
Growth Factor Dependent Regulation of Centrosome Function and Genomic Instability by HuR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Centrosome Amplification Evoked by Growth Factor Stimulation

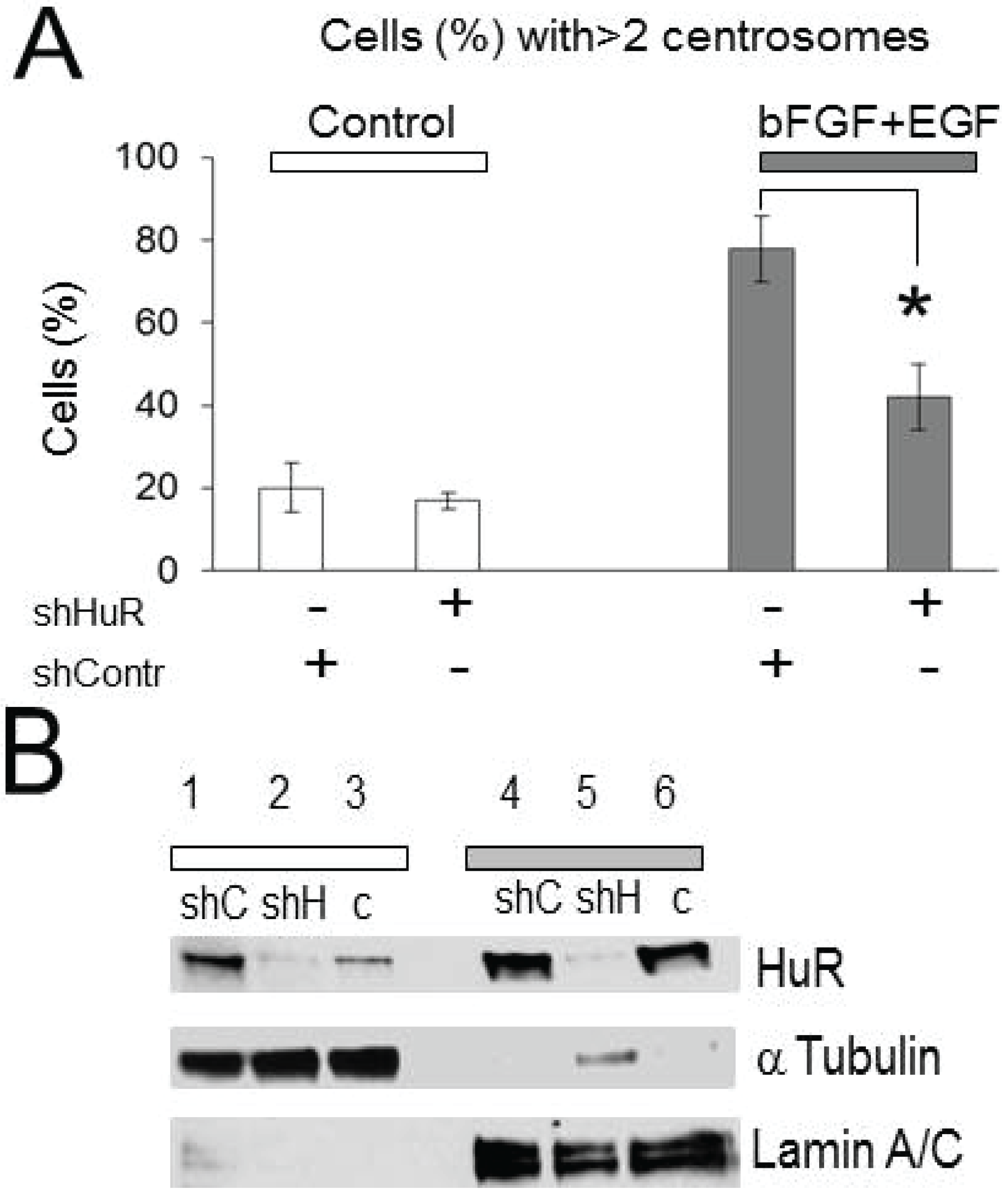
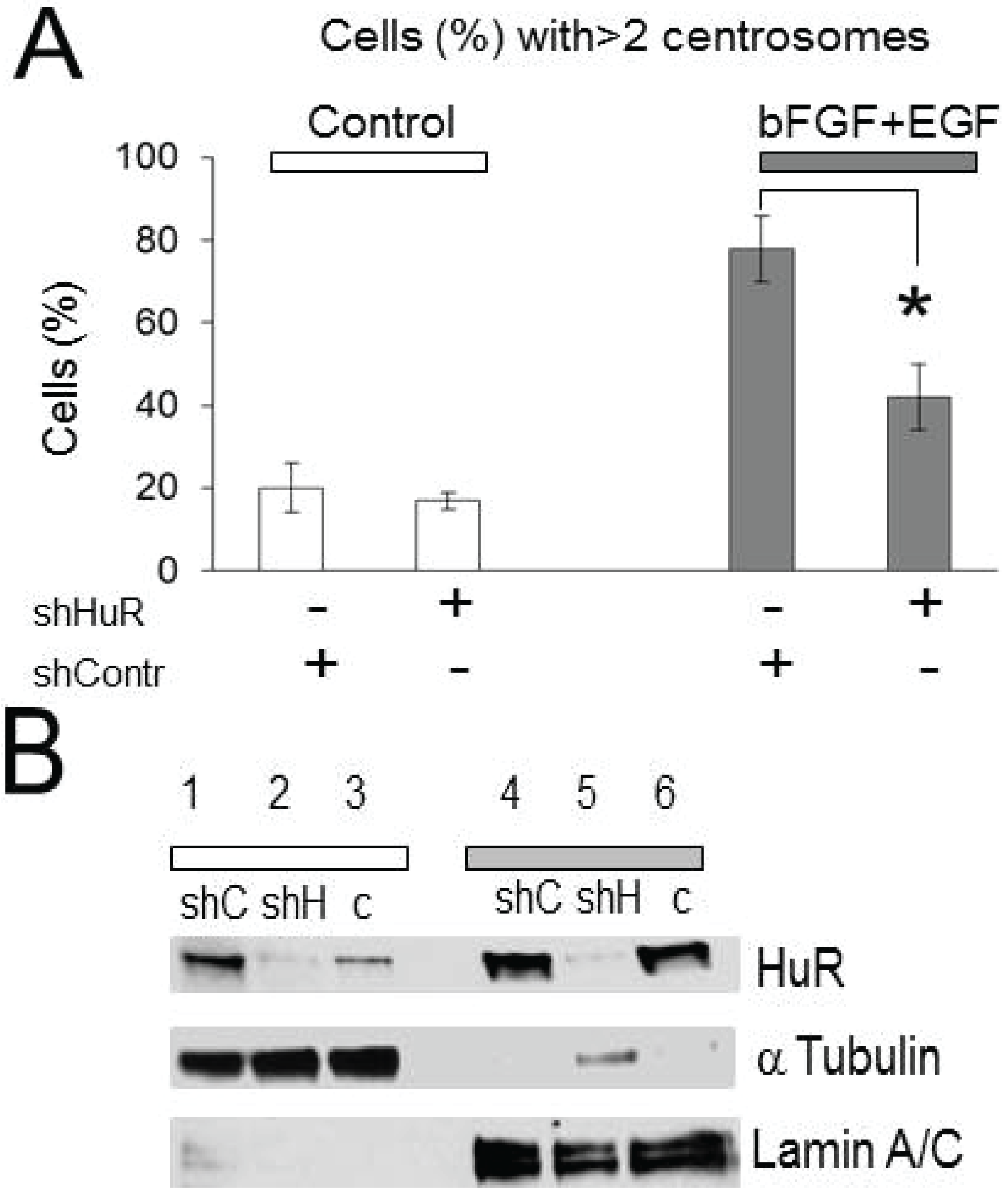
2.2. Centrosomes Amplification Induced by Growth Factor Stimulation Is HuR-Dependent
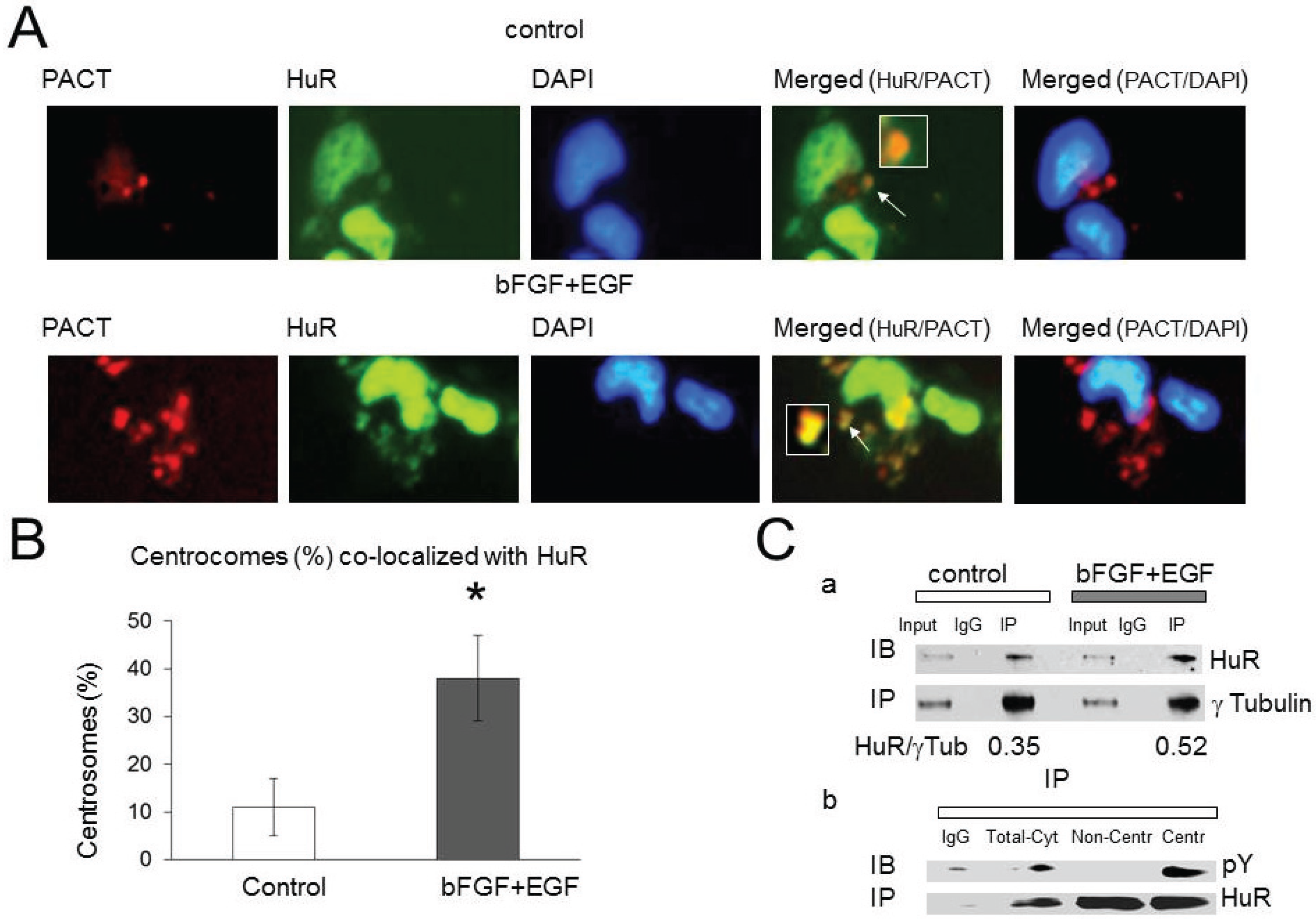
2.3. HuR Association with Pericentriolar Matrix (PCM) Is Enhanced by Growth Factor Stimulation
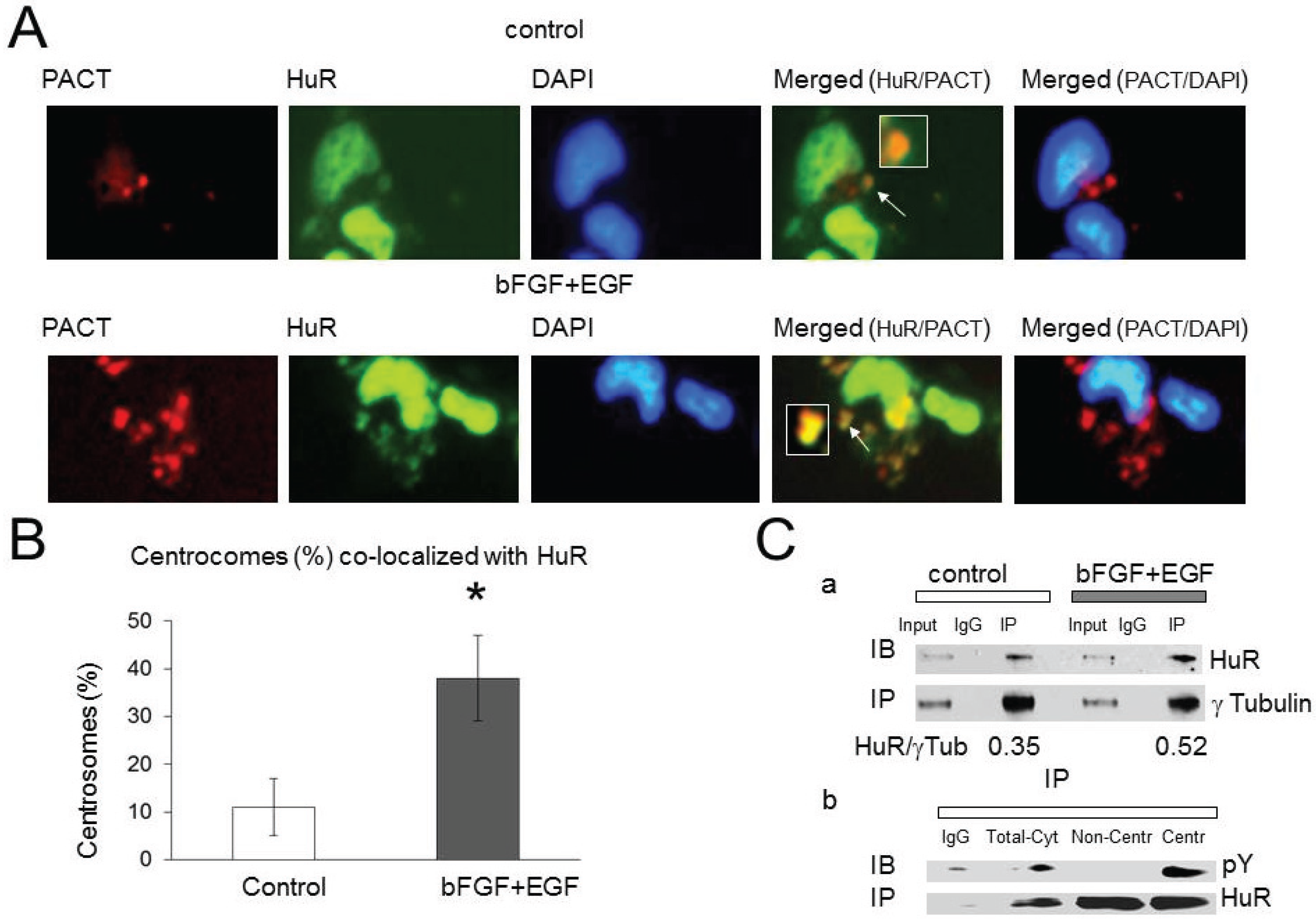
2.4. Abl-1 and SRC Kinases Are Possible Regulators of HuR in the PCM
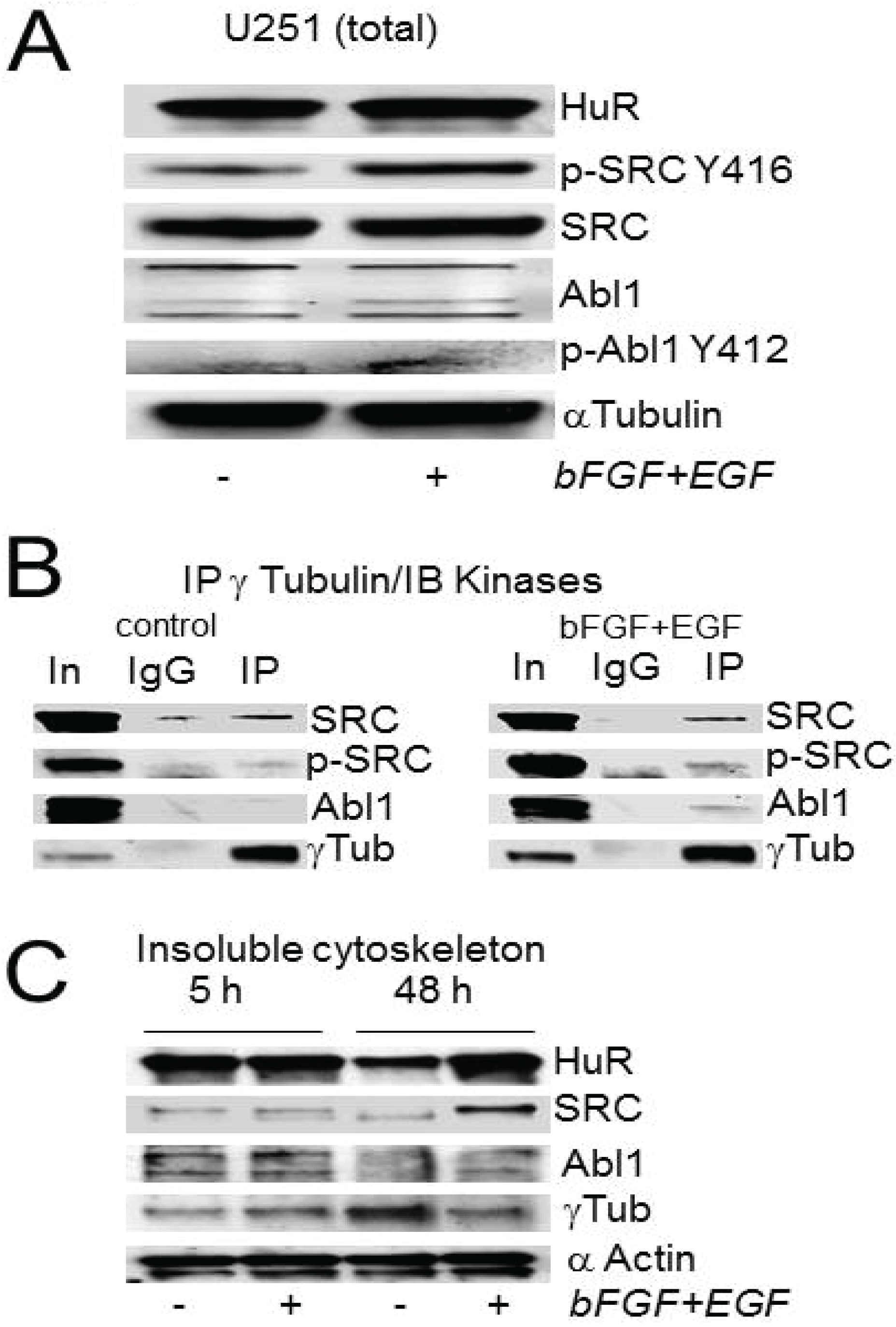
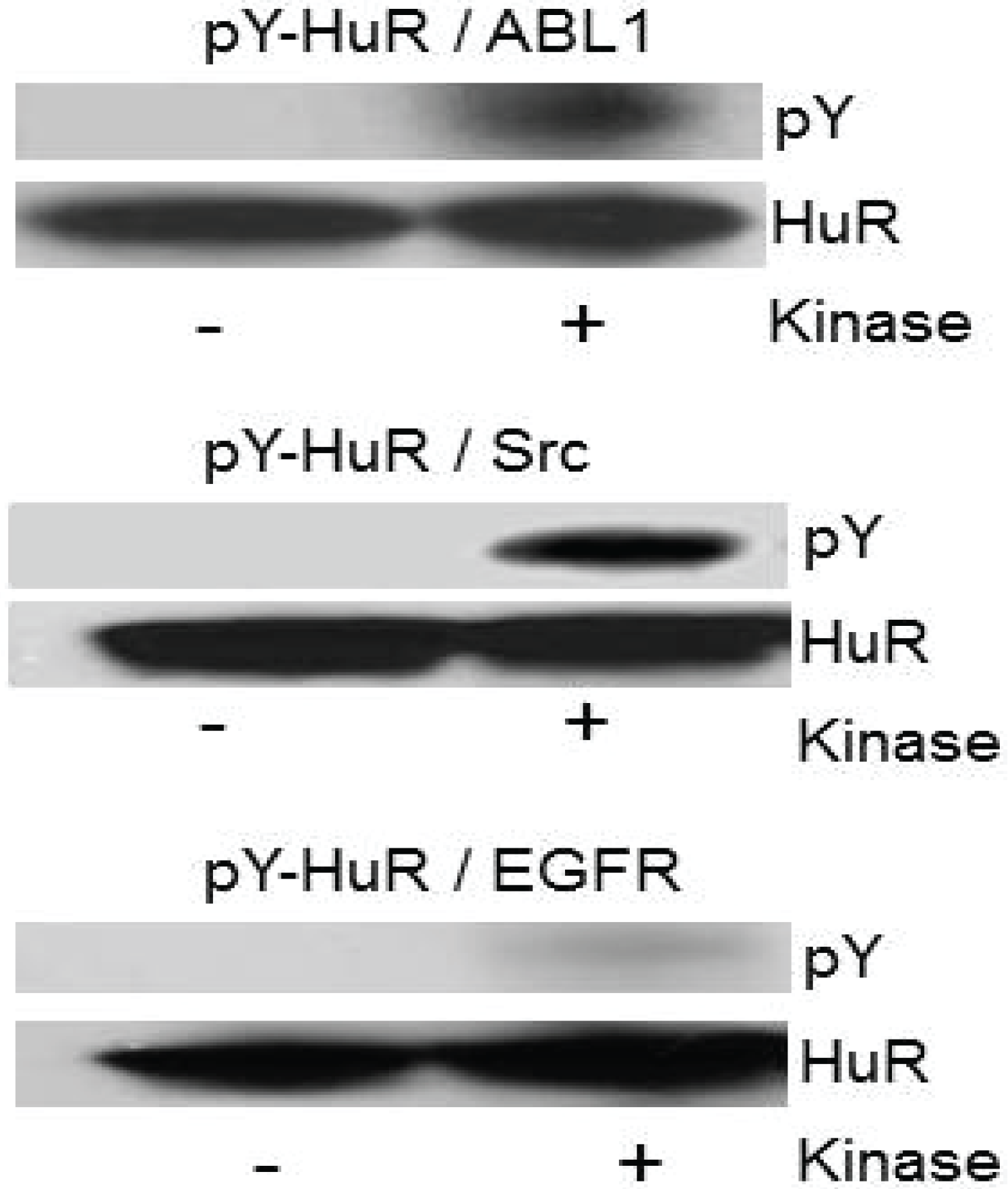
2.5. Recombinant HuR Is Phosphorylated by Abl-1 and SRC Kinases


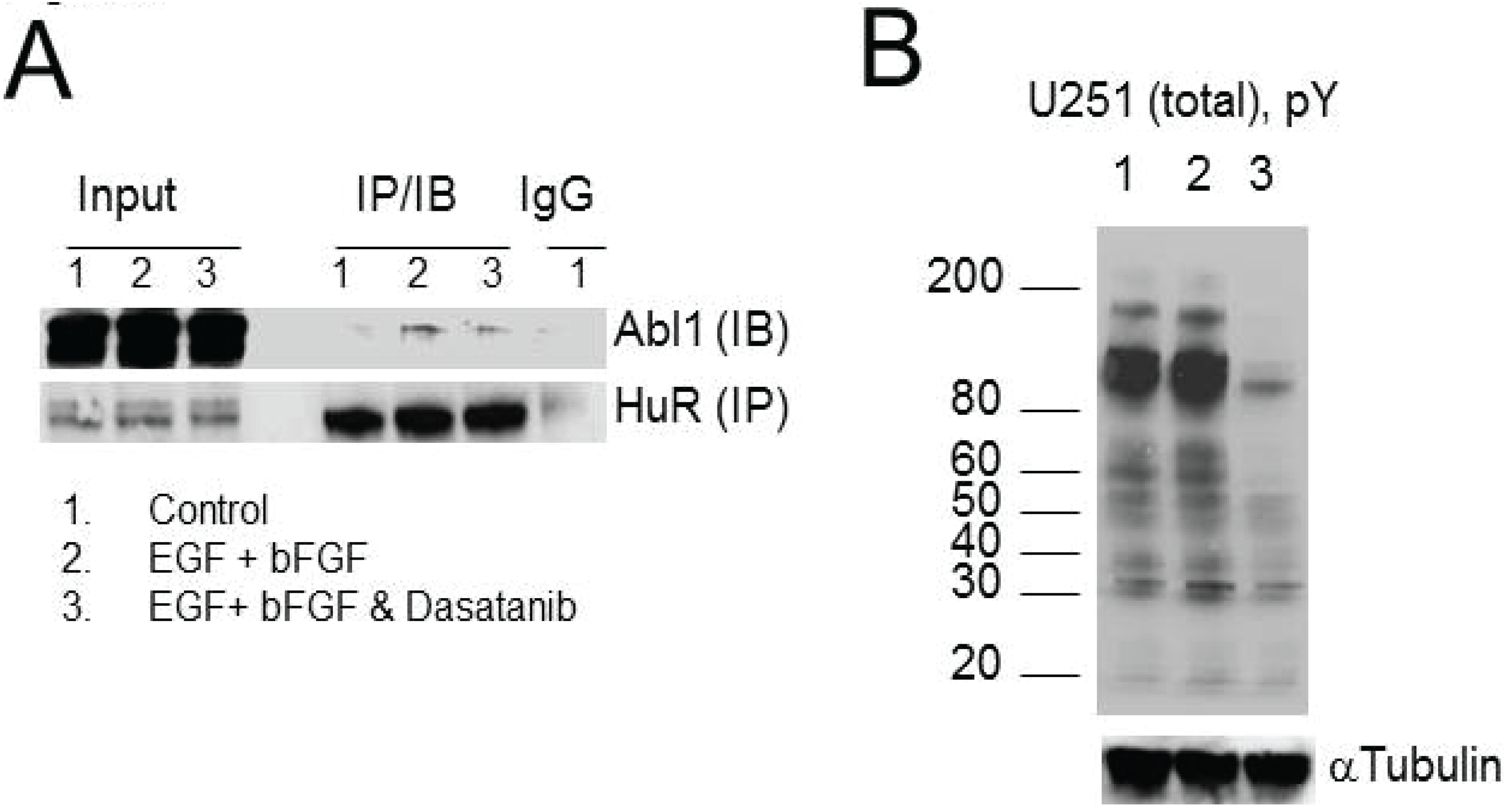
2.6. HuR Interaction with Abl-1 Kinase Is Growth Factor Dependent
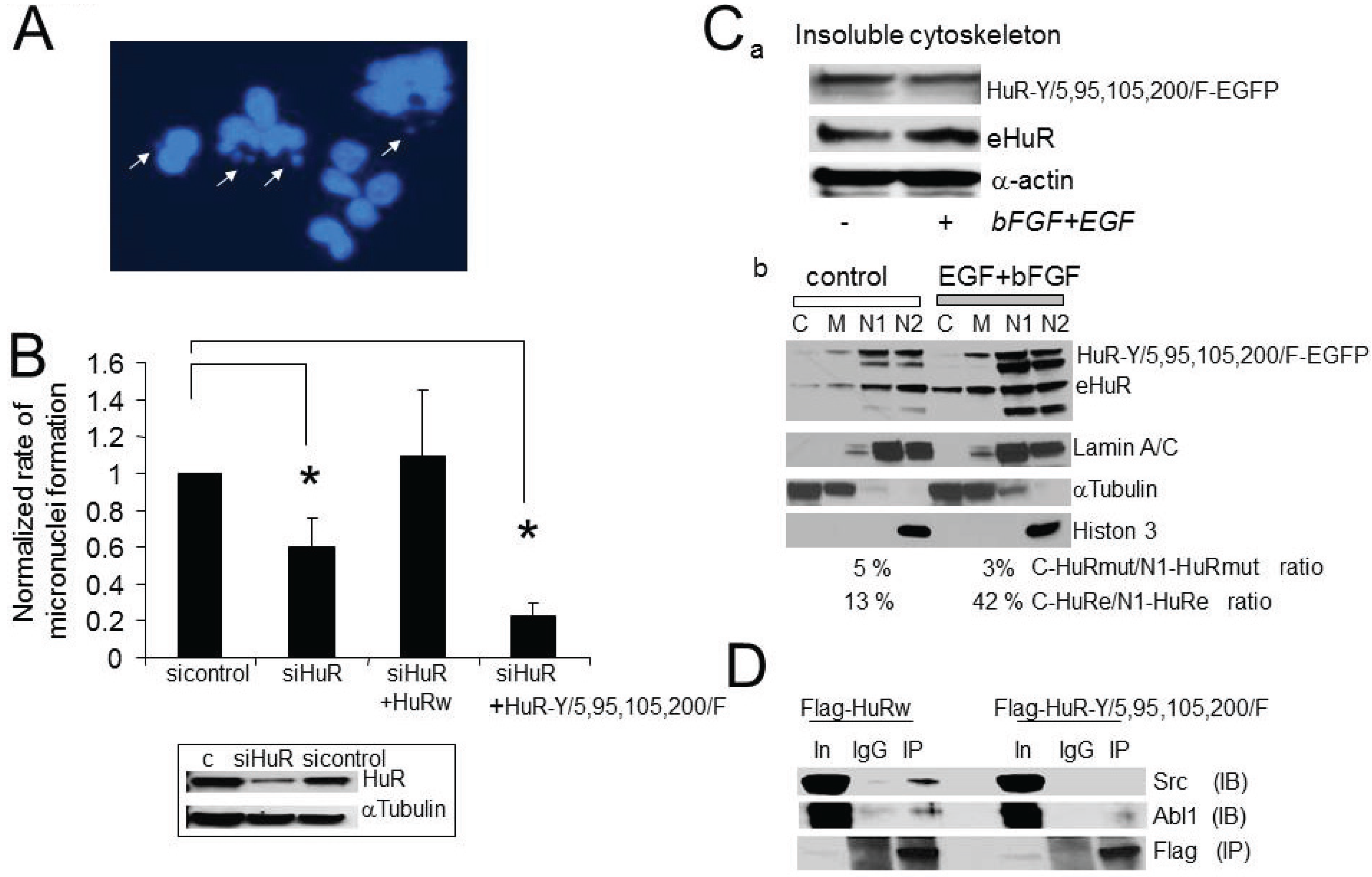
2.7. The Consequences of Aberrant Growth Factor Dependent HuR Phosphorylation at Tyrosine Residues
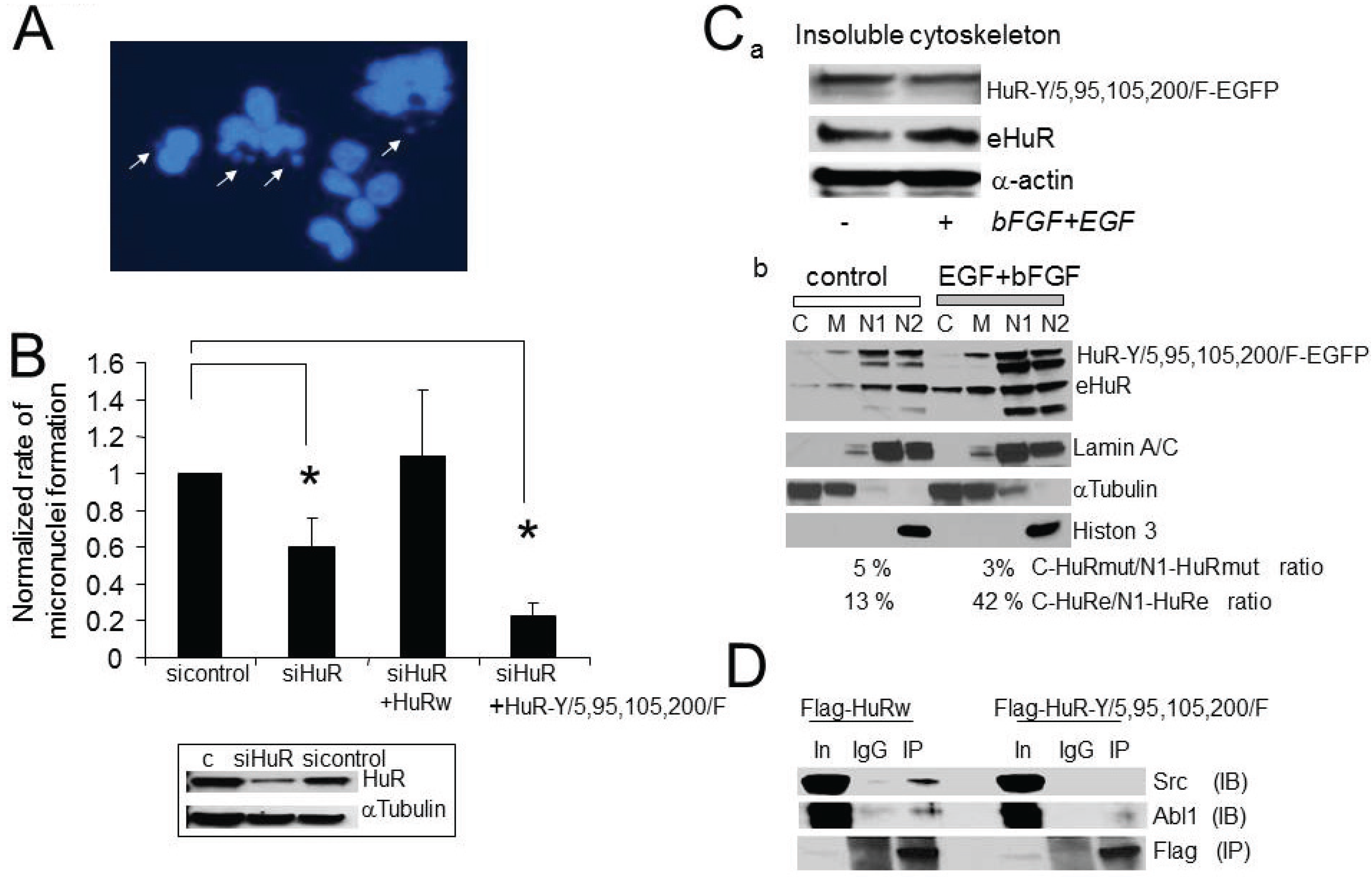
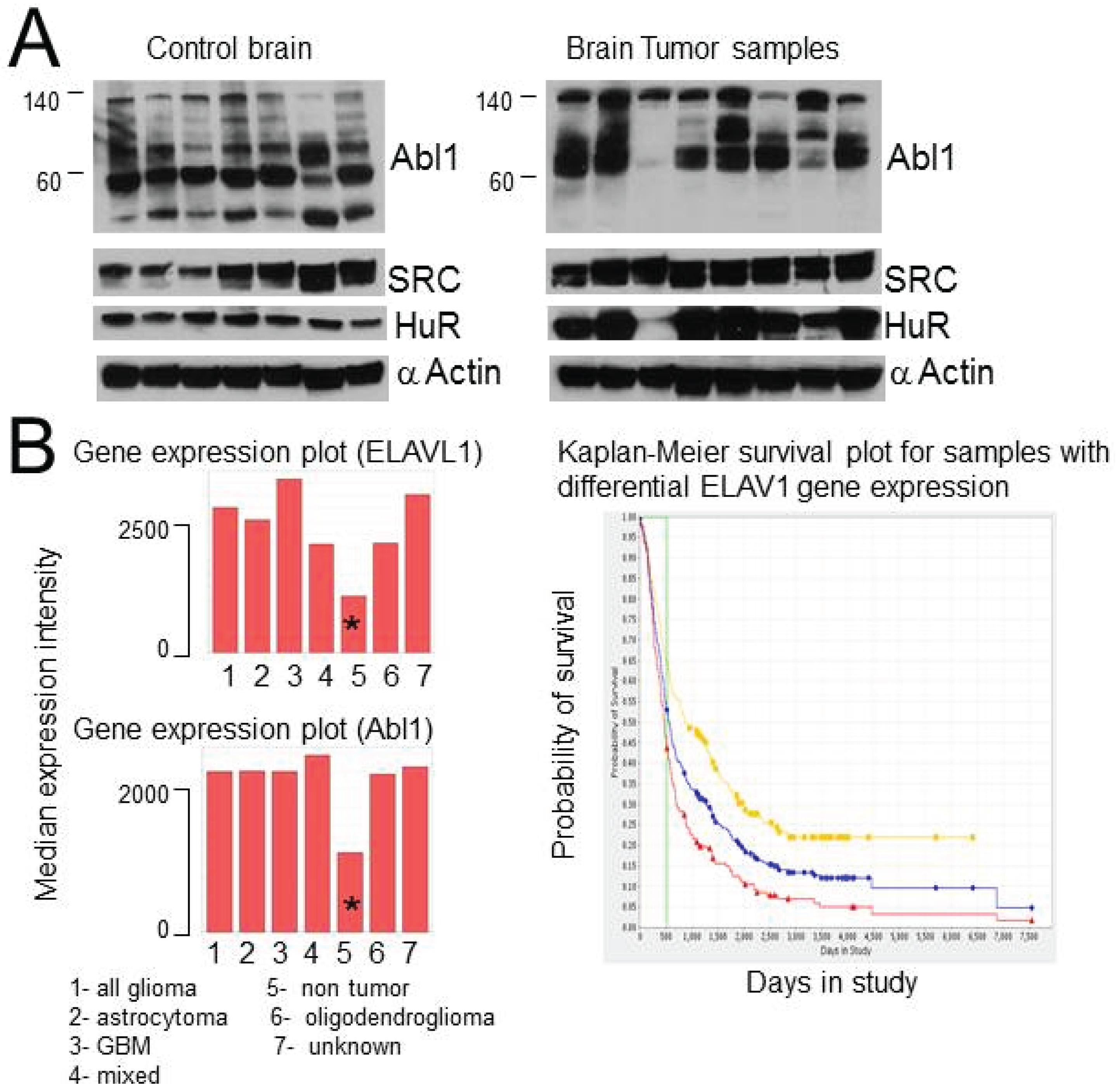
2.8. HuR, Abl-1 and SRC Expression in Tumor Samples and Glioma Cell Lines

3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urbani, L.; Stearns, T. The centrosome. Curr. Biol. 1999, 9, 315–317. [Google Scholar] [CrossRef]
- Dawe, H.R.; Farr, H.; Gull, K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J. Cell Sci. 2007, 120, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Cammer, M.; Lehman, J.; Nielsen, S.K.; Guerra, C.F.; Veland, I.R.; Stock, C.; Hoffmann, E.K.; Yoder, B.K.; Schwab, A.; et al. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cell. Physiol. Biochem. 2010, 25, 279–292. [Google Scholar]
- Badano, J.L.; Teslovich, T.M.; Katsanis, N. The centrosome in human genetic disease. Nat. Rev. Genet. 2005, 6, 194–205. [Google Scholar] [CrossRef] [PubMed]
- D’Assoro, A.B.; Lingle, W.L.; Salisbury, J.L. Centrosome amplification and the development of cancer. Oncogene 2002, 21, 6146–6153. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Colello, D.; Reverte, C.G.; Ward, R.; Jones, C.W.; Magidson, V.; Khodjakov, A.; LaFlamme, S.E. Androgen and Src signaling regulate centrosome activity. J. Cell Sci. 2010, 123, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 2007, 7, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Tseng, J.T.; Lee, Y.C.; Xia, W.Y.; Wang, Y.N.; Wu, M.L.; Chuang, Y.H.; Lai, C.H.; Chang, W.C. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008, 36, 4337–4351. [Google Scholar] [CrossRef] [PubMed]
- Kabil, A.; Silva, E.; Kortenkamp, A. Estrogens and genomic instability in human breast cancer cells—Involvement of Src/Raf/Erk signaling in micronucleus formation by estrogenic chemicals. Carcinogenesis 2008, 29, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dutra, A.; Pak, E.; Labrie, J.E., 3rd; Gerstein, R.M.; Pandolfi, P.P.; Recht, L.D.; Ross, A.H. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro Oncol. 2009, 11, 9–21. [Google Scholar]
- Li, J.J.; Weroha, S.J.; Lingle, W.L.; Papa, D.; Salisbury, J.L.; Li, S.A. Estrogen mediates Aurora-A overexpression, centrosome amplification, chromosomal instability, and breast cancer in female ACI rats. Proc. Natl. Acad. Sci. USA 2004, 101, 18123–18128. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, P.P.; Wang, S.Y.; Hulderman, T.; Luster, M.I. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic—Role in carcinogenesis. J. Biol. Chem. 2002, 277, 2945–2950. [Google Scholar] [CrossRef] [PubMed]
- Yih, L.H.; Tseng, Y.Y.; Wu, Y.C.; Lee, T.C. Induction of centrosome amplification during arsenite-induced mitotic arrest in CGL-2 cells. Cancer Res. 2006, 66, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Filippova, N.; Yang, X.; King, P.; Nabors, L.B. Phosphoregulation of the RNA-binding protein Hu antigen R (HuR) by Cdk5 affects centrosome function. J. Biol. Chem. 2012, 287, 32277–32287. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Gorospe, M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 2010, 1, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Lal, A.; Kim, H.H.; Gorospe, M. Posttranscriptional orchestration of an anti-apoptotic program by HuR. Cell Cycle 2007, 6, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Filippova, N.; Yang, X.; Wang, Y.; Gillespie, G.Y.; Langford, C.; King, P.H.; Wheeler, C.; Nabors, L.B. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer Res. 2011, 9, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 2008, 65, 3168–3181. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Hagner, P.R.; Corl, S.; Srikantan, S.; Wood, W.H.; Becker, K.G.; Gorospe, M.; Keene, J.D.; Levenson, A.S.; Gartenhaus, R.B. Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene 2008, 27, 6151–6163. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Cadenas, R.; Abascal, F.; Diez-Perez, J.; Carazo, J.M.; Pascual Montano, A. CentrosomeDB: A human centrosomal proteins database. Nucleic Acids Res. 2009, 37, 175–180. [Google Scholar] [CrossRef]
- Uren, P.J.; Burns, S.C.; Ruan, J.H.; Singh, K.K.; Smith, A.D.; Penalva, L.O.F. Genomic analyses of the RNA-binding protein Hu antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. J. Biol. Chem. 2011, 286, 37063–37066. [Google Scholar] [CrossRef] [PubMed]
- Gergely, F.; Basto, R. Multiple centrosomes: Together they stand, divided they fall. Genes Dev. 2008, 22, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Fabarius, A.; Giehl, M.; Rebacz, B.; Kramer, A.; Frank, O.; Haferlach, C.; Duesberg, P.; Hehlmann, R.; Seifarth, W.; Hochhaus, A. Centrosome aberrations and G1 phase arrest after in vitro and in vivo treatment with the SRC/ABL inhibitor dasatinib. Haematologica 2008, 93, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Bergalet, J.; Fawal, M.; Lopez, C.; Desjobert, C.; Lamant, L.; Delsol, G.; Morello, D.; Espinos, E. HuR-mediated control of C/EBPbeta mRNA stability and translation in ALK-positive anaplastic large cell lymphomas. Mol. Cancer Res. 2011, 9, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Fawal, M.; Armstrong, F.; Ollier, S.; Dupont, H.; Touriol, C.; Monsarrat, B.; Delsol, G.; Payrastre, B.; Morello, D. A “liaison dangereuse” between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood 2006, 108, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Fawal, M.; Espinos, E.; Jean-Jean, O.; Morello, D. Looking for the functions of RNA granules in ALK-transformed cells. Bioarchitecture 2011, 1, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Giehl, M.; Fabarius, A.; Frank, O.; Hochhaus, A.; Hafner, M.; Hehlmann, R.; Seifarth, W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005, 19, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Arlinghaus, R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene 2008, 27, 4385–4391. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Matsui, Y.; Takeda, Y.; Okamoto, M.; Abe, K.; Fukumoto, Y.; Yamaguchi, N. c-Src but not Fyn promotes proper spindle orientation in early prometaphase. J. Biol. Chem. 2012, 287, 24905–24915. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Gordon, M.Y. Abnormal centrosome-centriole cycle in chronic myeloid leukaemia? Br. J. Haematol. 2009, 146, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar]
- Perez-Garcia, A.; Carrion-Navarro, J.; Bosch-Fortea, M.; Lazaro-Ibanez, E.; Prat-Acin, R.; Ayuso-Sacido, A. Genomic instability of surgical sample and cancer-initiating cell lines from human glioblastoma. Front. Biosci. 2012, 17, 1469–1479. [Google Scholar] [CrossRef]
- Merlo, A. Genes and pathways driving glioblastomas in humans and murine disease models. Neurosurg. Rev. 2003, 26, 145–158. [Google Scholar] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Silanes, I.; Zhan, M.; Lal, A.; Yang, X.; Gorospe, M. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. USA 2004, 101, 2987–2992. [Google Scholar] [CrossRef] [PubMed]
- Thery, M.; Racine, V.; Pepin, A.; Piel, M.; Chen, Y.; Sibarita, J.B.; Bornens, M. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 2005, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Osherov, N.; Levitzki, A. Epidermal-growth-factor-dependent activation of the Src-family kinases. Eur. J. Biochem. 1994, 225, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Plattner, R.; Kadlec, L.; DeMali, K.A.; Kazlauskas, A.; Pendergast, A.M. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999, 13, 2400–2411. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, A.; Benistant, C.; Roche, S. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol. Cell 2008, 100, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Abdelmohsen, K.; Gorospe, M. Regulation of HuR by DNA Damage Response Kinases. J. Nucleic Acids 2010. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, C.J.; Chen, G.Y.; Yao, S.Q. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J. Am. Chem. Soc. 2012, 134, 3001–3014. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Bono, P.; Narko, K.; Chang, S.H.; Lundin, J.; Joensuu, H.; Furneaux, H.; Hla, T.; Haglund, C.; Ristimaki, A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005, 65, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Lim, S.J.; Kim, Y.W. Expression of HuR, COX-2, and survivin in lung cancers; cytoplasmic HuR stabilizes cyclooxygenase-2 in squamous cell carcinomas. Mod. Pathol. 2011, 24, 1336–1347. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Xu, X.; Bansbach, C.E.; Glick, G.G.; Zhao, R.X.; Ye, F.; Sirbu, B.M.; Titus, L.C.; Shyr, Y.; Cortez, D. Functional genomic screens identify CINP as a genome maintenance protein. Proc. Natl. Acad. Sci. USA 2009, 106, 19304–19309. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar]
- De Nadal, E.; Ammerer, G.; Posas, F. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011, 12, 833–845. [Google Scholar] [PubMed]
- Zhou, B.B.S.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Tebaldi, T.; Viero, G.; Sparta, A.M.; Quattrone, A.; Provenzani, A. Downregulation of HuR as a new mechanism of doxorubicin resistance in breast cancer cells. Mol. Cancer 2012. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippova, N.; Yang, X.; Nabors, L.B. Growth Factor Dependent Regulation of Centrosome Function and Genomic Instability by HuR. Biomolecules 2015, 5, 263-281. https://doi.org/10.3390/biom5010263
Filippova N, Yang X, Nabors LB. Growth Factor Dependent Regulation of Centrosome Function and Genomic Instability by HuR. Biomolecules. 2015; 5(1):263-281. https://doi.org/10.3390/biom5010263
Chicago/Turabian StyleFilippova, Natalia, Xiuhua Yang, and Louis Burt Nabors. 2015. "Growth Factor Dependent Regulation of Centrosome Function and Genomic Instability by HuR" Biomolecules 5, no. 1: 263-281. https://doi.org/10.3390/biom5010263