guidi, R.; Slot, I.; Callegari, S.; Sompallae, R.; Pickett, C.L.; Astrom, S.; Eisele, F.; Wolf, D.; Sjogren, C.; et al. Bacterial genotoxin triggers fen1-dependent RhoA activation, cytoskeleton remodeling and cell survival. J. Cell Sci. 2011, 124, 2735–2742. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases rock and lim-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Carr, H.S.; Morris, C.A.; Menon, S.; Song, E.H.; Frost, J.A. Rac1 controls the subcellular localization of the Rho guanine nucleotide exchange factor net1a to regulate focal adhesion formation and cell spreading. Mol. Cell Biol. 2013, 33, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear f-actin formation and reorganization upon cell spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef]
- Navarro-Lerida, I.; Pellinen, T.; Sanchez, S.A.; Guadamillas, M.C.; Wang, Y.; Mirtti, T.; Calvo, E.; del Pozo, M.A. Rac1 nucleocytoplasmic shuttling drives nuclear shape changes and tumor invasion. Dev. Cell 2015, 32, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yuan, Y.; Maestas, A.; Shen, Z. Recovery from DNA damage-induced G2 arrest requires actin-binding protein filamin-A/actin-binding protein 280. J. Biol. Chem. 2004, 279, 6098–6105. [Google Scholar] [CrossRef] [PubMed]
- Andrin, C.; McDonald, D.; Attwood, K.M.; Rodrigue, A.; Ghosh, S.; Mirzayans, R.; Masson, J.Y.; Dellaire, G.; Hendzel, M.J. A requirement for polymerized actin in DNA double-strand break repair. Nucleus 2012, 3, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mazzanti, M.; Mistrik, M.; Kosar, M.; Beznoussenko, G.V.; Mironov, A.A.; Garre, M.; Parazzoli, D.; Shivashankar, G.V.; Scita, G.; et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 2014, 158, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Sethi, R.; Doanes, A.M.; Johnson, T.M.; Pracyk, J.B.; Kirby, M.; Irani, K.; Goldschmidt-Clermont, P.J.; Finkel, T. Rac1 is required for cell proliferation and G2/M progression. Biochem. J. 1997, 326, 17–20. [Google Scholar] [CrossRef]
- Zuo, Y.; Oh, W.; Frost, J.A. Controlling the switches: Rho GTPase regulation during animal cell mitosis. Cell Signal 2014, 26, 2998–3006. [Google Scholar] [CrossRef]
- Chircop, M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases 2014. [Google Scholar] [CrossRef]
- Yoshizaki, H.; Ohba, Y.; Kurokawa, K.; Itoh, R.E.; Nakamura, T.; Mochizuki, N.; Nagashima, K.; Matsuda, M. Activity of Rho-family GTPases during cell division as visualized with fret-based probes. J. Cell Biol. 2003, 162, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Fromigue, O.; Hamidouche, Z.; Marie, P.J. Statin-induced inhibition of 3-hydroxy-3-methyl glutaryl coenzyme a reductase sensitizes human osteosarcoma cells to anticancer drugs. J. Pharmacol. Exp. Ther. 2008, 325, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Feleszko, W.; Mlynarczuk, I.; Balkowiec-Iskra, E.Z.; Czajka, A.; Switaj, T.; Stoklosa, T.; Giermasz, A.; Jakobisiak, M. Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clin. Cancer Res. 2000, 6, 2044–2052. [Google Scholar] [PubMed]
- Bourgier, C.; Haydont, V.; Milliat, F.; Francois, A.; Holler, V.; Lasser, P.; Bourhis, J.; Mathe, D.; Vozenin-Brotons, M.C. Inhibition of Rho kinase modulates radiation induced fibrogenic phenotype in intestinal smooth muscle cells through alteration of the cytoskeleton and connective tissue growth factor expression. Gut 2005, 54, 336–343. [Google Scholar] [PubMed]
- Haydont, V.; Bourgier, C.; Vozenin-Brotons, M.C. Rho/rock pathway as a molecular target for modulation of intestinal radiation-induced toxicity. Br. J. Radiol. 2007, 80, 32–40. [Google Scholar] [CrossRef]
- Iseri, S.; Ercan, F.; Gedik, N.; Yuksel, M.; Alican, I. Simvastatin attenuates cisplatin-induced kidney and liver damage in rats. Toxicology 2007, 230, 256–264. [Google Scholar]
- An, Y.; Xin, H.; Yan, W.; Zhou, X. Amelioration of cisplatin-induced nephrotoxicity by pravastatin in mice. Exp. Toxicol. Pathol. 2011, 63, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, W.; Wang, J.; Xie, L.; Li, T.; He, Y.; Deng, Y.; Peng, Q.; Li, S.; Qin, X. Association between statin use and colorectal cancer risk: A meta-analysis of 42 studies. Cancer Causes Control 2014, 25, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, T.; Fukaura, Y.; Terada, K.; Umezawa, A.; Tanida, N.; Yazawa, K.; Ishikawa, C. Prevention of 1,2-dimethylhydrazine-induced colon tumorigenesis by HMG-coA reductase inhibitors, pravastatin and simvastatin, in icr mice. Carcinogenesis 1994, 15, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. Cd28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 2003, 111, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The biology of the cytolethal distending toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Oswald, E.; Sugai, M. Cytolethal distending toxin: A bacterial bullet targeted to nucleus. J. Biochem. 2004, 136, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Bezine, E.; Vignard, J.; Mirey, G. The cytolethal distending toxin effects on mammalian cells: A DNA damage perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dorsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Lara-Tejero, M.; Galan, J.E. Cytolethal distending toxin: Limited damage as a strategy to modulate cellular functions. Trends Microbiol. 2002, 10, 147–152. [Google Scholar] [CrossRef]
- Sugihara, K.; Nakatsuji, N.; Nakamura, K.; Nakao, K.; Hashimoto, R.; Otani, H.; Sakagami, H.; Kondo, H.; Nozawa, S.; Aiba, A.; et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 1998, 17, 3427–3433. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritz, G.; Henninger, C. Rho GTPases: Novel Players in the Regulation of the DNA Damage Response? Biomolecules 2015, 5, 2417-2434. https://doi.org/10.3390/biom5042417
Fritz G, Henninger C. Rho GTPases: Novel Players in the Regulation of the DNA Damage Response? Biomolecules. 2015; 5(4):2417-2434. https://doi.org/10.3390/biom5042417
Chicago/Turabian StyleFritz, Gerhard, and Christian Henninger. 2015. "Rho GTPases: Novel Players in the Regulation of the DNA Damage Response?" Biomolecules 5, no. 4: 2417-2434. https://doi.org/10.3390/biom5042417