Zinc-Binding Cysteines: Diverse Functions and Structural Motifs

Abstract
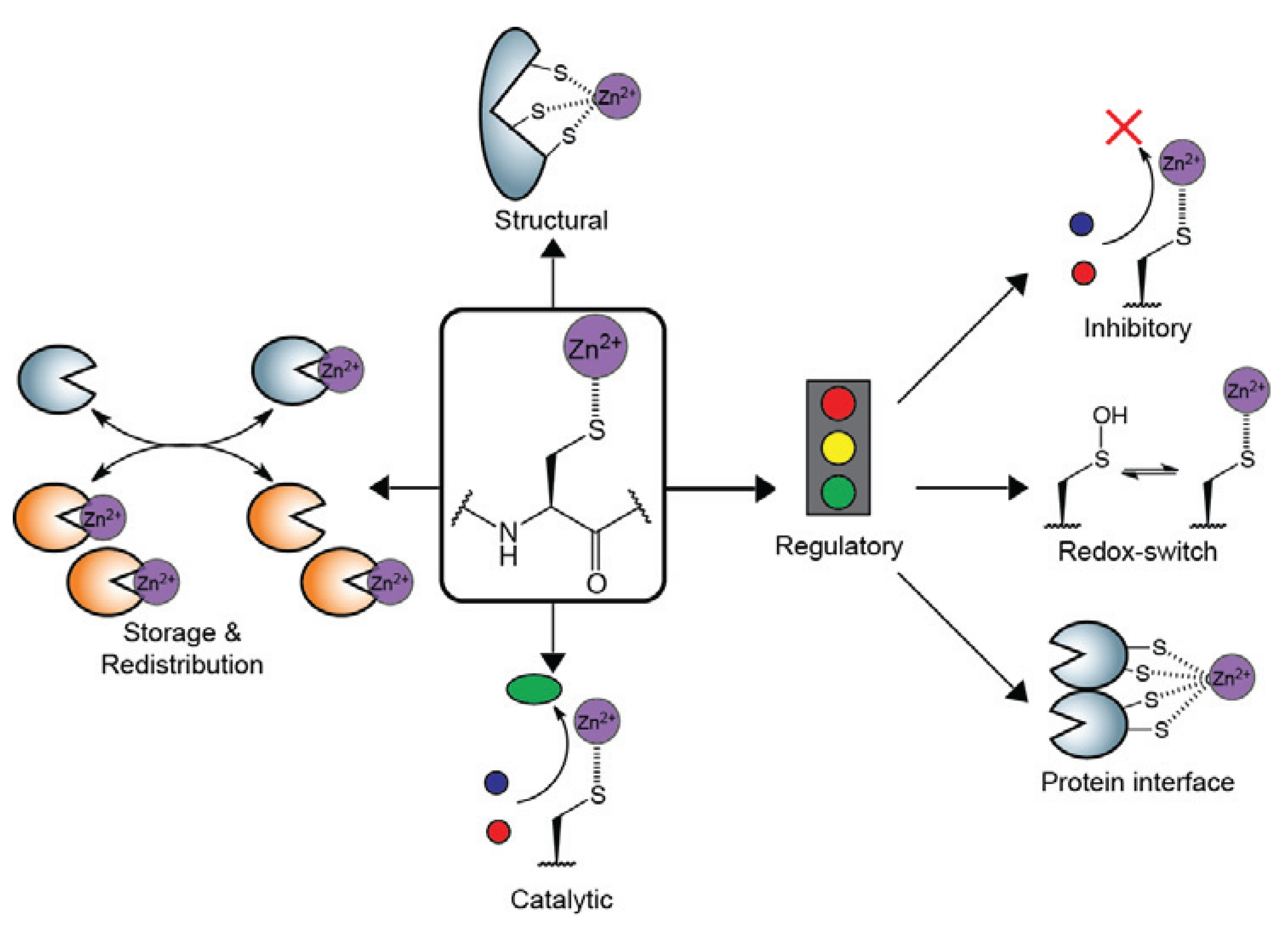
:1. Introduction
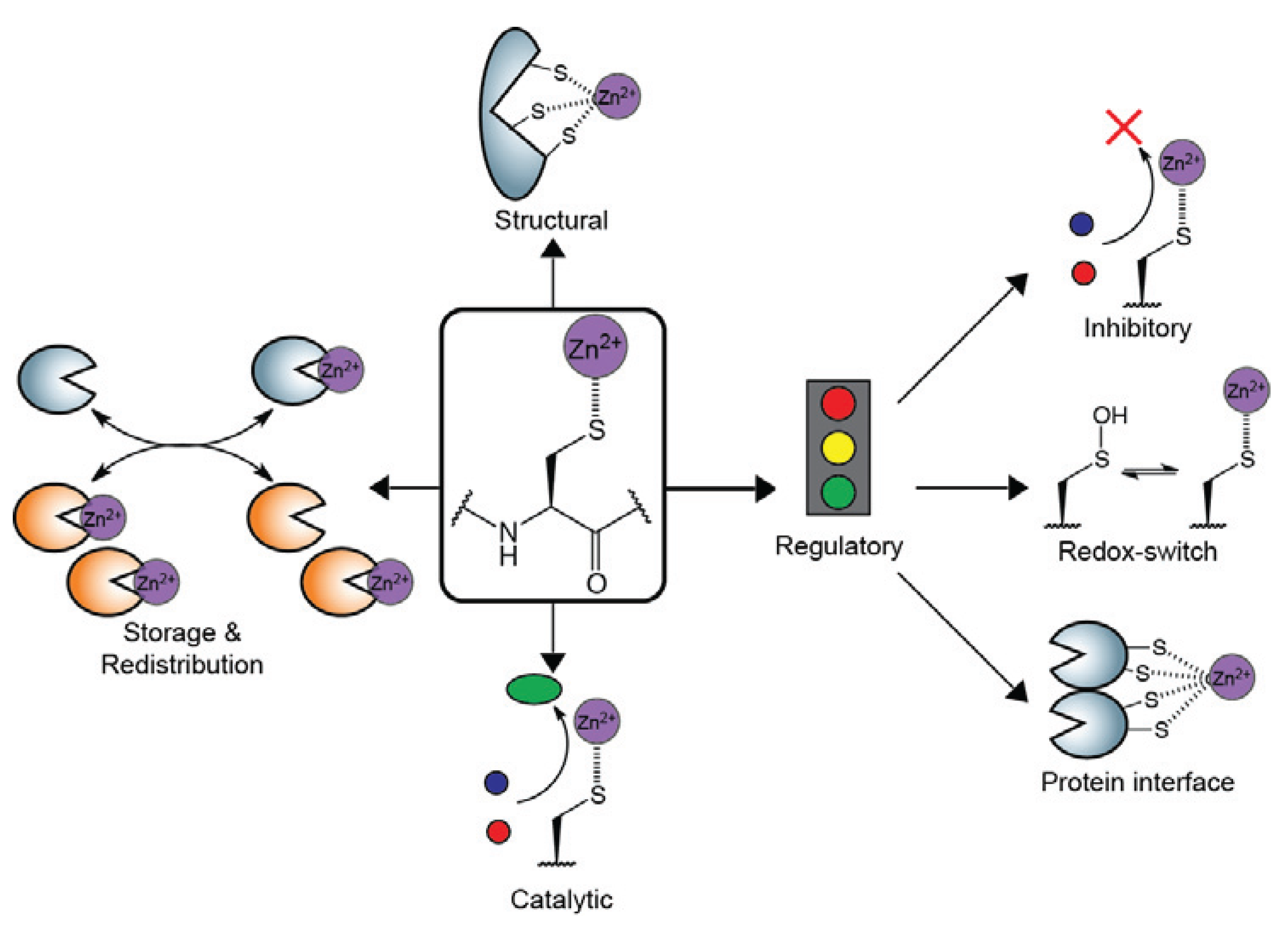
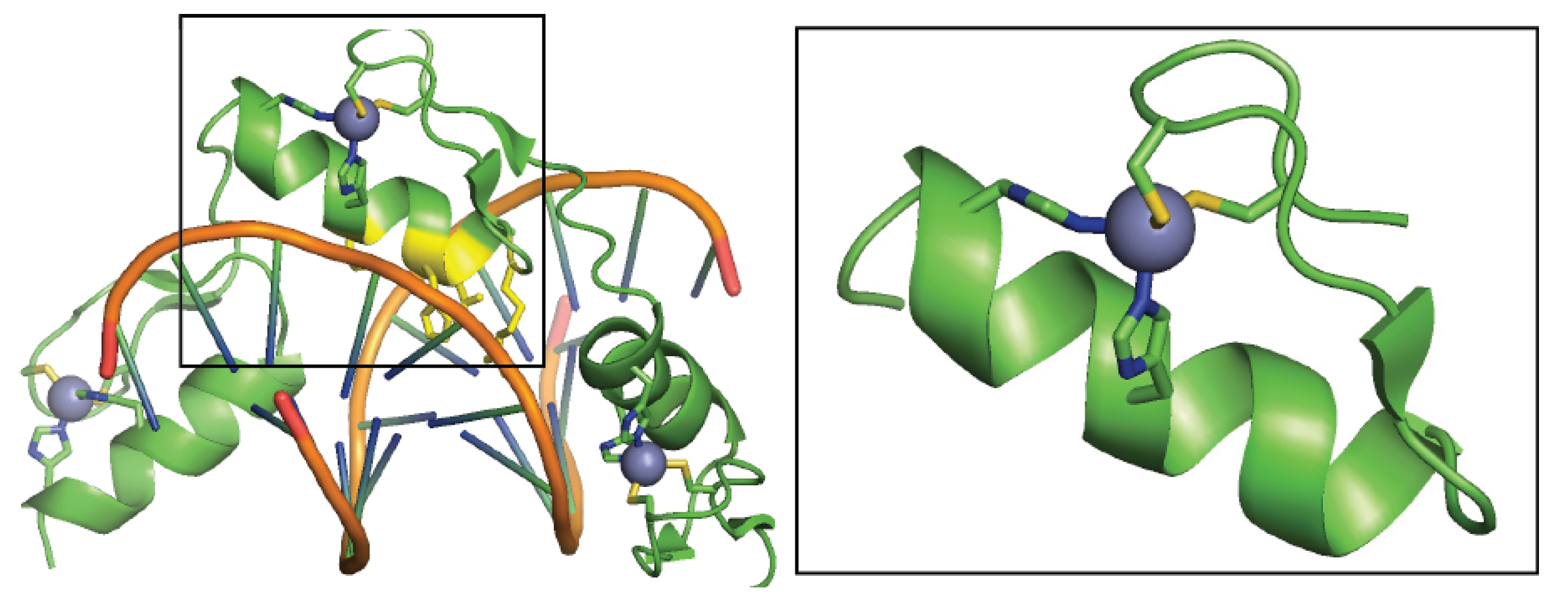
2. Structural Zn2+-Cysteine Complexes: Zinc Fingers

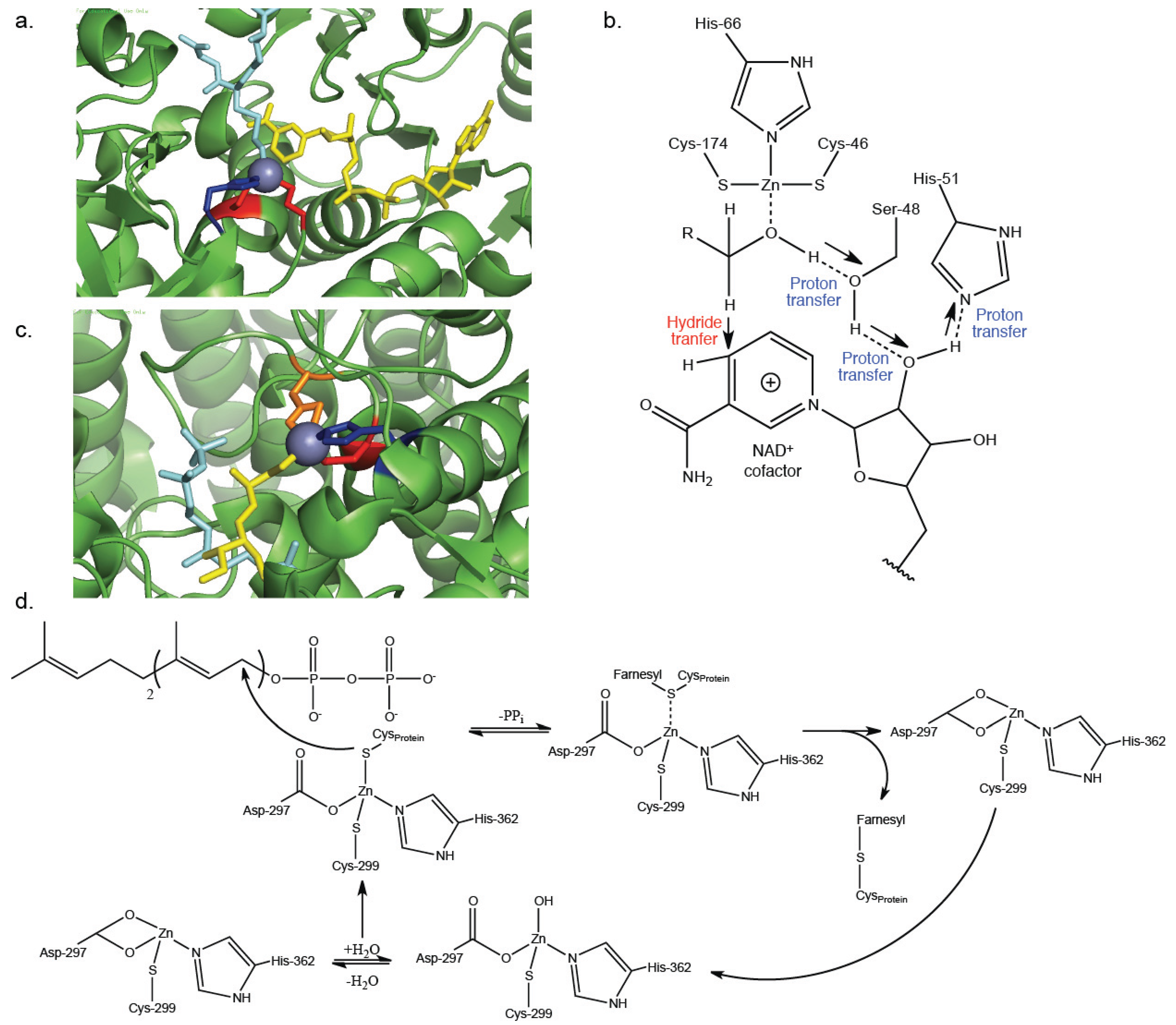
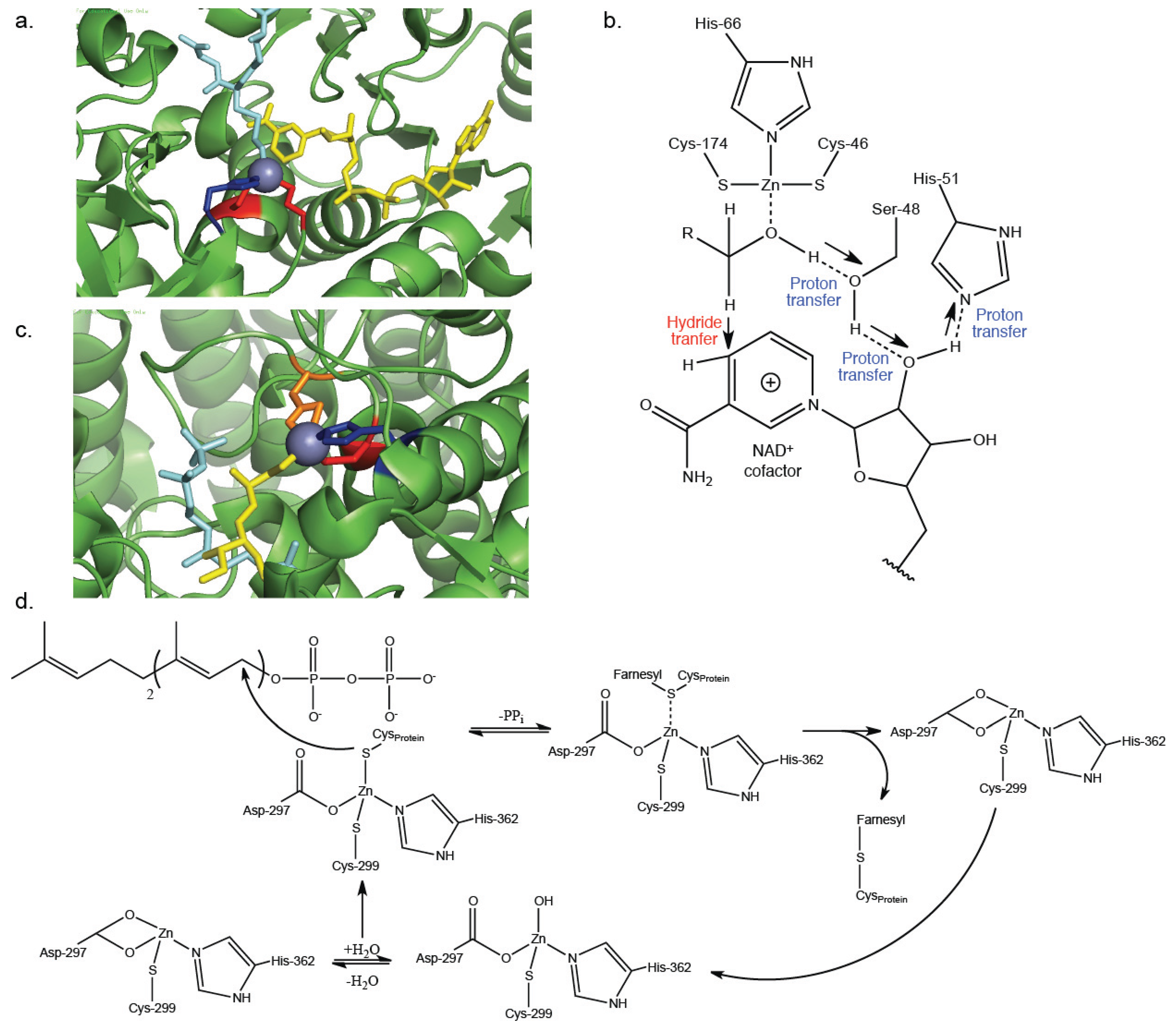
3. Catalytic Zn2+-Cysteine Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Enzyme Class | Function | Mechanism | PDB Structure |
|---|---|---|---|---|
| Alcohol dehydrogenase | Oxidoreductase | Interconverts alcohols to aldehydes and ketones | Zn2+-coordination of substrate [23] | 1MC5 [25] |
| Sorbitol dehydrogenase | Oxidoreductase | Reversible conversion of sorbitol to fructose | Zn2+-activation of nucleophilic water molecule [27] | 1PL7 [27] |
| Cytidine deaminase | Hydrolase | Irreversible hydrolytic deamination of cytidine to uridine | Zn2+-activation of nucleophilic water molecule [28,29] | 2KEM [30] |
| GTP cyclohydrolase I | Hydrolase | Converts GTP to dihydroneopterin triphosphate | Zn2+-activation of nucleophilic water molecule [31] | 1FB1 [31] |
| Betain-homocysteine methyltransferase | Transferase | Transfers methyl group from betaine to homocysteine, forming dimethyl glycine and methionine | Zn2+-activation of thiol of homocysteine substrate [32] | 1LT8 [32] |
| Protein farnesyltransferase | Transferase | Post-translational addition of farnesyl to cysteine residues within proteins | Zn2+-activation of thiol on target protein [33,34] | 1JCQ [35] |
4. Regulatory Zn2+-Cysteine Complexes
| Protein | Enzyme Class | Function | Mechanism | PDB Structure |
|---|---|---|---|---|
| Dimethylarginine dimethylaminohydrolase | Hydrolase | Converts N-omega,N-omega-methyl-L-arginine to dimethylamine and L-citrulline | Inhibitory [39] | 2CI7 [40] |
| Ornithine transcarbamoylase | Transferase | Converts carbamoyl phosphate and ornithine to citrulline and phosphate | Inhibitory [41] | 1EP9 [42] |
| Cathepsin S | Protease | Lysosomal cysteine protease | Inhibitory [8,43] | 2HH5 [43] |
| Caspase 3 | Protease | Cysteine protease | Inhibitory [44,45] | - |
| Caspase 6 | Protease | Cysteine protease | Inhibitory [46] | 4FXO [46] |
| Caspase 9 | Protease | Cysteine protease | Inhibitory [47] | 1JXQ [47] |
| Aconitase 2 | Isomerase | Converts citrate to iso-citrate | Inhibitory [48] | - |
| Glutathione S-transferase omega | Transferase | Conjugates glutathione to a variety of electrophiles | Inhibitory [49] | - |
| Betain-homocysteine methyltransferase | Transferase | Transfers methyl group from betaine to homocysteine, forming dimethyl glycine and methionine | Redox-switch [32] | 1LT7, 1LT8 [32] |
| Protein kinase C | Kinase | Phosphorylates serines and threonines | Redox-switch [50] | 3PFQ [51] |
| Nitric oxide synthase | Oxidoreductase | Produces nitric oxide from arginine | Protein interface[52]; Redox-switch [53] | 3NOS [52] |
| Apo2L/TRAIL | Cytokine | Induces signaling pathways to trigger apoptosis | Protein interface [54] | 1DG6 [54] |
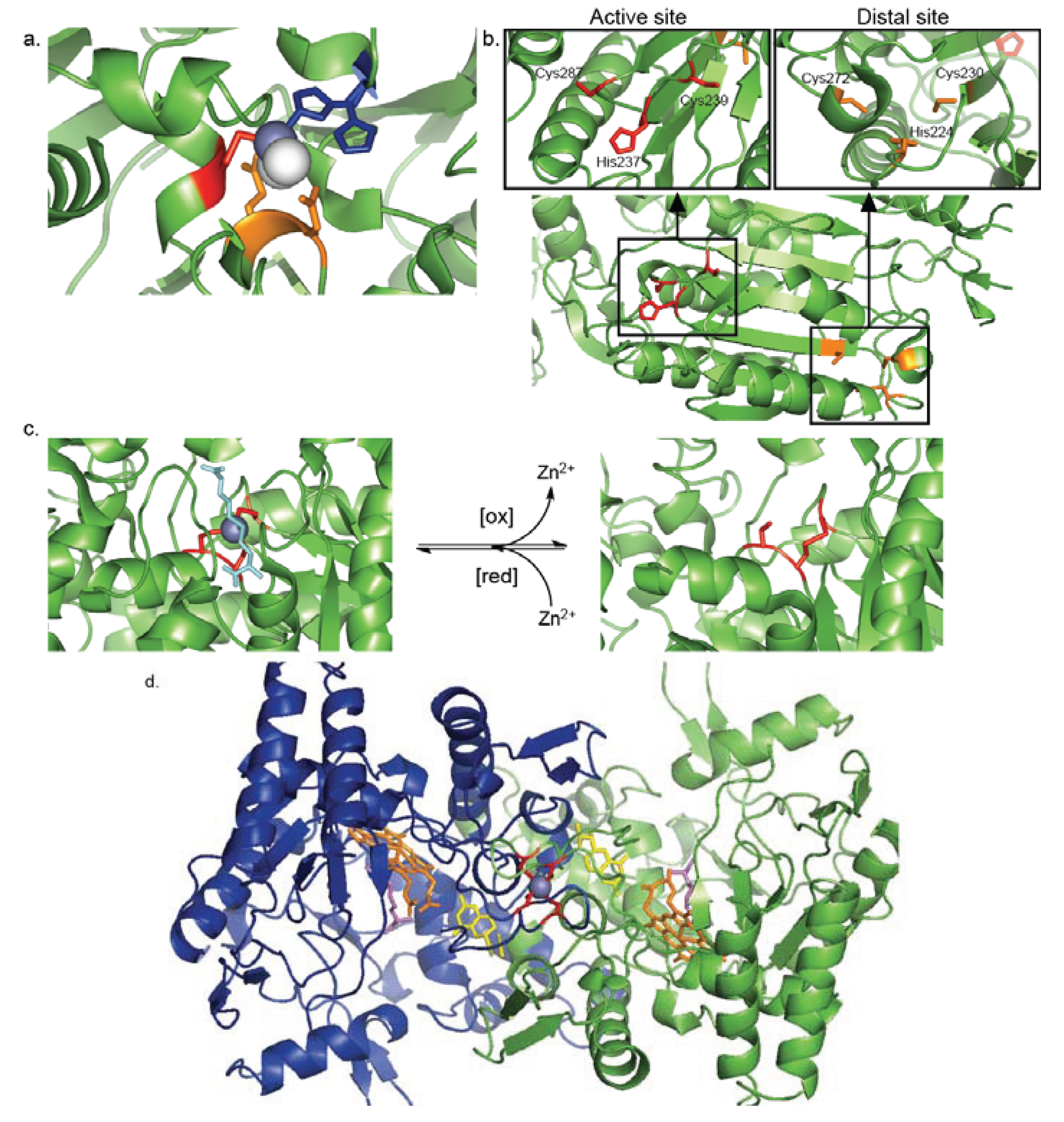
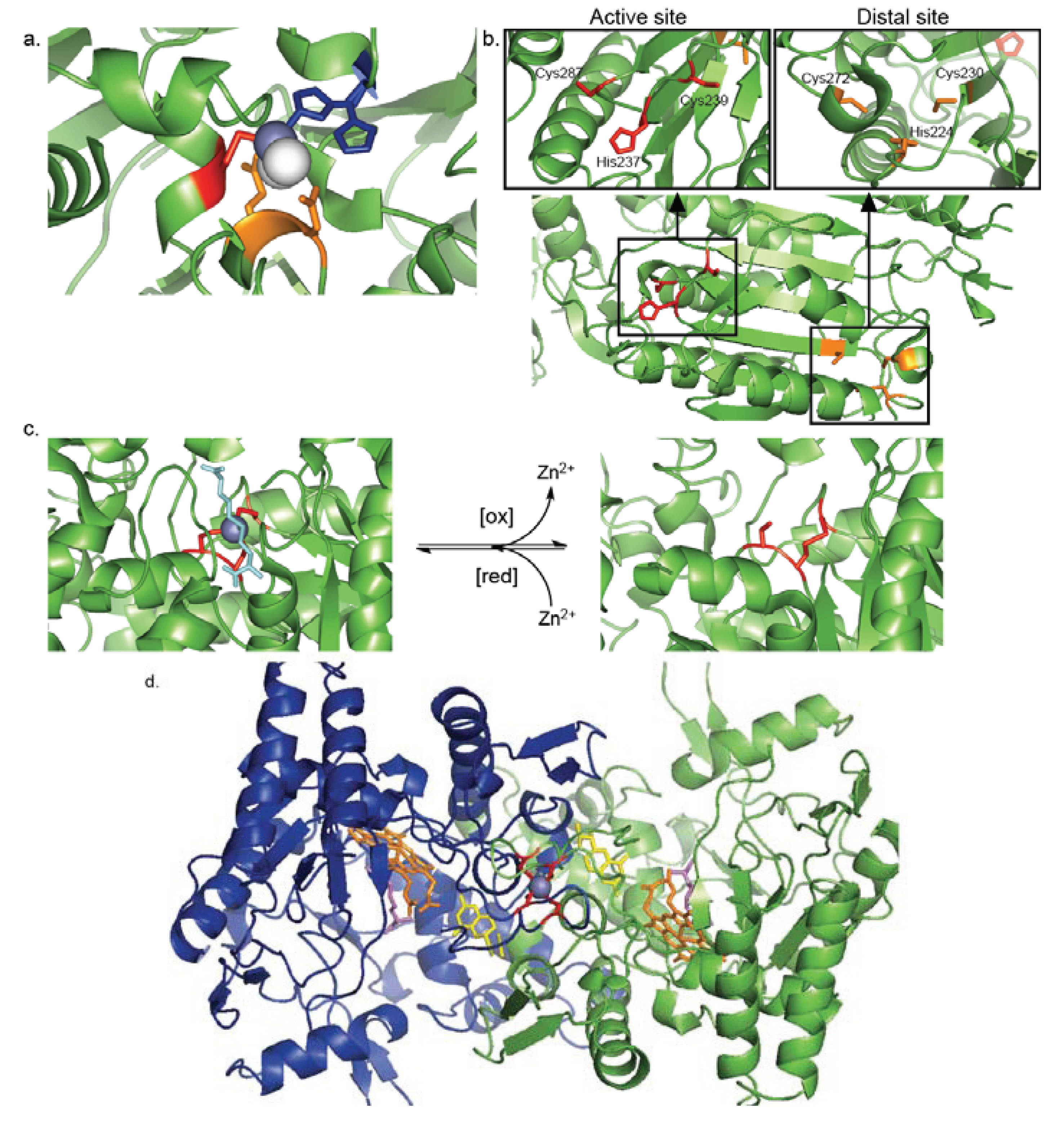
4.1. Inhibitory Zn2+-Cysteine Complexes
4.2. Redox-Switch Zn2+-Cysteine Complexes
4.3. Protein Interface Zn2+-Cysteine Complexes
5. Zn2+-Cysteine Complexes for Zn2+ Transfer & Cellular Redistribution
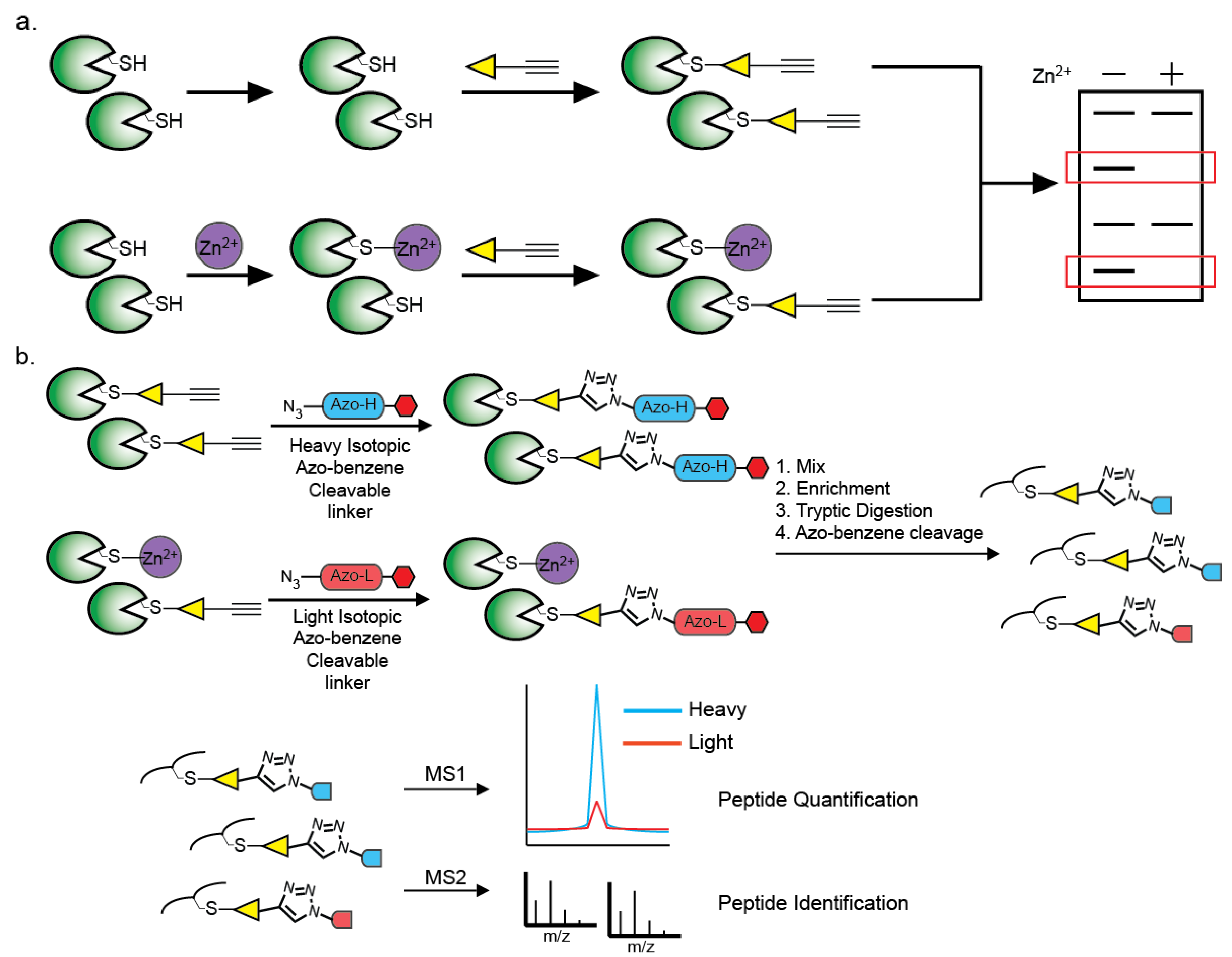
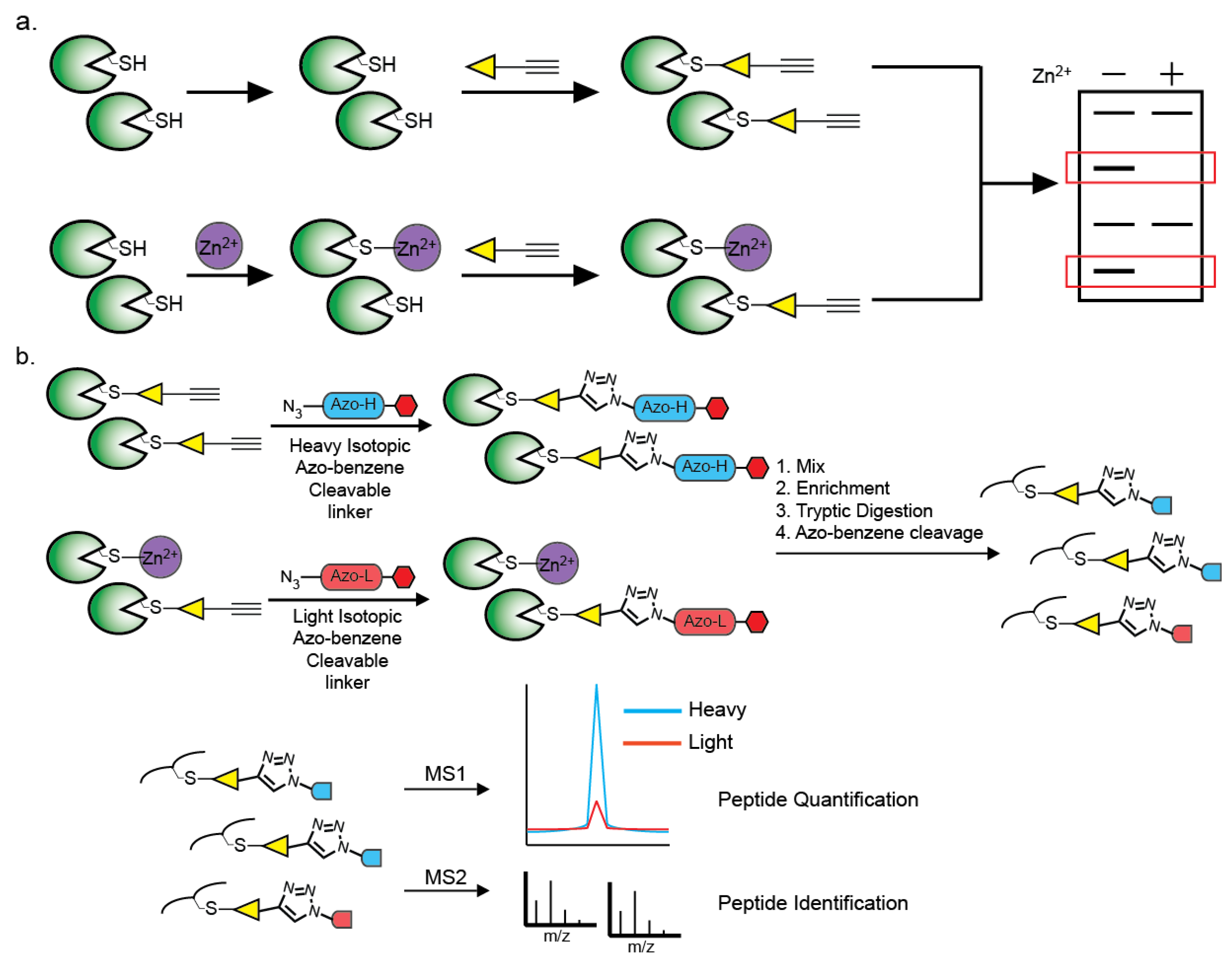
6. Methods of Identification of Zn2+-Cysteine Complexes
7. Perspective and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bulaj, G.; Kortemme, T.; Goldenberg, D.P. Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 1998, 37, 8965–8972. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pka values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Giles, N.M.; Watts, A.B.; Giles, G.I.; Fry, F.H.; Littlechild, J.A.; Jacob, C. Metal and redox modulation of cysteine protein function. Chem. Biol. 2003, 10, 677–693. [Google Scholar]
- Tainer, J.A.; Roberts, V.A.; Getzoff, E.D. Metal-binding sites in proteins. Curr. Opin. Biotechnol. 1991, 2, 582–591. [Google Scholar]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor iiia from xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar]
- Klug, A. The discover of zinc fingers and their application in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef]
- Razin, S.V.; Borunova, V.V.; Maksimenko, O.G.; Kantidze, O.L. Cys2his2 zinc finger protein family: Classification, functions, and major members. Biochemistry 2011, 77, 217–226. [Google Scholar]
- Maret, W. Inhibitory zinc sites in enzymes. Biometals 2013, 2, 197–204. [Google Scholar] [CrossRef]
- Maret, W. Zinc coordination environments in proteins determine zinc functions. J. Trace Elem. Med. Biol. 2005, 19, 7–12. [Google Scholar] [CrossRef]
- Maret, W. New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 2012, 111, 110–116. [Google Scholar] [CrossRef]
- Maret, W.; Yetman, C.A.; Jiang, L.-J. Enzyme regulation by reversible zinc inhibition: Glycerol phosphate dehydrogenase as an example. Chem. Biol. Interact. 2001, 130–132, 891–901. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Lim, C. Physical basis of structural and catalytic Zn-binding sites in proteins. J. Mol. Biol. 2008, 379, 545–553. [Google Scholar]
- Krishna, S.S.; Majumdar, I.; Grishin, N.V. Structural classification of zinc fingers: Survey and summary. Nucleic Acids Res. 2003, 31, 532–550. [Google Scholar] [CrossRef]
- Lee, M.S.; Gippert, G.P.; Soman, K.V.; Case, D.A.; Wright, P.E. Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science 1989, 245, 635–637. [Google Scholar]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. Dna recognitions by cys2his2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 1999, 3, 183–212. [Google Scholar]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., II. ZFN, TALEN, ADN CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F., III. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5525–5530. [Google Scholar]
- Beerli, R.R.; Segal, D.J.; Birgit, D.; Barbas, C.F.I. Toward controlling gene expression at will: Specific regulation of the erB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. USA 1998, 95, 14628–14633. [Google Scholar]
- Beerli, R.R.; Dreier, B.; Barbas, C.F., III. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA 1999, 97, 1495–1500. [Google Scholar]
- Theorell, H.; McKinley McKee, J.S. Mechanism of action of liver alcohol dehydrogenase. Nature 1961, 192, 47–50. [Google Scholar] [CrossRef]
- Hoog, J.-O.; Ostberg, L.J. Mammalian alcohol dehydrogenases—A comparative investigation at gene and protein levels. Chem. Biol. Interact. 2011, 191, 2–7. [Google Scholar] [CrossRef]
- Klinman, J.P. Probes of mechanism and transition-state structure in the alcohol dehydrogenase reaction. Crit. Rev. Biochem. Mol. Biol. 1981, 10, 39–78. [Google Scholar] [CrossRef]
- Pettersson, G. Liver alcohol dehydrogenase. Crit. Rev. Biochem. Mol. Biol. 1987, 21, 349–388. [Google Scholar] [CrossRef]
- Sanghani, P.C.; Bosron, W.F.; Hurley, T.D. Human glutathione-dependent formaldehyde dehydrogenase. Structural changes associated with ternary complex formation. Biochemistry 2002, 41, 15189–15194. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Benkovic, S.J. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006, 75, 519–541. [Google Scholar] [CrossRef]
- Pauly, T.A.; Ekstrom, J.L.; Beebe, D.A.; Chrunyk, B.; Cunningham, D.; Griffor, M.; Kamath, A.; Lee, S.E.; Madura, R.; Mcguire, D.; et al. X-ray crystallographic and kinetic studies of human sorbitol dehydrogenase. Structure 2003, 11, 1072–1085. [Google Scholar]
- Carter, C.W., Jr. The nucleoside deaminases for cytidine and adenosine: Structure, transistion state stabilization, mechanism, and evolution. Biochimie 1995, 77, 92–98. [Google Scholar] [CrossRef]
- Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W., Jr. Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry 1995, 34, 4516–4523. [Google Scholar]
- Harjes, E.; Gross, P.J.; Chen, K.-M.; Lu, Y.; Shindo, K.; Nowarski, R.; Gross, J.D.; Kotler, M.; Harris, R.S.; Matsuo, H. An extended structure of the apobec3g catalytic domain suggests a unqiue holoenzyme model. J. Mol. Biol. 2009, 389, 819–832. [Google Scholar] [CrossRef]
- Auerbach, G.; Herrmann, A.; Bracher, A.; Bader, G.; Gutlich, M.; Fischer, M.; Neikamm, M.; Garrido-Franco, M.; Richarson, J.; Nar, H.; et al. Zinc plays a key role in human and bacterial gtp cyclohydrolase I. Proc. Natl. Acad. Sci. USA 2000, 97, 13567–13572. [Google Scholar] [CrossRef]
- Evans, J.C.; Huddler, D.P.; Jiracek, J.; Castro, C.; Millian, N.S.; Garrow, T.A.; Ludwig, M.L. Betain-homocysteine methyltransferase: Zinc in a distored barrel. Structure 2002, 10, 1159–1171. [Google Scholar] [CrossRef]
- Long, S.B.; Casey, P.J.; Beese, L.S. Reaction path of protein farnesyltransferase at atomic resolution. Nature 2002, 419, 645–650. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Unraveling the mechanism of the farnesyltransferase enzyme. J. Biol. Inorg. Chem. 2004, 10, 3–10. [Google Scholar]
- Long, S.B.; Hancock, P.J.; Kral, A.M.; Hellinga, H.W.; Beese, L.S. The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics. Proc. Natl. Acad. Sci. USA 2001, 98, 12948–12953. [Google Scholar]
- Zhang, F.L.; Casey, P.J. Protein prenylation: Molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996, 65, 241–269. [Google Scholar] [CrossRef]
- Ashar, H.R.; James, L.; Gray, K.; Carr, D.; Black, S.; Armstrong, L.; Bishop, W.R.; Kirschmeier, P. Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of the CENP-E with microtubules. J. Biol. Chem. 2000, 275, 30451–30457. [Google Scholar]
- Zverina, E.A.; Lamphear, C.L.; Wright, E.N.; Fierke, C.A. Recent advances in protein prenyltransferases: Substrate identification, regulation, and disease interventions. Curr. Opin. Chem. Biol. 2012, 16, 544–552. [Google Scholar] [CrossRef]
- Knipp, M.; Charnock, J.M.; Garner, C.D.; Vasak, M. Structural and functional characterization of the Zn(II) site in dimethylargininase-1 (DDAH-1) from bovine brain. J. Biol. Chem. 2001, 276, 40449–40456. [Google Scholar]
- Frey, D.; Braun, O.; Briand, C.; Vasak, M.; Grutter, M.G. Structure of the mammalian NOS regulator dimethylarginine dimethylaminohydrolase: A basis for the design of specific inhibitors. Structure 2006, 14, 901–911. [Google Scholar] [CrossRef]
- Lee, S.; Shen, W.-H.; Miller, A.; Kuo, L.C. Zn2+ regulation of ornithine transcarbamoylase. J. Mol. Biol. 1990, 211, 255–269. [Google Scholar] [CrossRef]
- Shi, D.; Morizono, H.; Tong, L.; Allewell, N.M.; Tuchman, M. Human ornithine transcarbamylase: Crystallographic insights into substrate recognition and conformational changes. Biochem. J. 2001, 354, 501–509. [Google Scholar]
- Tully, D.C.; Liu, H.; Chatterjee, A.K.; Alper, P.B.; Epple, R.; Williams, J.A.; Roberts, M.J.; Woodmansee, D.H.; Masick, B.T.; Tumanut, C.; et al. Synthesis and SAR of arylaminoethyl amides as noncovalent inhibitors of cathepsin S: P3 cyclic ethers. Bioorg. Med. Chem. Lett. 2006, 16, 5112–5117. [Google Scholar] [CrossRef]
- Perry, D.K.; Smyth, M.J.; Stennicke, H.R.; Salvesen, G.S.; Duriez, P.; Poirier, G.G.; Hannun, Y.A. Zinc is a potent inhibitor of the apoptotic protease, caspase-3: A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem. 1997, 272, 18530–18533. [Google Scholar]
- Peterson, Q.P.; Goode, D.R.; West, D.C.; Ramsey, K.N.; Lee, J.J.Y.; Hergenrother, P.J. Pac-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J. Mol. Biol. 2009, 388, 144–158. [Google Scholar] [CrossRef]
- Velazquez-Delgado, E.M.; Hardy, J.A. Zinc-mediated allosteric inhibition of caspase-6. J. Biol. Chem. 2012, 287, 36000–36011. [Google Scholar] [CrossRef]
- Huber, K.L.; Hardy, J.A. Mechanism of zinc-mediated inhibition of caspase-9. Protein Sci. 2012, 21, 1056–1065. [Google Scholar] [CrossRef]
- Costello, L.C.; Liu, Y.; Franklin, R.B.; Kennedy, M.C. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J. Biol. Chem. 1997, 272, 28875–28881. [Google Scholar]
- Pace, N.J.; Weerapana, E. A competitive chemical-proteomic platform to identify zinc-binding cysteines. ACS Chem. Biol. 2014, 9, 258–265. [Google Scholar] [CrossRef]
- Korichneva, I.; Hoyos, B.; Chua, R.; Levi, E.; Hammerling, U. Zinc release from protein kinase c as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 2002, 277, 44327–44331. [Google Scholar]
- Leonard, T.A.; Rozycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystal structure and allosteric activation of protein kinase C bii. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef]
- Fischmann, T.O.; Hruza, A.; da Niu, X.; Fossetta, J.D.; Lunn, C.A.; Dolphin, E.; Prongay, A.J.; Reichert, P.; Lundell, D.J.; Narula, S.K.; et al. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nat. Struct. Biol. 1999, 6, 233–242. [Google Scholar] [CrossRef]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; O’Connell, M.P.; Ultsch, M.H.; Hurst, A.; Totpal, K.; Ashkenazi, A.; de Vos, A.M.; Kelley, R.F. A unique zinc-binding site revealed by a high-resolution X-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 2000, 39, 633–640. [Google Scholar] [CrossRef]
- MacAllister, R.J.; Parry, H.; Kimoto, M.; Ogawa, T.; Russell, R.J.; Hodson, H.; Whitley, G.S.J.; Vallance, P. Regulation of nitric oxide by dimethylarginine dimethylaminohydrolase. Br. J. Pharmacol. 1996, 119, 1533–1540. [Google Scholar] [CrossRef]
- Wang, Y.; Monzingo, A.F.; Hu, S.; Schaller, T.H.; Robertus, J.D.; Fast, W. Developing dual and specific inhibitors of dimethylarginine dimethylaminohydrolase-1 and nitric oxide synthase: Toward a targeted polypharmacology to control nitric oxide. Biochemistry 2009, 48, 8624–8635. [Google Scholar]
- Thornberry, N.A. The caspase family of cysteine proteases. Br. Med. Bull. 1997, 53, 478–490. [Google Scholar]
- Zalewski, P.D.; Forbes, I.J.; Betts, W.H. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II). Biochem. J. 1993, 296, 403–408. [Google Scholar]
- Paulsen, C.E.; Carroll, K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Pajares, M.A.; Perez-Sala, D. Betaine homocysteine S-methyltransferase: Just a regulator of homocysteine metabolism? Cell. Mol. Life Sci. 2006, 63, 2792–2803. [Google Scholar] [CrossRef]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1999, 1411, 217–230. [Google Scholar]
- Irving, H.; Williams, R.J.P. Order of stability of metal complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Krezel, A.; Maret, W. Dual nanomolar and picomolar Zn(II) binding properties of metallothionein. J. Am. Chem. Soc. 2007, 129, 10911–10921. [Google Scholar] [CrossRef]
- Krezel, A.; Maret, W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 2006, 11, 1049–1062. [Google Scholar] [CrossRef]
- Romero-Isart, N.; Vasak, M. Advances in the structure and chemistry of metallothioneins. J. Inorg. Biochem. 2002, 88, 388–396. [Google Scholar] [CrossRef]
- Maret, W.; Larsen, K.S.; Vallee, B.L. Coordination dynamics of biological zinc “Clusters” in metallothioneins and in the DNA-binding domain of transcription factor Gal4. Proc. Natl. Acad. Sci. USA 1997, 94, 2233–2237. [Google Scholar] [CrossRef]
- Heinz, U.; Kiefer, M.; Tholey, A.; Adolph, H.-W. On the competition for available zinc. J. Biol. Chem. 2005, 280, 3197–3207. [Google Scholar]
- Babula, P.; Masarik, M.; Adam, V.; Eckschlager, T.; Stiborova, M.; Trnkova, L.; Skutkova, H.; Provaznik, I.; Hubalek, J.; Kizek, R. Mammalian metallothioneins: Properties and functions. Metallomics 2012, 4, 739–750. [Google Scholar] [CrossRef]
- Maret, W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef]
- Bertini, I.; Decaria, L.; Rosato, A. The annotation of full zinc proteomes. J. Biol. Inorg. Chem. 2010, 15, 1071–1078. [Google Scholar] [CrossRef]
- Kornhaber, G.J.; Snyder, D.; Moseley, H.N.; Montelione, G.T. Identification of zinc-ligated cysteine residues based on 13Cα and 13Cβ chemical shift data. J. Biomol. NMR 2006, 34, 259–269. [Google Scholar] [CrossRef]
- Qian, Y.; Martell, J.; Pace, N.J.; Ballard, T.E.; Johnson, D.S.; Weerapana, E. An isotopically tagged azobenzene-based cleavable linker for quantitative proteomics. ChemBioChem 2013, 14, 1410–1414. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pace, N.J.; Weerapana, E. Zinc-Binding Cysteines: Diverse Functions and Structural Motifs. Biomolecules 2014, 4, 419-434. https://doi.org/10.3390/biom4020419
Pace NJ, Weerapana E. Zinc-Binding Cysteines: Diverse Functions and Structural Motifs. Biomolecules. 2014; 4(2):419-434. https://doi.org/10.3390/biom4020419
Chicago/Turabian StylePace, Nicholas J., and Eranthie Weerapana. 2014. "Zinc-Binding Cysteines: Diverse Functions and Structural Motifs" Biomolecules 4, no. 2: 419-434. https://doi.org/10.3390/biom4020419
APA StylePace, N. J., & Weerapana, E. (2014). Zinc-Binding Cysteines: Diverse Functions and Structural Motifs. Biomolecules, 4(2), 419-434. https://doi.org/10.3390/biom4020419