Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding

Abstract
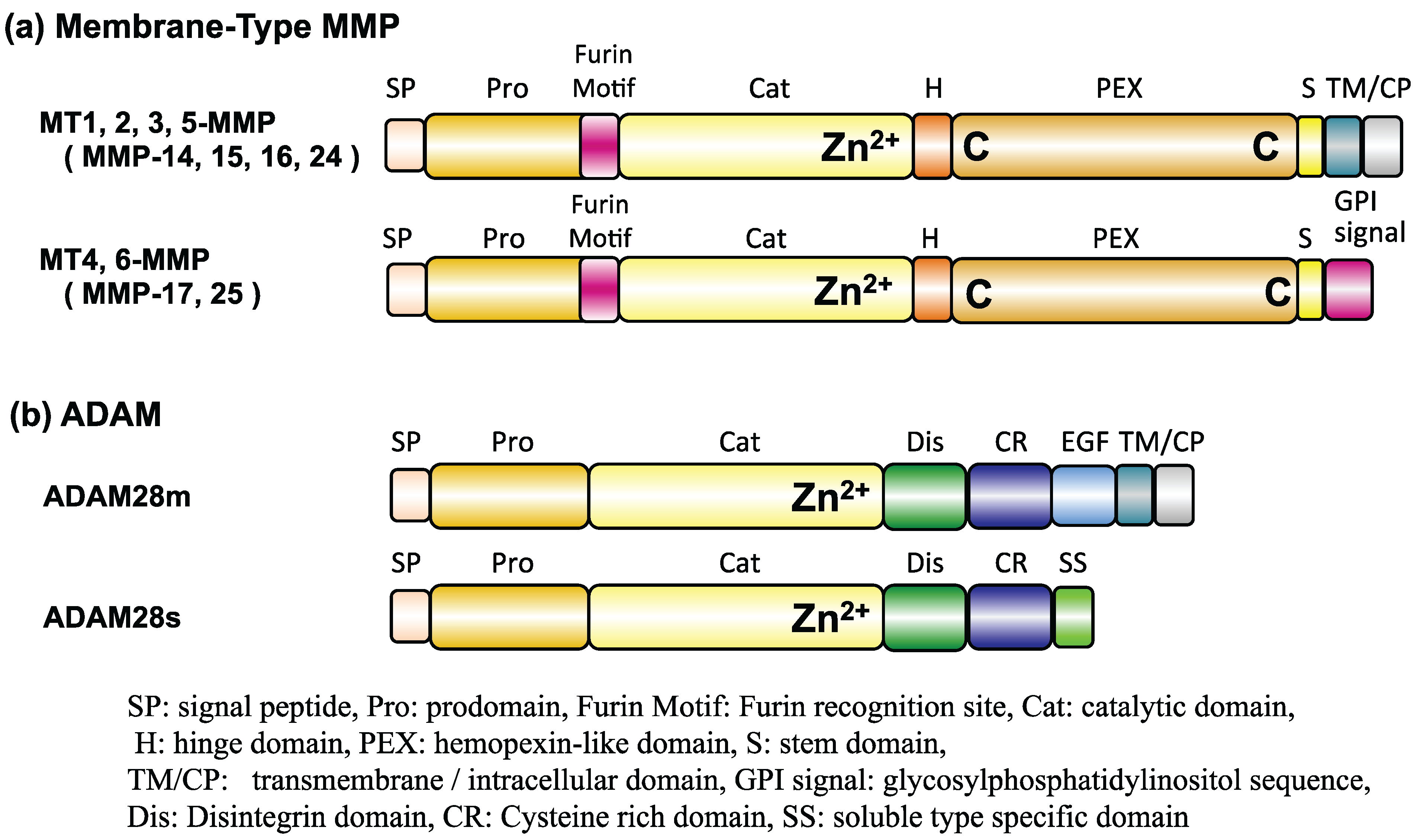
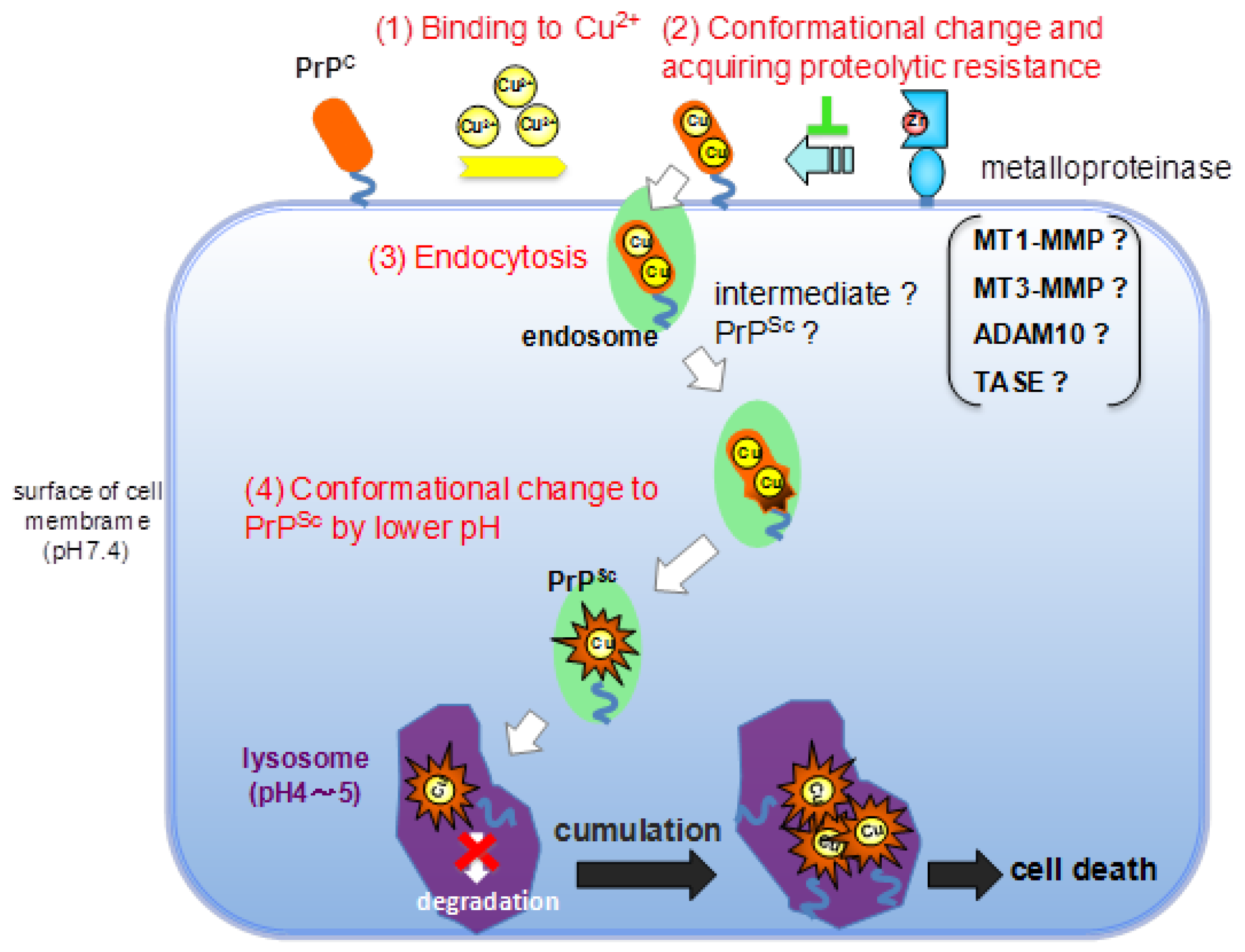
:1. Introduction
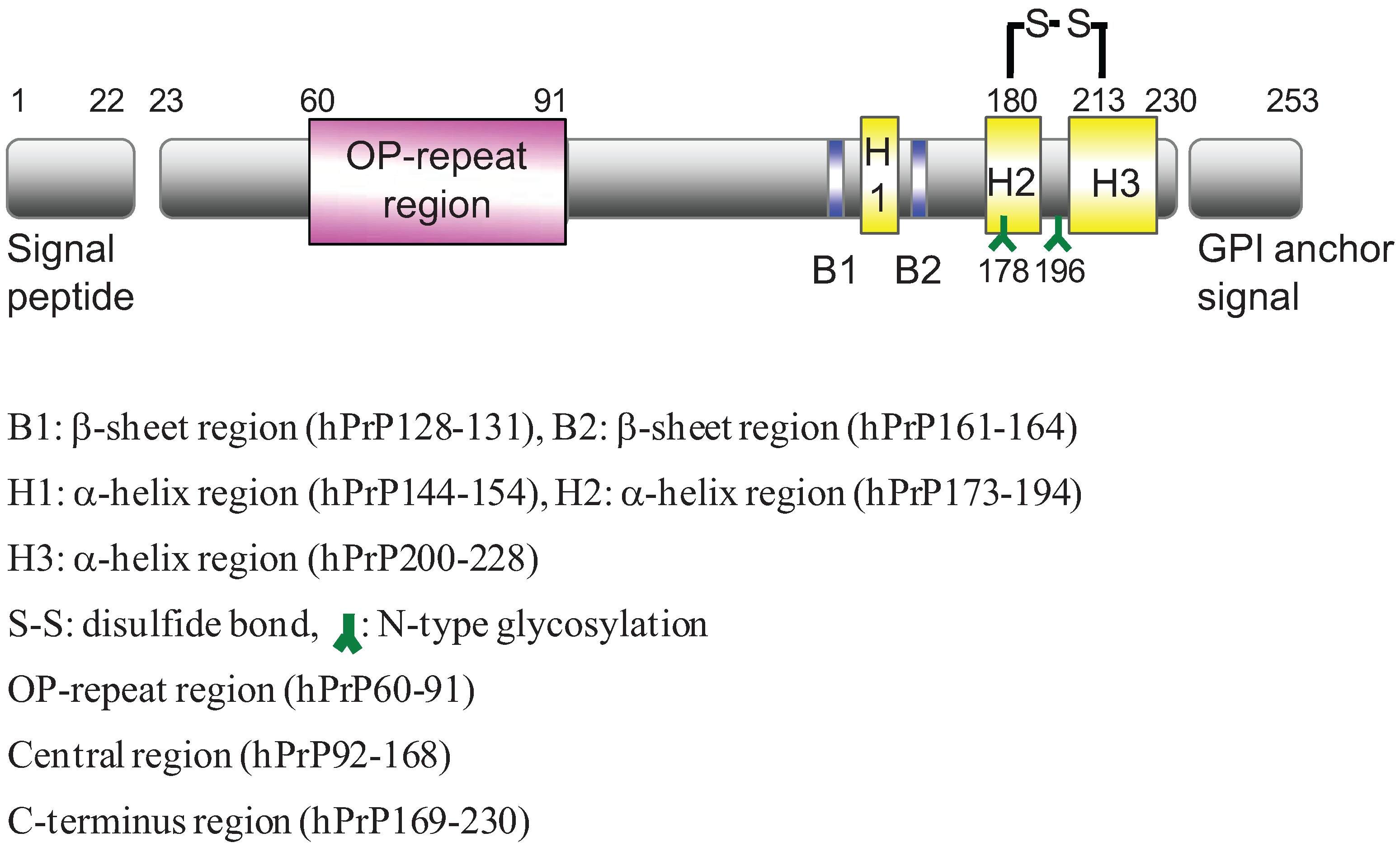
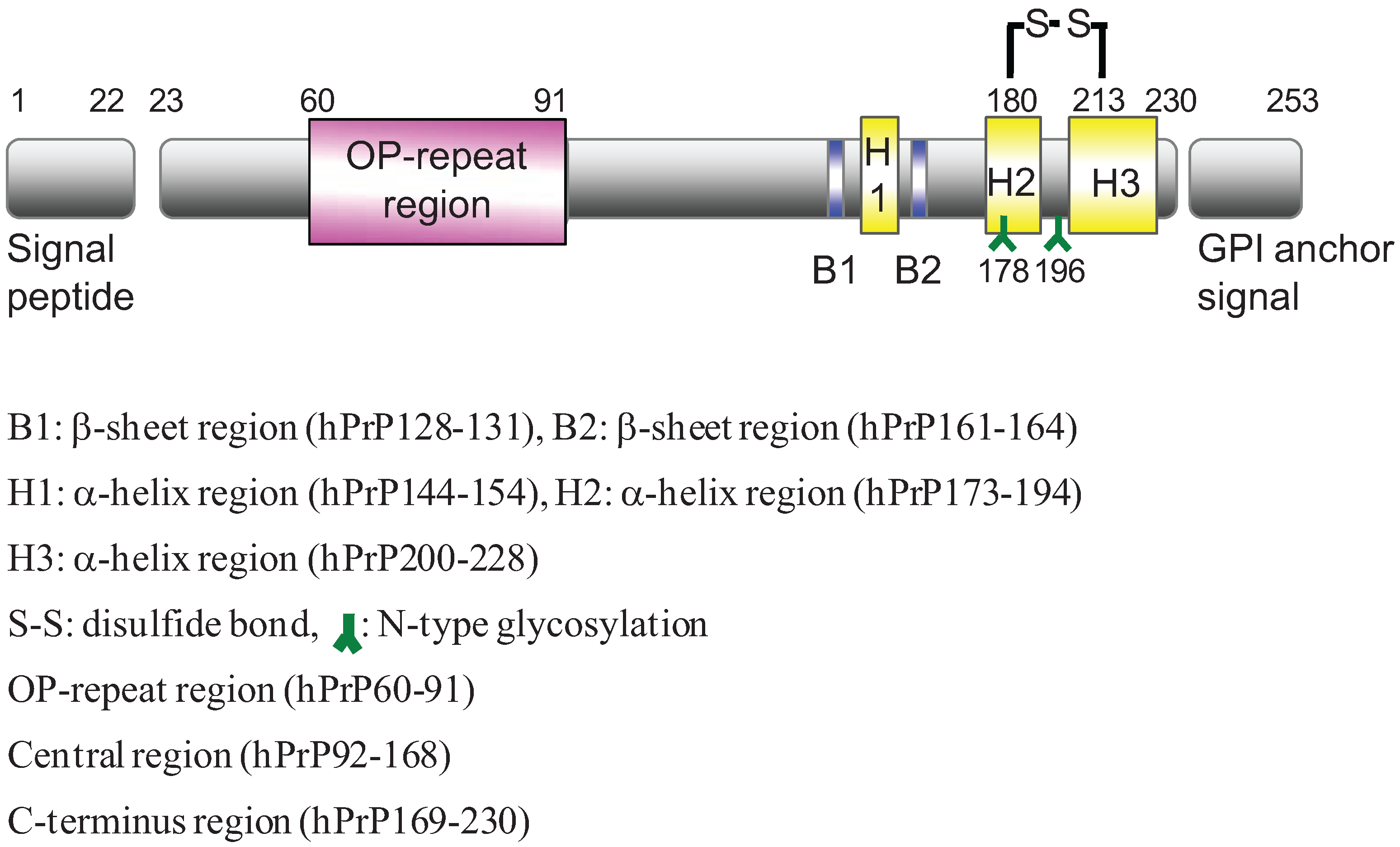
2. Results and Discussion
2.1. Preparation of Synthetic Fragment Peptides
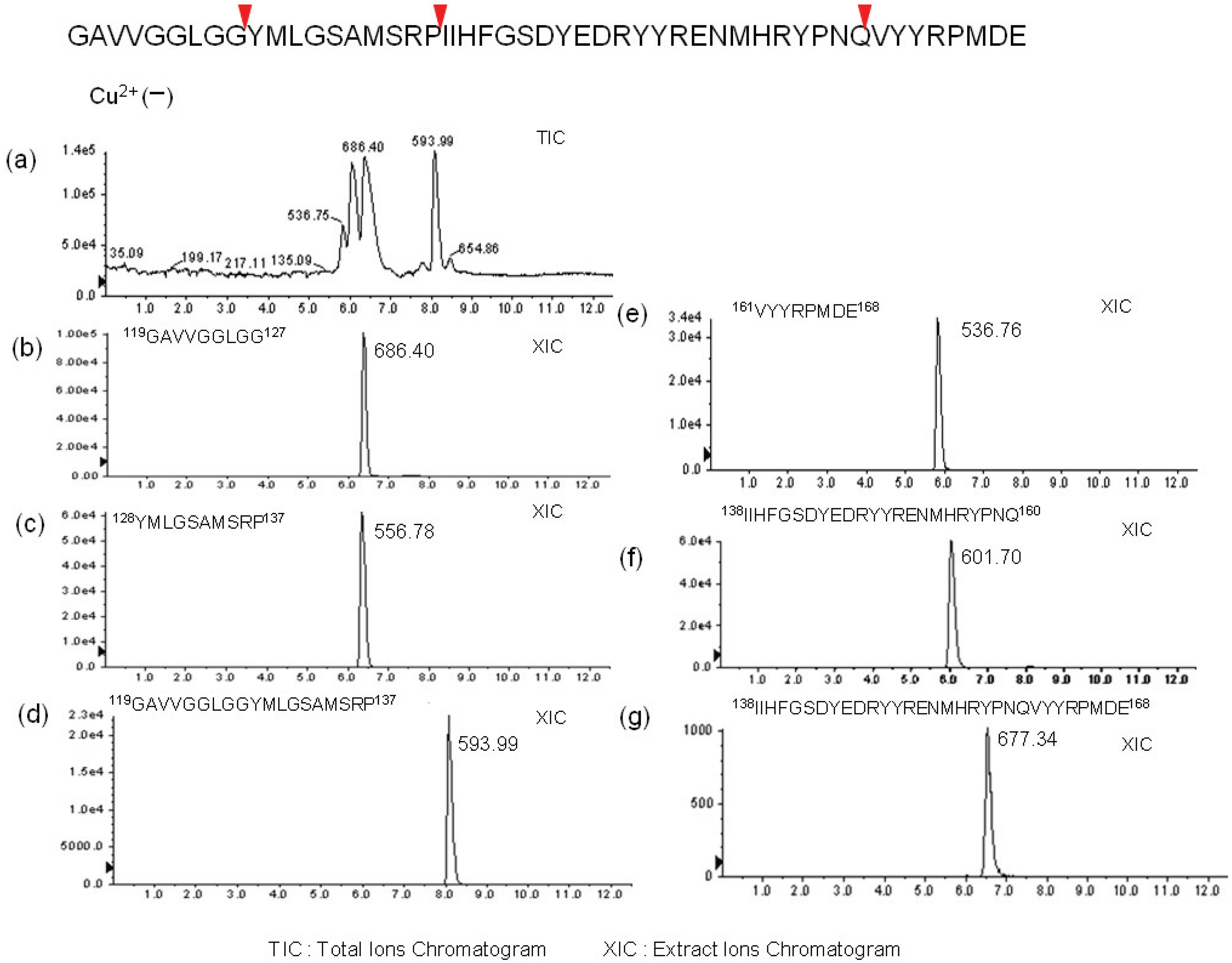
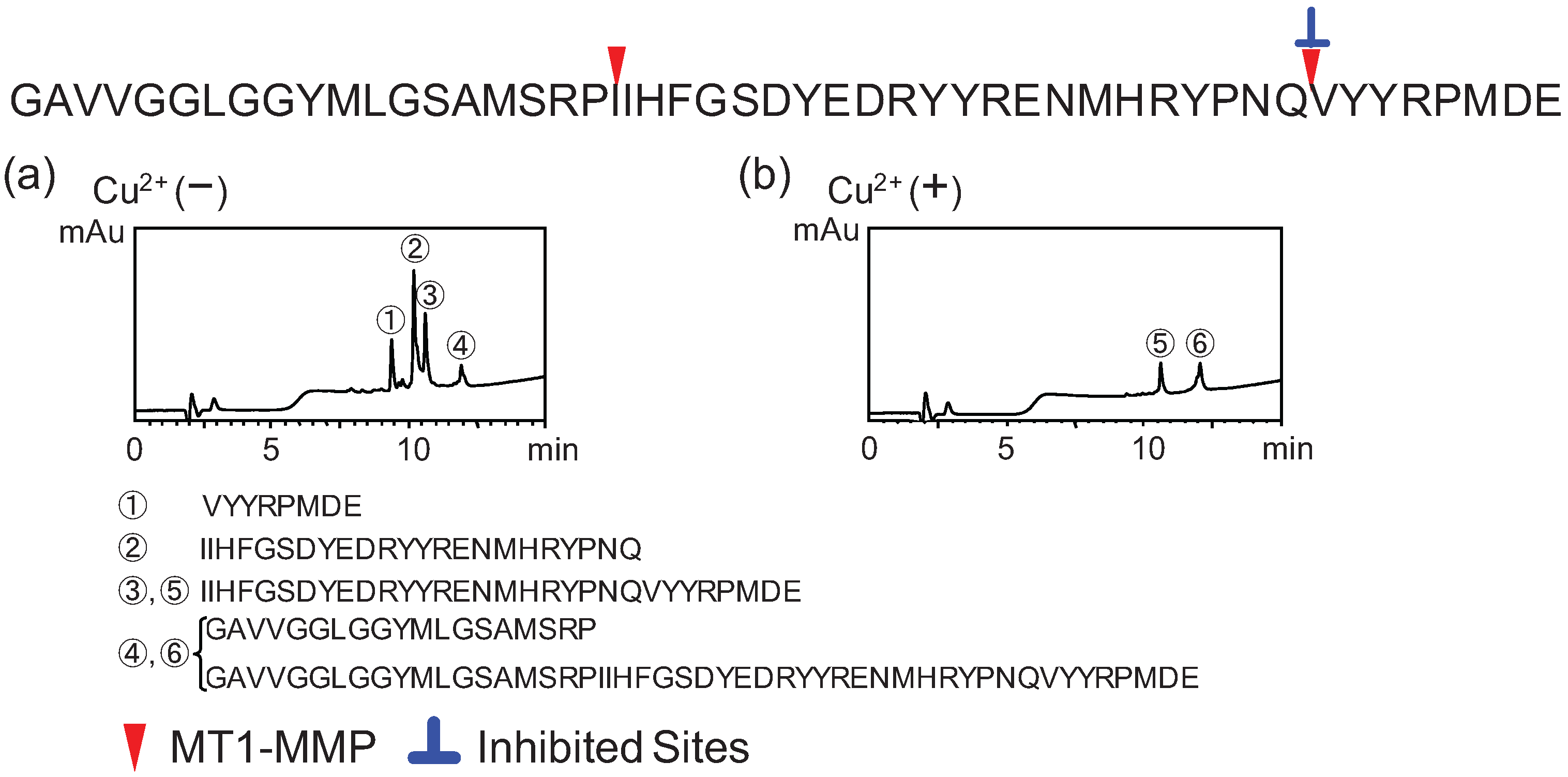
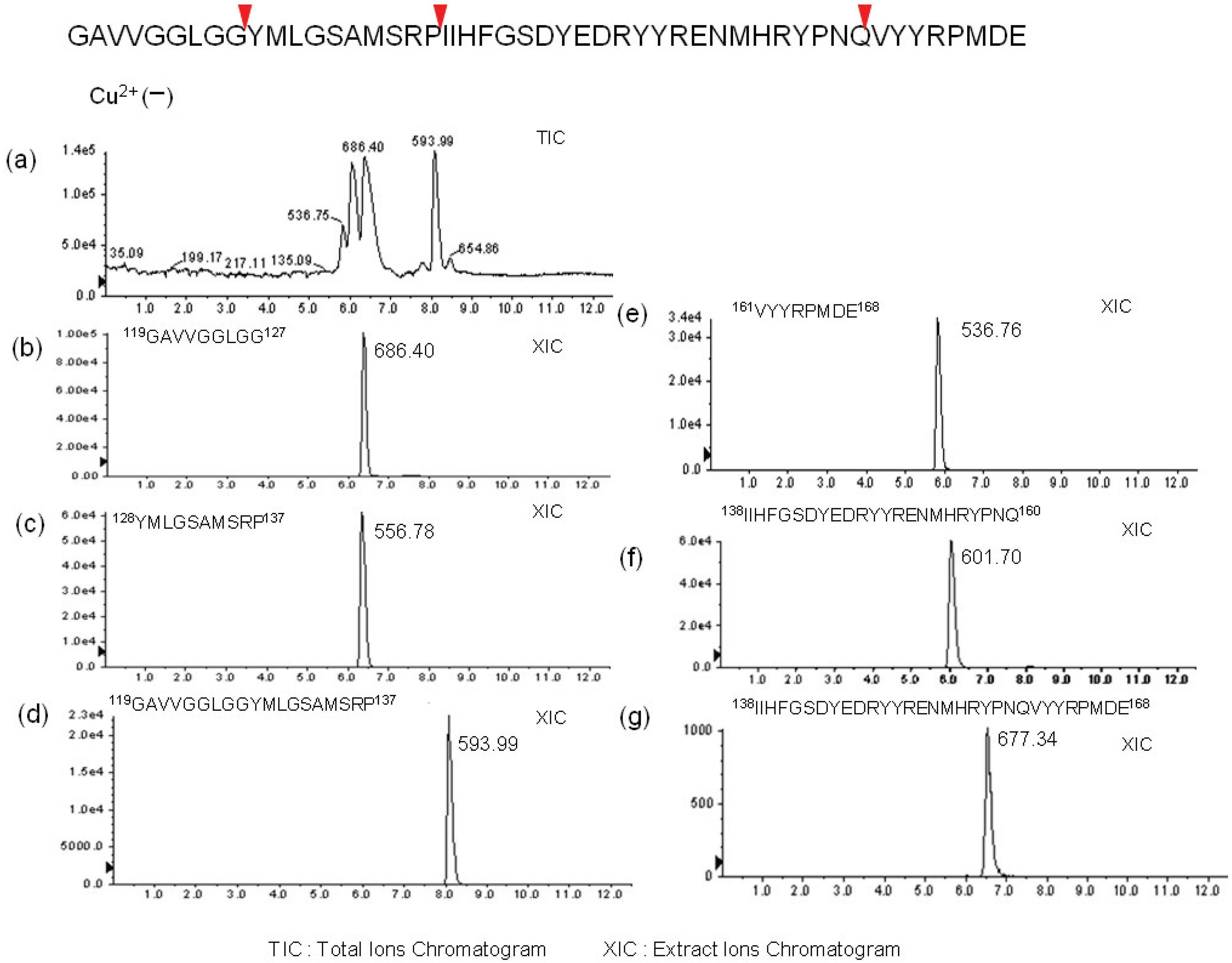
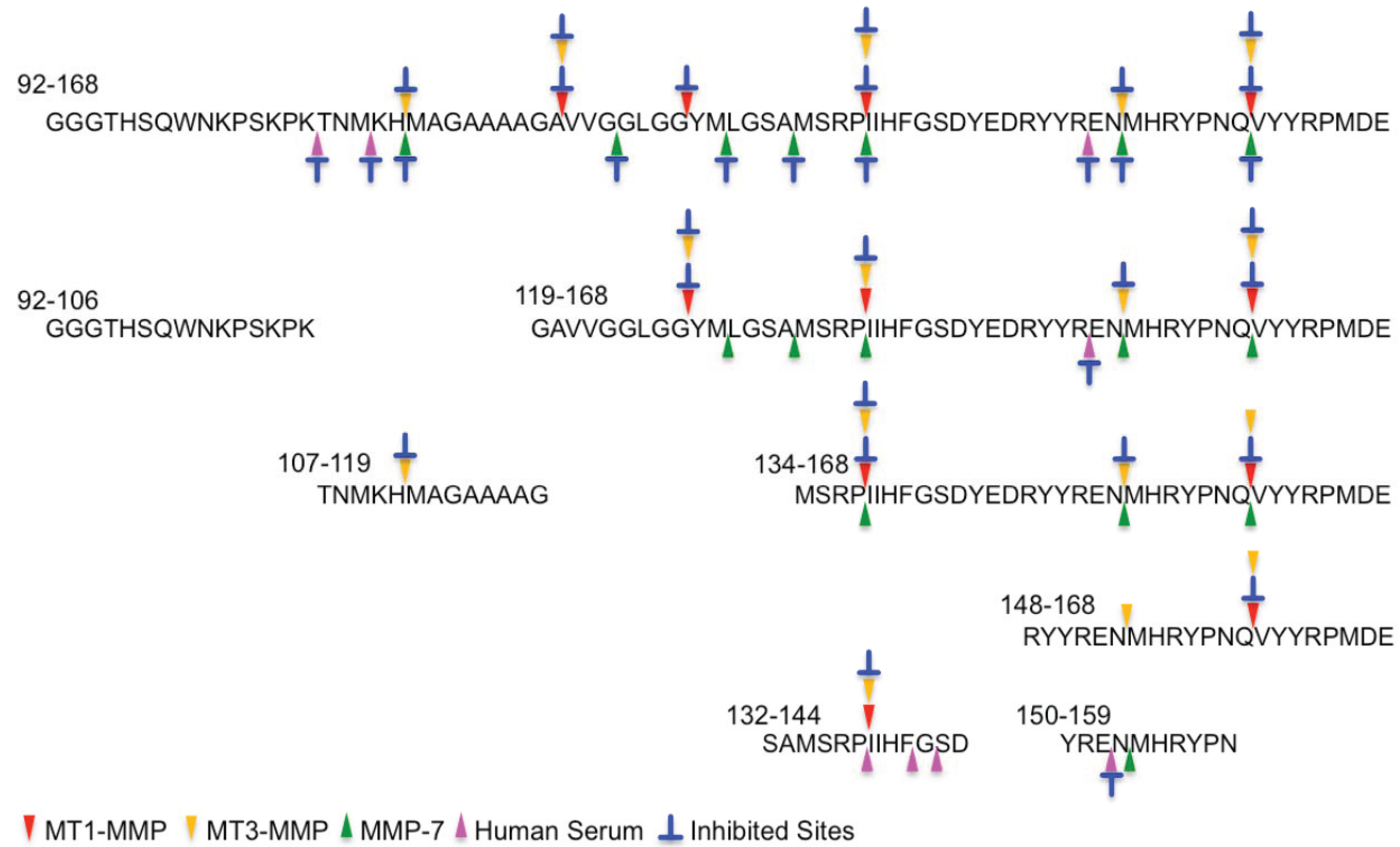
2.2. Determination of Cleavage Sites

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| 60–91(OP-4) | PHGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQ |
| 60–83(OP-3) | PHGGGWGQPHGGGWGQPHGGGWGQ |
| 60–75(OP-2) | PHGGGWGQPHGGGWGQ |
| 60–67(OP–1) | PHGGGWGQ |
| OP-1/HA | PAGGGWGQ |
| 92–168 | GGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDE |
| 119–168 | GAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDE |
| 134–168 | MSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDE |
| 148–168 | RYYRENMHRYPNQVYYRPMDE |
| 92–106 | GGGTHSQWNKPSKPK |
| 107–119 | TNMKHMAGAAAAG |
| 132–144 | SAMSRPIIHFGSD |
| 150–159 | YRENMHRYPN |
| 169–192 | YSNQNNFVHDCVNITIKQHTVTTT |
| 169–179 | YSNQNNFVHDC |
| 169–178 | YSNQNNFVHD |
| 175–189 | FVHDCVNITIKQHTV |
| 180–192 | VNITIKQHTVTTT |
| 180–192/HA | VNITIKQATVTTT |
| 193–230 | TKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGS |
| 215–230 | ITQYERESQAYYQRGS |

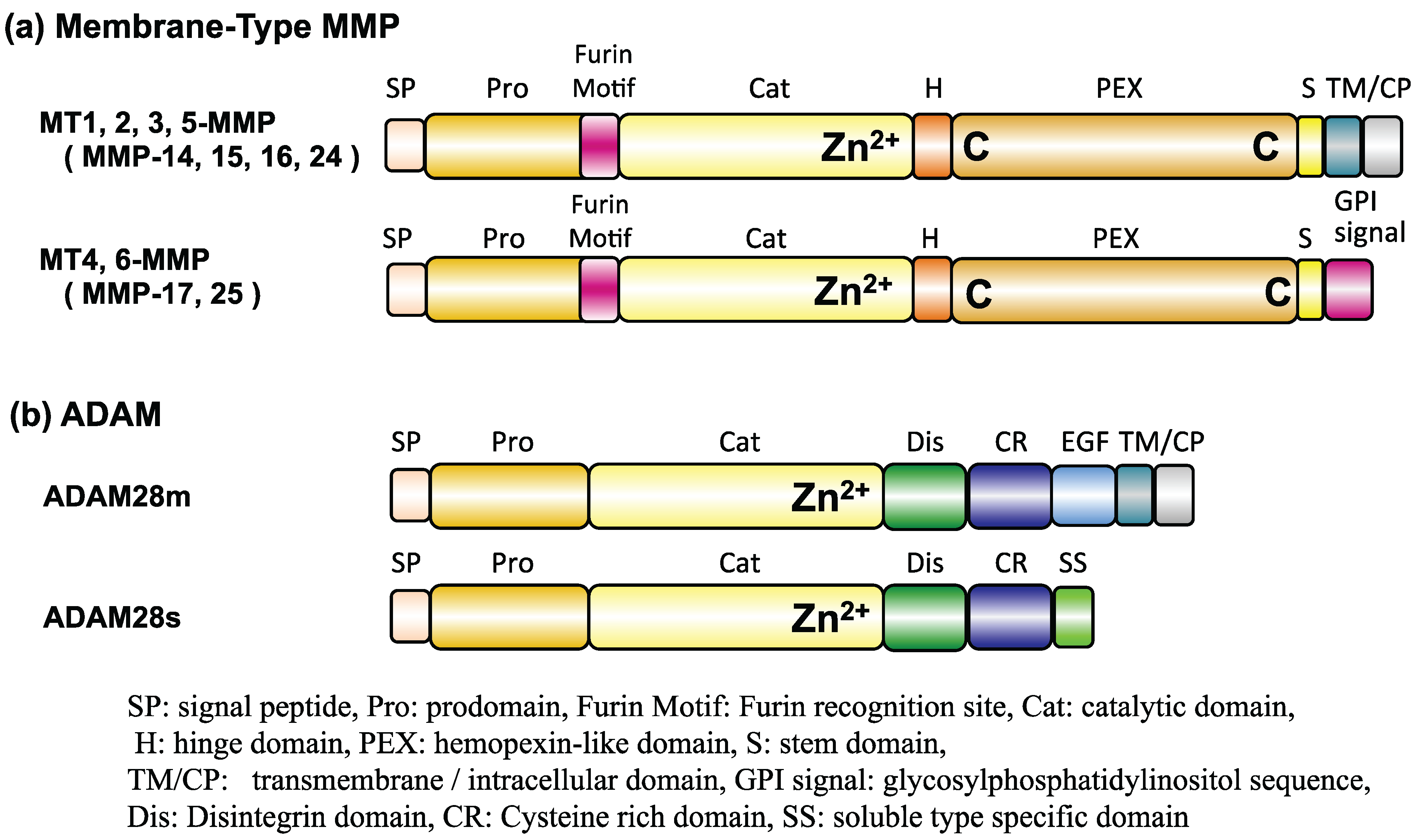
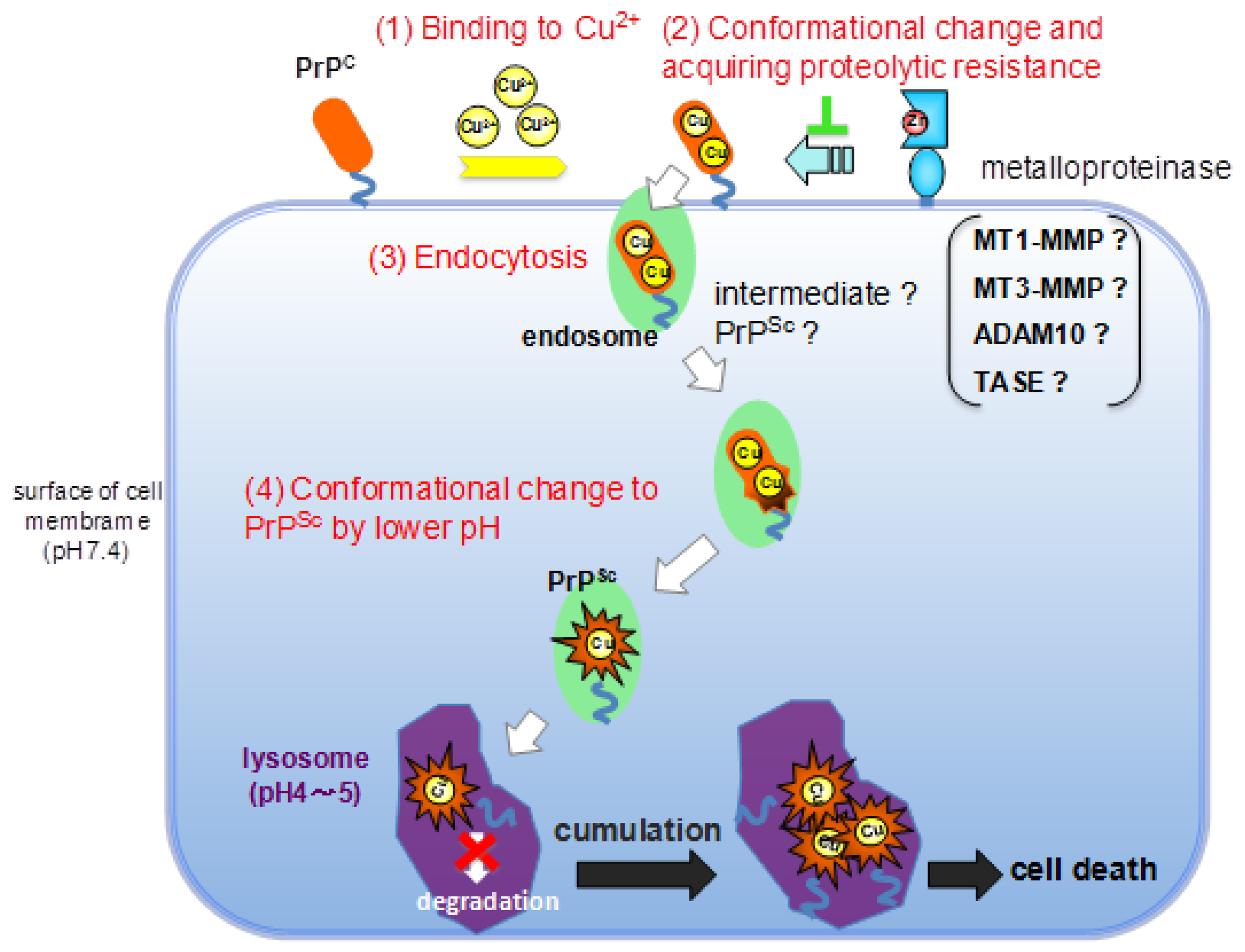
3. Experimental
3.1. Preparation of Recombinant Human MMPs
3.2. Materials
3.3. Preparation of Synthetic Fragment Peptide
3.4. Analytical HPLC
3.5. Determination of Cleavage Sites
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Chazot, G.; Broussolle, E.; Lapras, C.; Blattler, T.; Aguzzi, A.; Kopp, N. New variant of Creutzfeldt-Jakob disease in a 26-year-old French man. Lancet 1996, 347, 1181. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prion diseases and the BSE crisis. Science 1997, 278, 245–251. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of α-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Safar, J.; Roller, P.P.; Gajdusek, D.C.; Gibbs, C.J., Jr. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 1993, 268, 20276–20284. [Google Scholar]
- Cohen, F.E.; Pan, K.M.; Huang, Z.; Baldwin, M.; Fletterick, R.J.; Prusiner, S.B. Structural clues to prion replication. Science 1994, 264, 530–531. [Google Scholar]
- Telling, G.C.; Scott, M.; Mastrianni, J.; Gabizon, R.; Torchia, M.; Cohen, F.E.; DeArmond, J.; Prusiner, S.B. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995, 83, 79–90. [Google Scholar] [CrossRef]
- Kaneko, K.; Zulianello, L.; Scott, M.; Cooper, C.M.; Wallace, A.C.; James, T.L.; Cohen, F.E.; Prusiner, S.B. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 10069–10074. [Google Scholar] [CrossRef]
- Harris, D.A.; Huber, M.T.; van Dijken, P.; Shyng, S.L.; Chait, B.T.; Wang, R. Processing of a cellular prion protein: Identification of N- and C-terminal cleavage sites. Biochemistry 1993, 32, 1009–1016. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar] [CrossRef]
- Selvaggini, C.; de Gioia, L.; Cantu, L.; Ghibaudi, E.; Diomede, L.; Passerini, F.; Forloni, G.; Bugiani, O.; Tagliavini, F.; Salmona, M. Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem. Biophys. Res. Commun. 1993, 194, 1380–1386. [Google Scholar] [CrossRef]
- Brown, D.R. Prion protein peptide neurotoxicity can be mediated by astrocytes. J. Neurochem. 1999, 73, 1105–1113. [Google Scholar] [CrossRef]
- Ettaiche, M.; Pichot, R.; Vincent, J.P.; Chabry, J. In vivo cytotoxicity of the prion protein fragment 106–126. J. Biol. Chem. 2000, 275, 36487–36490. [Google Scholar] [CrossRef]
- O’Donovan, C.N.; Tobin, D.; Cotter, T.G. Prion protein fragment PrP-(106–126) induces apoptosis via mitochondrial disruption in human neuronal SH-SY5Y cells. J. Biol. Chem. 2001, 276, 43516–43523. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Localization matrix metalloproteinase activities in the pericellular enviroment. FEBS J. 2011, 278, 2–15. [Google Scholar]
- Oku, N.; Matsukawa, M.; Yamakawa, S.; Asai, T.; Yahara, S.; Hashimoto, F.; Akizawa, T. Inhibitory effect of green tea polyphenols on membrane-type 1 matrix metalloproteinase, MT1-MMP. Biol. Pharm. Bull. 2003, 26, 1235–1238. [Google Scholar] [CrossRef]
- Millar, A.W.; Brown, P.D.; Moore, J.; Galloway, W.A.; Cornish, A.G.; Lenehan, T.J.; Lynch, K.P. Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br. J. Clin. Pharmacol. 1998, 45, 21–26. [Google Scholar]
- Zitka, O.; Kukacka, J.; Krizkova, S.; Huska, D.; Adam, V.; Masarik, M.; Prusa, R.; Kizek, R. Matrix metalloproteinases. Curr. Med. Chem. 2010, 17, 3751–3768. [Google Scholar] [CrossRef]
- Butler, G.S.; Overall, C.M. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry 2009, 48, 10830–10845. [Google Scholar] [CrossRef]
- Rodriguez, D.; Morrison, J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Scott, M.R.; DeArmond, S.J.; Cohen, F.E. Prion protein biology. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Prox, J.; Puig, B.; Dohler, F.; Falker, C.; Krasemann, S.; Glatzel, M. Roles of endoproteolytic a-cleavage and shedding of the prion protein in neurodegeration. FEBS J. 2013, 280, 4338–4347. [Google Scholar] [CrossRef]
- Vincent, B.; Paitel, E.; Saftig, P.; Frobert, Y.; Hartmann, D.; de Strooper, B.; Grassi, J.; Lopez-Perez, E.; Checler, F. The disintegrins ADAN10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cedllular prion protein. J. Biol. Chem. 2001, 276, 37743–37746. [Google Scholar]
- Liang, J.; Wang, W.; Sorensen, D.; Medina, S.; Ilchenko, S.; Kiselar, J.; Surewicz, W.K.; Booth, S.A.; Kong, Q. Cellular prion protein regulates ist own alpha-cleabage through ADAM8 in skeletal muscle. J. Biol. Chem. 2012, 287, 16510–16520. [Google Scholar] [CrossRef]
- Beland, M.; Motard, J.; Barbarin, A.; Roucou, X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 2012, 32, 13255–13263. [Google Scholar] [CrossRef]
- Wik, L.; Klingeborn, M.; Willander, H.; Linner, T. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 2012, 6, 498–509. [Google Scholar] [CrossRef]
- Cisse, M.A.; Sunyach, C.; Lefrance-Jullien, S.; Postina, R.; Vincent, B.; Checler, F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J. Biol. Chem. 2005, 280, 40624–40631. [Google Scholar]
- Tousseyn, T.; Thathiah, A.; Jorissen, E.; Snellinx, A.; Serneels, L.; Nyabi, O.; Annaert, W.; Saftig, P.; Hartmann, D.; de Strooper, B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J. Biol. Chem. 2009, 284, 11738–11747. [Google Scholar] [CrossRef]
- Moss, M.L.; Powell, G.; Miller, M.A.; Edwards, L.; Qi, B.; Sang, Q.X.; de Strooper, B.; Tesseur, I.; Lichtenthaler, S.F.; Taverna, M.; et al. ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protein. J. Biol. Chem. 2011, 286, 40443–40451. [Google Scholar] [CrossRef] [Green Version]
- Yong, V.W.; Power, C.; Forsyth, P.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001, 2, 502–511. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Ibi, D.; Takuma, K.; Toth, E.; Sato, J.; Itohara, S.; Nabeshima, T.; Yamada, K. Alterations of emotional and cognitive behaviors in matrix metalloproteinase-2 and -9-deficient mice. Open Behav. Sci. J. 2010, 4, 19–25. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Yamada, K.; Nabeshima, T. Matrix metalloproteinases contribute to neuronal dysfunction in animal models of drug dependence, Alzheimer’s disease, and epilepsy. Biochem. Res. Int. 2011. [Google Scholar] [CrossRef]
- Stöckel, J.; Yamada, K.; Safar, J.; Wallace, A.C.; Cohen, F.E.; Prusiner, S.B. Prion protein selectively binds copper(II) Ions. Biochemistry 1998, 37, 7185–7193. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar]
- Miura, T.; Hori-i, A.; Takeuchi, H. Metal-dependent alpha-helix formation promoted by the glycine-rich octapeptide region of prion protein. FEBS Lett. 1996, 396, 248–252. [Google Scholar] [CrossRef]
- Walter, E.D.; Chattopadhyay, M.; Millhauser, G.L. The affinity of copper binding to the prion protein octarepeat domain: Evidence for negative cooperativity. Biochemistry 2006, 45, 13083–13092. [Google Scholar] [CrossRef]
- Wells, M.A.; Jackson, G.S.; Jones, S.; Hosszu, L.L.; Craven, C.J.; Clarke, A.R.; Collinge, J.; Waltho, J.P. A reassessment of copper (II) binding in the full-length prion protein. Biochem. J. 2006, 399, 435–444. [Google Scholar] [CrossRef]
- Wells, M.A.; Jelinska, C.; Hosszu, L.L.; Craven, C.J.; Clarke, A.R.; Collinge, J.; Waltho, J.P.; Jackson, G.S. Multiple forms of copper (II) co-ordination occur throughout the disordered N-terminal region of the prion protein at pH 7.4. Biochem. J. 2006, 400, 501–510. [Google Scholar] [CrossRef]
- Klewpatinond, M.; Davies, P.; Bowen, S.; Brown, D.R.; Viles, J.H. Deconvoluting the Cu2+ binding modes of full-length prion protein. J. Biol. Chem. 2008, 283, 1870–1881. [Google Scholar] [CrossRef]
- Ronga, L.; Palladino, P.; Saviano, G.; Tancredi, T.; Benedetti, E.; Ragone, R.; Rossi, F. NMR structure and CD titration with metal cations of human prion alpha2-helix-related peptides. Bioinorg. Chem. Appl. 2007. [Google Scholar] [CrossRef]
- Aronoff-Spencer, E.; Burns, C.S.; Avdievich, N.I.; Gerfen, G.J.; Peisach, J.; Antholine, W.E.; Ball, H.L.; Cohen, F.E.; Prusiner, S.B.; Millhauser, G.L. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry 2000, 39, 13760–13771. [Google Scholar] [CrossRef]
- Burns, C.S.; Aronoff-Spencer, E.; Legname, G.; Prusiner, S.B.; Antholine, W.E.; Gerfen, G.L.; Peisach, J.; Millhauser, G.L. Copper coordination in the full-length, recombinant prion protein. Biochemistry 2003, 42, 6794–6803. [Google Scholar] [CrossRef]
- Bonomo, R.P.; Imperllizzeri, G.; Pappalardo, G.; Rizzarelli, E.; Tabbi, G. Copper(II) binding modes in the prion octapeptide PHGGGWGQ: A spectroscopic and voltammetric study. Chemistry 2000, 6, 4195–4202. [Google Scholar] [CrossRef]
- Viles, J.H.; Cohen, F.E.; Prusiner, S.B.; Goodin, D.B.; Wright, P.E.; Dyson, H.J. Copper binding to the prion protein: Structural implications of four identical cooperative binding sites. Proc. Natl. Acad. Sci. USA 1999, 96, 2042–2047. [Google Scholar]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M.; Lakey, J.H. Copper binding to the N-terminal tandem repeat region of mammalian and avian prion protein: Structural studies using synthetic peptides. Biochem. Biophys. Res. Commun. 1995, 214, 993–999. [Google Scholar] [CrossRef]
- Burns, C.S.; Aronoff-Spencer, E.; Dunham, C.M.; Lario, P.; Avdievich, N.I.; Antholine, W.E.; Olmstead, M.M.; Vrielink, A.; Gerfen, G.L.; Peisach, J.; et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 2002, 41, 3991–4001. [Google Scholar] [CrossRef]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.P.; Gibbs, N.; Walltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8532–8535. [Google Scholar]
- Jones, C.E.; Klewpatinond, M.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: The unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J. Mol. Biol. 2005, 346, 1393–1407. [Google Scholar] [CrossRef]
- Qin, K.F.; Yang, Y.; Mastrangelo, P.; Westaway, D. Mapping Cu(II) binding sites in prion proteins by diethyl pyrocarbonate modification and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectromeric footprinting. J. Biol. Chem. 2002, 277, 1981–1990. [Google Scholar] [CrossRef]
- Whittal, R.M.; Ball, H.L.; Cohen, F.E.; Burlingame, A.L.; Prusiner, S.B.; Baldwin, M.A. Copper binding to octarepeat peptides of the prion protein monitored by mass spectrometry. Protein Sci. 2000, 9, 332–343. [Google Scholar]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M. Copper binding to the N-terminal tandem repeat regions of mammalian and avian prion protein. Biochem. Biophys. Res. Commun. 1995, 207, 621–629. [Google Scholar] [CrossRef]
- Miura, T.; Hori-i, A.; Mototani, H.; Takeuchi, H. Raman spectroscopic study on the copper(II) binding mode of prion octapeptide and its pH dependence. Biochemistry 1999, 38, 11560–11569. [Google Scholar] [CrossRef]
- Miura, T.; Sasaki, S.; Toyama, A.; Takeuch, H. Copper reduction by the octapeptide repeat region of prion protein: pH dependence and implicatios in cellular copper uptake. Biochemistry 2005, 44, 8712–8720. [Google Scholar] [CrossRef]
- Gustiananda, M.; Haris, P.I.; Milburm, P.J.; Gready, J.E. Copper-induced conformational change in a marsupial prion protein repeat peptide probed using FTIR spectroscopy. FEBS Lett. 2002, 512, 38–42. [Google Scholar] [CrossRef]
- Valensin, D.; Luczkozlowski, M.; Mancini, F.M.; Legowska, A.; Gaggelli, E.; Valensin, G.; Rolka, K.; Kozlowski, H. The dimeric and tetrameric octarepeat fragments of prion protein behave differently to its monomeric unit. Dalton Trans. 2004, 7, 1284–1293. [Google Scholar]
- Yu, X.; Wojciechowski, M.; Fenselau, C. Assessment of metals in reconstituted metallothioneins by electrospray mass spectrometry. Anal. Chem. 1993, 65, 1355–1359. [Google Scholar] [CrossRef]
- Lim, J.; Vachet, R.W. Using mass spectrometry to study copper-protein binding under native and non-native conditions: Beta-2-microglobulin. Anal. Chem. 2004, 76, 3498–3504. [Google Scholar] [CrossRef]
- Kojima, A.; Konishi, M.; Akizawa, T. Metal-binding ability of peptides originating from prion protein by a column switch HPLC. Pept. Sci. 2010, 46, 223–226. [Google Scholar]
- Kojima, A.; Mabuchi, Y.; Konishi, M.; Okihara, R.; Nagano, M.; Akizawa, T. Metal-binding ability of human prion protein fragment peptides analyzed by column switch HPLC. Chem. Pharm. Bull. 2011, 59, 965–971. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion in health and disease. Am. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar]
- Mizoguchi, H.; Yamada, K. Roles of matrix metalloproteinases and their targets in epileptogenesis and seizures. Clin. Psychopharmacol. Neurosci. 2013, 11, 45–52. [Google Scholar] [CrossRef]
- Goldfarb, L.G.; Brown, P.; McCombie, W.R.; Goldgaber, D.; Swergold, G.D.; Wills, P.R.; Cervenakova, L.; Baron, H.; Gibbs, C.J., Jr.; Gajdusek, D.C. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad Sci. USA 1991, 88, 10926–10930. [Google Scholar] [CrossRef]
- Moore, R.A.; Herzog, C.; Errett, J.; Kocisko, D.A.; Arnold, K.M.; Hayes, S.F.; Priola, S.A. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006, 15, 609–619. [Google Scholar] [CrossRef]
- Li, B.; Qing, L.; Yan, J.; Kong, Q. Instability of the octarepeat region of the human prion protein gene. PLoS One 2011, 6, e26635. [Google Scholar]
- Fell, G.S.; Smith, H.; Howie, R.A. Neutron activation analysis OFR copper in biological material applied to Wilson’s disease. J. Clin. Pathol. 1968, 21, 8–11. [Google Scholar] [CrossRef]
- Sass-Kortsak, A. Copper metabolism. Adv. Clin. Chem. 1965, 8, 1–67. [Google Scholar] [CrossRef]
- Pal, A.; Kumar, A.; Prasad, R. Predictive association of copper metabolism proteins with Alzheimer’s disease and Parkinson’s disease: A preliminary perspective. Biometals 2014, 27, 25–31. [Google Scholar] [CrossRef]
- Constantinidis, J. Treatment of Alzheimer’s disease by zinc compounds. Drug Dev. Res. 1992, 27, 1–14. [Google Scholar] [CrossRef]
- Itoh, M.; Masuda, K.; Ito, Y.; Akizawa, T.; Yoshioka, M.; Imai, K.; Okada, Y.; Sato, H.; Seiki, M. Purification and refolding of recombinant human proMMP-7 (pro-matrilysin) expressed in Escherichia coli and its characterization. J. Biochem. 1996, 119, 667–673. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kojima, A.; Konishi, M.; Akizawa, T. Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding. Biomolecules 2014, 4, 510-526. https://doi.org/10.3390/biom4020510
Kojima A, Konishi M, Akizawa T. Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding. Biomolecules. 2014; 4(2):510-526. https://doi.org/10.3390/biom4020510
Chicago/Turabian StyleKojima, Aya, Motomi Konishi, and Toshifumi Akizawa. 2014. "Prion Fragment Peptides Are Digested with Membrane Type Matrix Metalloproteinases and Acquire Enzyme Resistance through Cu2+-Binding" Biomolecules 4, no. 2: 510-526. https://doi.org/10.3390/biom4020510