Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cotton Materials and In Vitro Ovule Culture
2.2. Lipid Extraction
2.3. Lipidomics
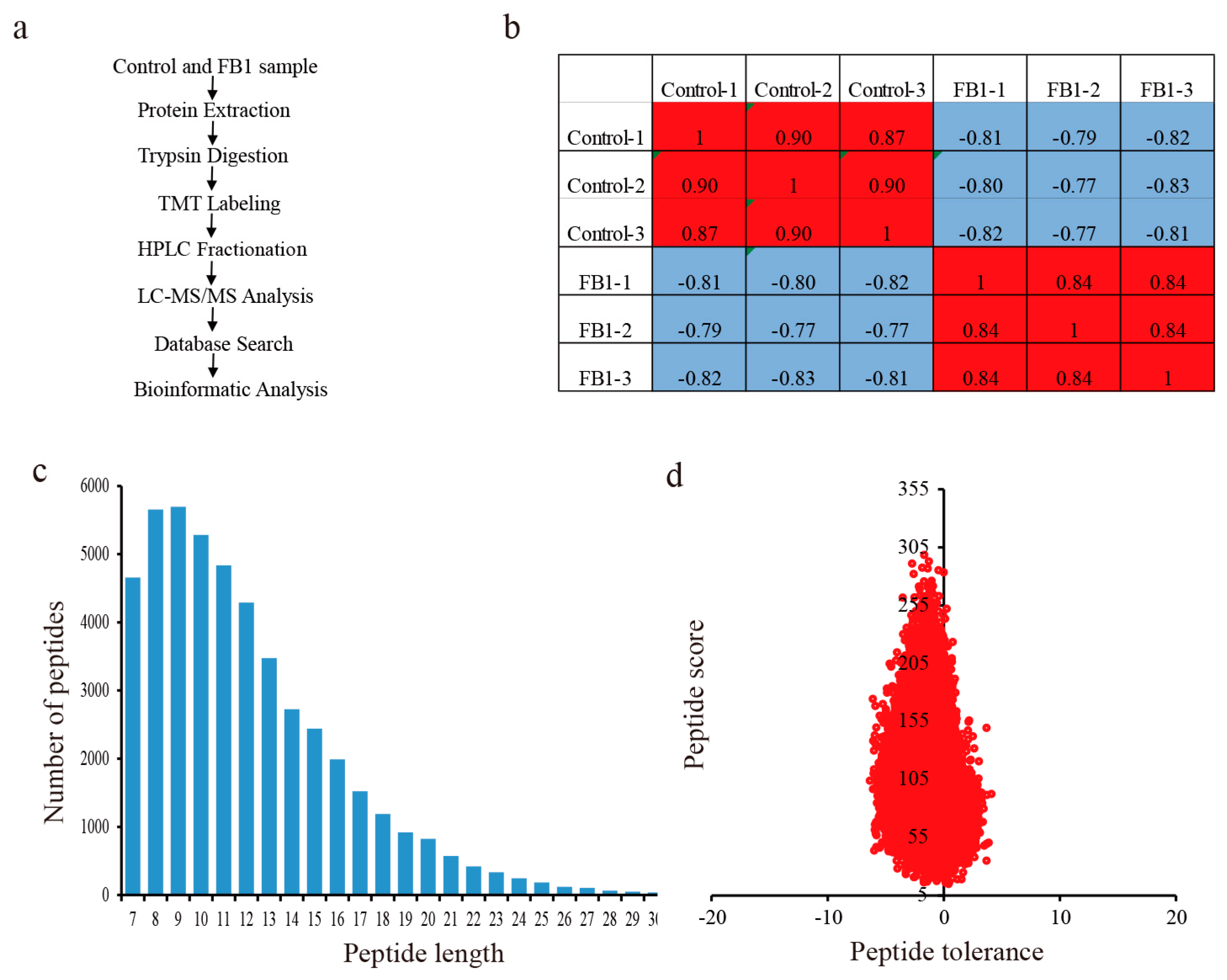
2.4. Protein Extraction and Digestion by Trypsin
2.5. Tandem Mass Tag (TMT) Labeling, HPLC Fractionation, and LC-MS/MS Analysis
2.6. Database Search and Bioinformatic Methods
2.7. RNA Extraction and Semi-Quantitative PCR
2.8. Enzyme Activity Determination
3. Results
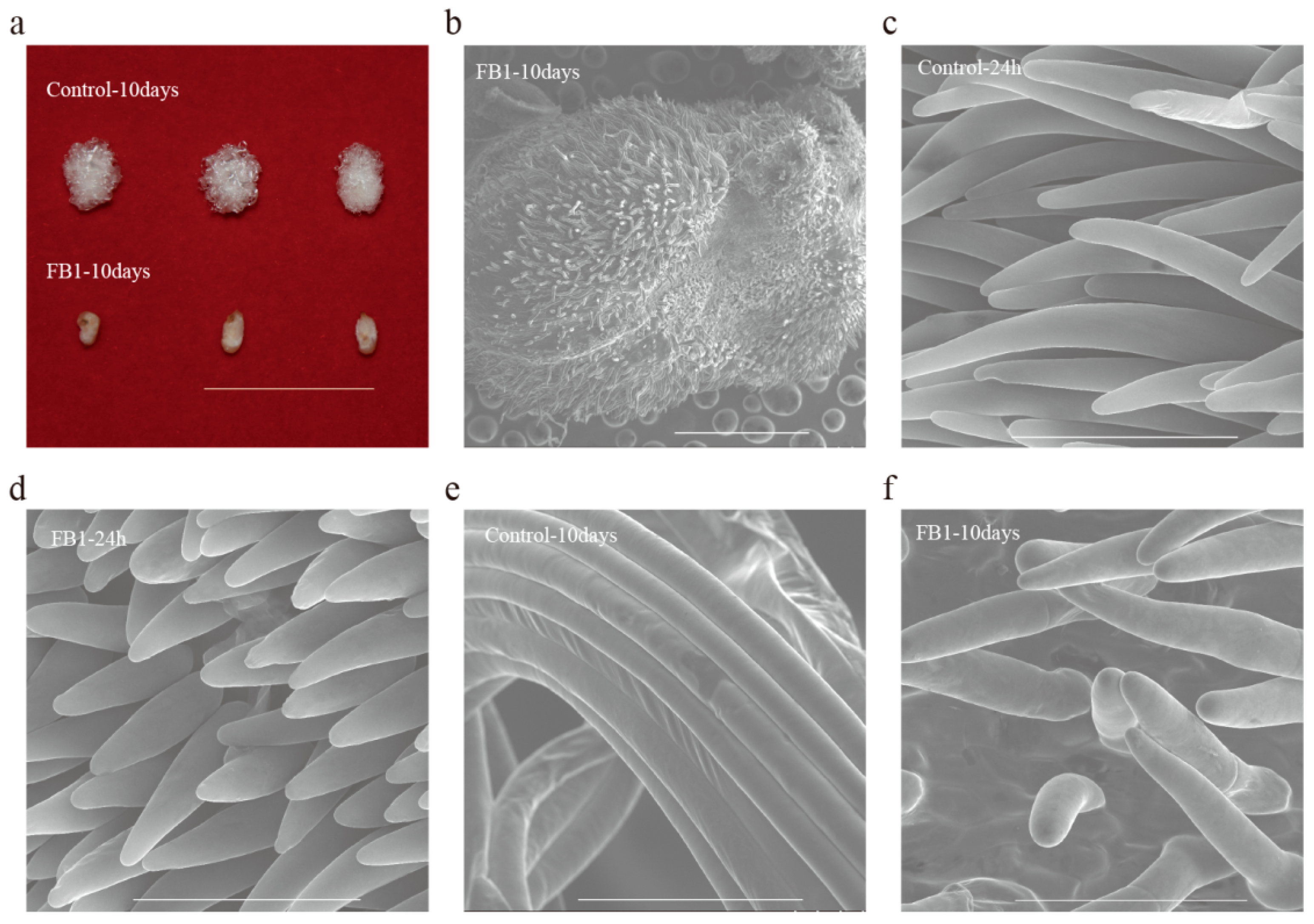
3.1. FB1 Blocked the Elongation of Cotton Fibers
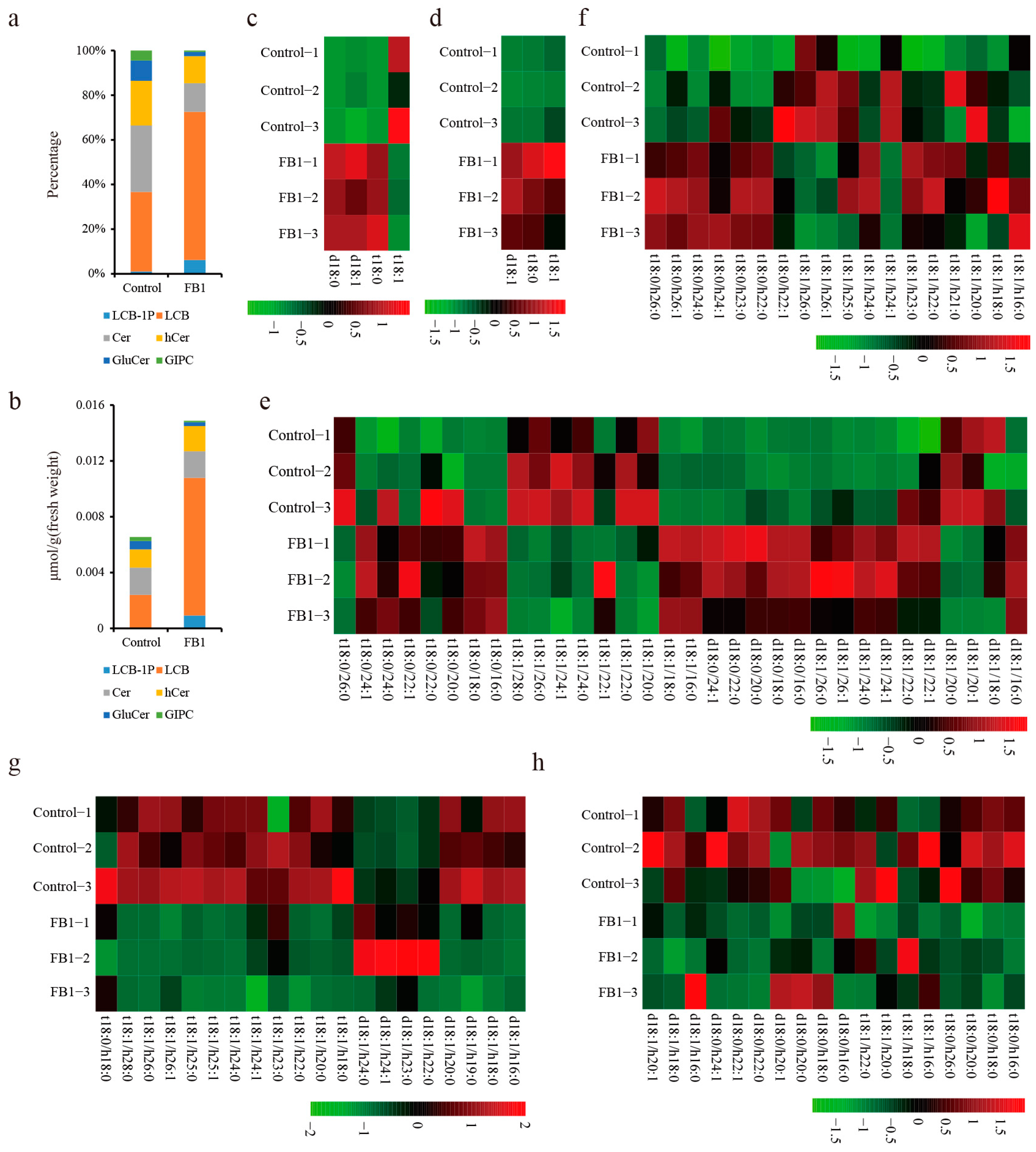
3.2. Sphingolipid Homeostasis Was Disrupted by FB1 in Cotton Fibers and Ovules
3.3. Quantitative Proteome Analysis and the Impacts of FB1 on the Global Proteome of Cotton Fibers and Ovules
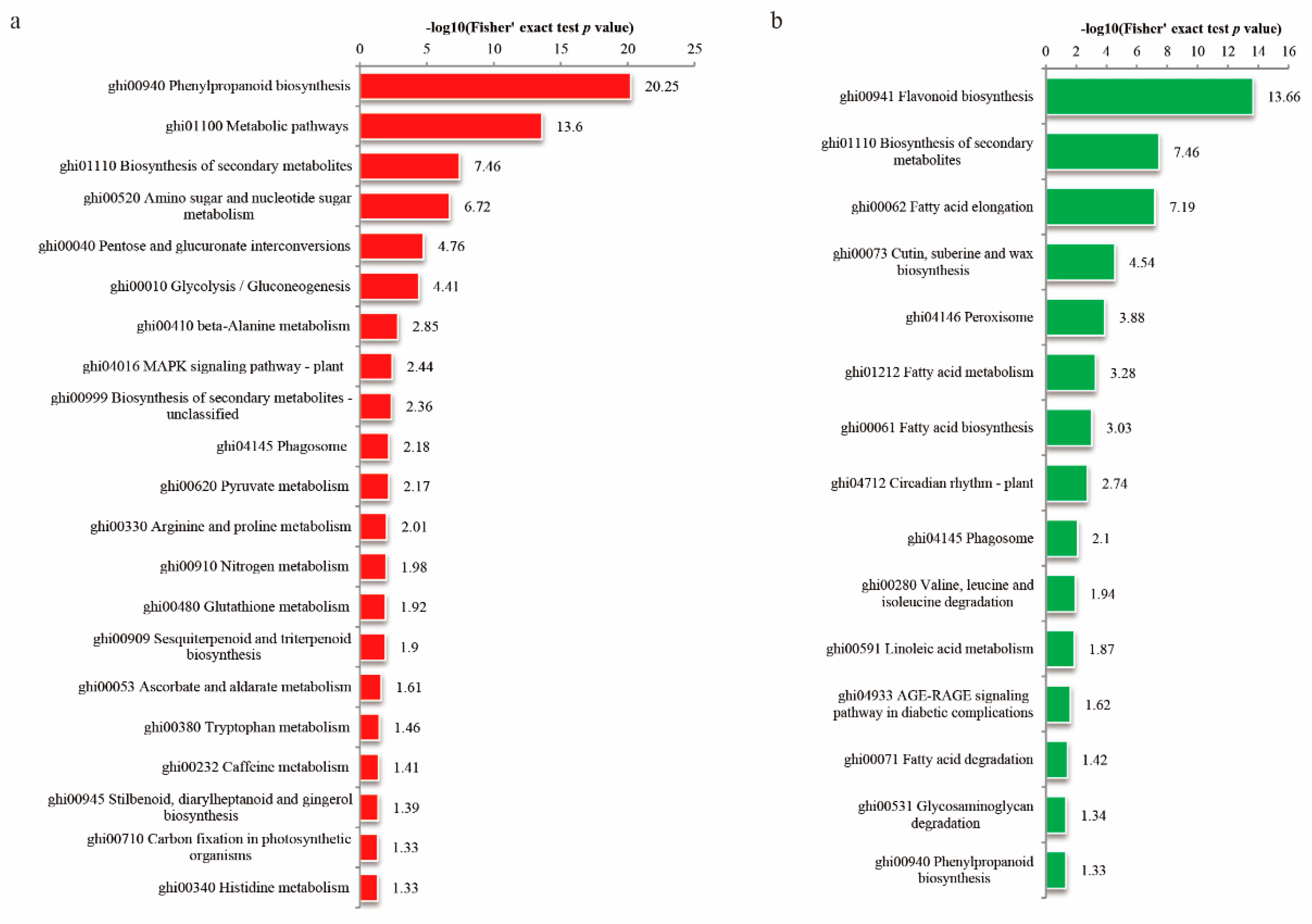
3.4. Enrichment Analysis of the DEPs in Cotton Fiber and Ovule under FB1
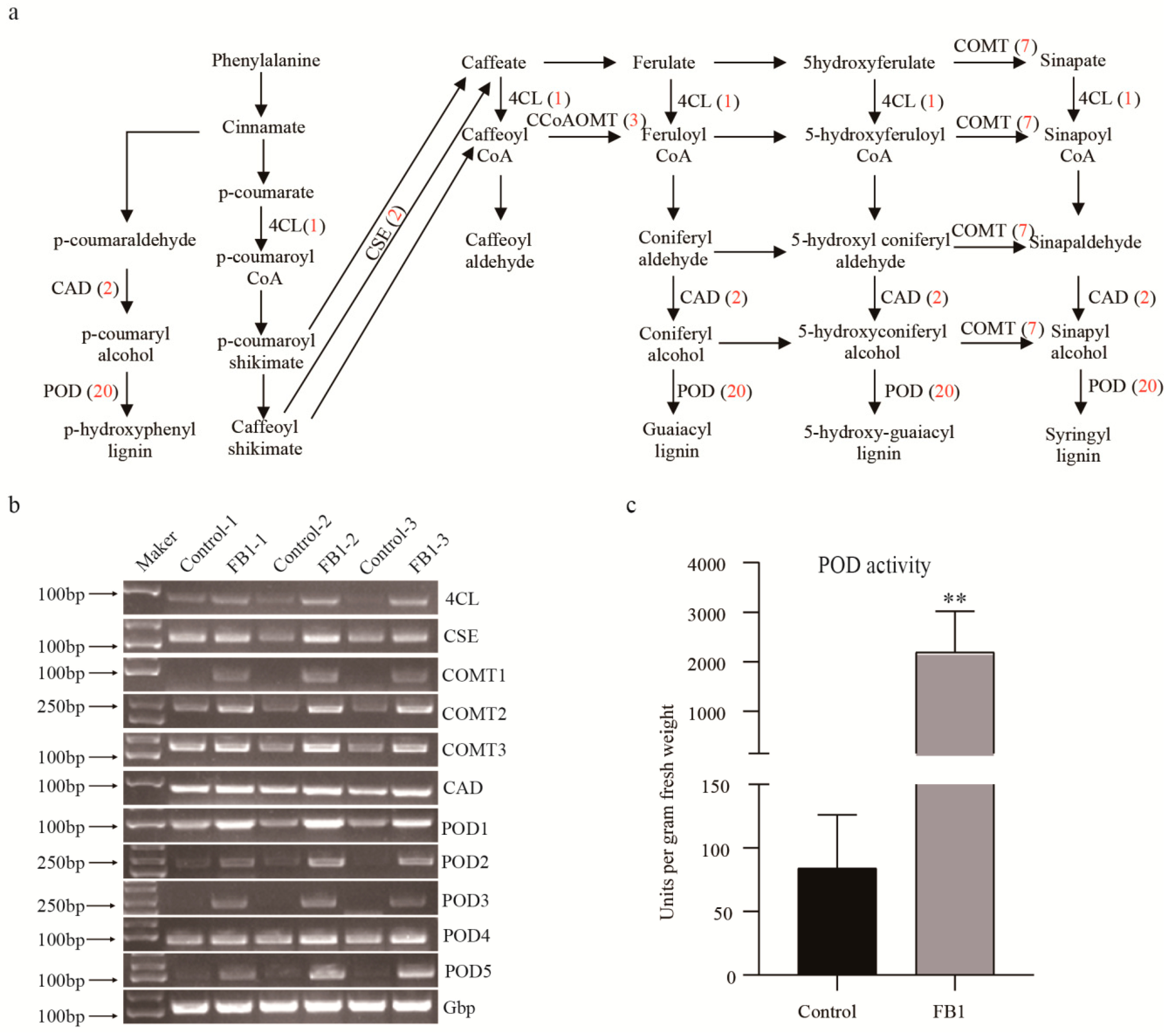
3.5. FB1 Altered the Phenylpropanoid Biosynthesis Pathway in Cotton Fibers and Ovules
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant. Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef]
- Zhao, B.; Cao, J.-F.; Hu, G.; Chen, Z.; Wang, L.-Y.; Shangguan, X.-X.; Wang, L.-J.; Mao, Y.-B.; Zhang, T.-Z.; Wendel, J.F.; et al. Core cis-element variation confers subgenome-biased expression of a transcription factor that functions in cotton fiber elongation. New Phytol. 2018, 218, 1061–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Gomez, M.C.; Runavot, J.-L.; Meulewaeter, F.; Knox, P. Developmental features of cotton fibre middle lamellae in relation to cell adhesion and cell detachment in cultivars with distinct fibre qualities. BMC Plant Boil. 2017, 17, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Suo, X.; Li, F.; Bao, C.; He, S.; Huang, L.; Luo, M. Membrane lipid raft organization during cotton fiber development. J. Cotton Res. 2020, 3, 1–9. [Google Scholar] [CrossRef]
- Chao, D.-Y.; Gable, K.; Chen, M.; Baxter, I.; Dietrich, C.R.; Cahoon, E.B.; Guerinot, M.L.; Lahner, B.; Lü, S.; Markham, J.E.; et al. Sphingolipids in the root play an important role in regulating the leaf ionome in Arabidopsis thaliana. Plant Cell 2011, 23, 1061–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, P.; Franke, S.; Lüthje, S.; Heinz, E. Are glucocerebrosides the predominant sphingolipids in plant plasma membranes? Plant Physiol. Biochem. 2005, 43, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Tjellström, H.; Hellgren, L.I.; Wieslander, Å.; Sandelius, A.S. Lipid asymmetry in plant plasma membranes: Phosphate deficiency-induced phospholipid replacement is restricted to the cytosolic leaflet. FASEB J. 2009, 24, 1128–1138. [Google Scholar] [CrossRef]
- Niu, Q.; Tan, K.; Zang, Z.; Xiao, Z.; Chen, K.; Hu, M.; Luo, M. Modification of phytosterol composition influences cotton fiber cell elongation and secondary cell wall deposition. BMC Plant Boil. 2019, 19, 208. [Google Scholar] [CrossRef]
- Heaver, S.L.; Johnson, E.L.; Ley, R. Sphingolipids in host–microbial interactions. Curr. Opin. Microbiol. 2018, 43, 92–99. [Google Scholar] [CrossRef]
- Marquês, J.T.; Marinho, H.S.; De Almeida, R.F.M.; De Almeida, R.F.M. Sphingolipid hydroxylation in mammals, yeast and plants—An integrated view. Prog. Lipid Res. 2018, 71, 18–42. [Google Scholar] [CrossRef]
- Liang, H.; Yao, N.; Song, J.T.; Luo, S.; Lü, H.; Greenberg, J.T. Ceramides modulate programmed cell death in plants. Genes Dev. 2003, 17, 2636–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Bielawski, J.; Mu, J.; Dong, H.; Teng, C.; Zhang, J.; Yang, X.; Tomishige, N.; Hanada, K.; A Hannun, Y.; et al. Involvement of sphingoid bases in mediating reactive oxygen intermediate production and programmed cell death in Arabidopsis. Cell Res. 2007, 17, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Ormancey, M.; Thuleau, P.; Van Der Hoorn, R.A.L.; Grat, S.; Testard, A.; Kamal, K.Y.; Boudsocq, M.; Cotelle, V.; Mazars, C. Sphingolipid-induced cell death in Arabidopsis is negatively regulated by the papain-like cysteine protease RD21. Plant Sci. 2019, 280, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-M.; Yang, X.; Tangchaiburana, S.; Ndeh, R.; Markham, J.E.; Tsegaye, Y.; Dunn, T.M.; Wang, G.-L.; Bellizzi, M.; Parsons, J.F.; et al. An inositolphosphorylceramide synthase is involved in regulation of plant programmed cell death associated with defense in Arabidopsis. Plant Cell 2008, 20, 3163–3179. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.; Zhang, L.; Zhang, Z.; Dong, S.; Li, J.; Wang, Y.; Zheng, X.-B. The LCB2subunit of the sphingolip biosynthesis enzyme serine palmitoyltransferase can function as an attenuator of the hypersensitive response and Bax-induced cell death. New Phytol. 2008, 181, 127–146. [Google Scholar] [CrossRef]
- Coursol, S.; Fan, L.-M.; Le Stunff, H.; Spiegel, S.; Gilroy, S.; Assmann, S.M. Sphingolipid signalling in Arabidopsis guard cells involves heterotrimeric G proteins. Nature 2003, 423, 651–654. [Google Scholar] [CrossRef]
- Ng, C.K.-Y.; Carr, K.; McAinsh, M.R.; Powell, B.; Hetherington, A.M. Drought-induced guard cell signal transduction involves sphingosine-1-phosphate. Nature 2001, 410, 596–599. [Google Scholar] [CrossRef]
- Worrall, D.; Liang, Y.-K.; Alvarez, S.; Holroyd, G.H.; Spiegel, S.; Panagopulos, M.; Gray, J.E.; Hetherington, A.M. Involvement of sphingosine kinase in plant cell signalling. Plant J. 2008, 56, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Peer, M.; Stegmann, M.; Mueller, M.J.; Waller, F. Pseudomonas syringae infection triggers de novo synthesis of phytosphingosine from sphinganine in Arabidopsis thaliana. FEBS Lett. 2010, 584, 4053–4056. [Google Scholar] [CrossRef] [Green Version]
- Dutilleul, C.; Benhassaine-Kesri, G.; Demandre, C.; Rézé, N.; Launay, A.; Pelletier, S.; Renou, J.-P.; Zachowski, A.; Baudouin, E.; Guillas, I. Phytosphingosine-phosphate is a signal for AtMPK6 activation and Arabidopsis response to chilling. New Phytol. 2012, 194, 181–191. [Google Scholar] [CrossRef]
- Cantrel, C.; Vazquez, T.; Puyaubert, J.; Rézé, N.; Lesch, M.; Kaiser, W.M.; Dutilleul, C.; Guillas, I.; Zachowski, A.; Baudouin, E. Nitric oxide participates in cold-responsive phosphosphingolipid formation and gene expression in Arabidopsis thaliana. New Phytol. 2010, 189, 415–427. [Google Scholar] [CrossRef]
- Krüger, F.; Krebs, M.; Viotti, C.; Langhans, M.; Schumacher, K.; Robinson, D.G. PDMP induces rapid changes in vacuole morphology in Arabidopsis root cells. J. Exp. Bot. 2012, 64, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-X.; Li, J.; Liu, Z.; Yin, J.; Chang, Z.-Y.; Rong, C.; Wu, J.-L.; Bi, F.-C.; Yao, N. The Arabidopsis ceramidase AtACER functions in disease resistance and salt tolerance. Plant J. 2015, 81, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Abbas, H.K.; Tanaka, T.; Duke, S.O.; Porter, J.K.; Wray, E.M.; Hodges, L.; Sessions, A.E.; Wang, E.; Merrill, A.H.; Riley, R.T.; et al. Fumonisin- and AAL-toxin-induced disruption of sphingolipid metabolism with accumulation of free sphingoid bases. Plant Physiol. 1994, 106, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttgeharm, K.D.; Cahoon, E.B.; Markham, J.E. Substrate specificity, kinetic properties and inhibition by fumonisin B1 of ceramide synthase isoforms from Arabidopsis. Biochem. J. 2016, 473, 593–603. [Google Scholar] [CrossRef]
- Stone, J.M.; Heard, J.E.; Asai, T.; Ausubel, F.M. Simulation of fungal-mediated cell death by fumonisin B1 and selection of fumonisin B1-resistant (fbr) Arabidopsis mutants. Plant Cell 2000, 12, 1811. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Liang, X.; Nekl, E.R.; Stiers, J.J. Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to fumonisin B1. Plant J. 2005, 41, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Beasley, C.A.; Ting, I.P.J.A.j.b. The effects of plant growth substances on in vitro fiber development from fertilized cotton ovules. Am. J. Bot. 1973, 60, 130–139. [Google Scholar] [CrossRef]
- Welti, R. Profiling Membrane Lipids in Plant Stress Responses. ROLE OF PHOSPHOLIPASE Dalpha IN FREEZING-INDUCED LIPID CHANGES IN ARABIDOPSIS. J. Boil. Chem. 2002, 277, 31994–32002. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.Q.; Sun, Y.B.; Ma, Z.M.; Ke, M.Y.; Cui, Y.; Chen, Z.C.; Chen, C.F.; Ji, C.Y.; Tran, T.M.; Yang, L.; et al. Salicylic acid-mediated plasmodesmal closure via Remorin-dependent lipid organization. Proc. Natl. Acad. Sci. USA 2020, 117, 8659. [Google Scholar] [CrossRef] [Green Version]
- Markham, J.E.; Jaworski, J.G. Rapid measurement of sphingolipids from Arabidopsis thaliana by reversed-phase high-performance liquid chromatography coupled to electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; A Dennis, E.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.; Wang, Z.; Wei, C.; Amo, A.; Ahmad, B.; Yang, X.; Zhang, X. Phenylpropanoid pathway engineering: An emerging approach towards plant defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sã¡nchez-Rangel, D.; Vicente, M.R.-S.; De La Torre-Hernã¡ndez, M.E.; Nã¡jera-Martã-nez, M.; Plasencia, J.; Nájera-Martínez, M. Deciphering the link between salicylic acid signaling and sphingolipid metabolism. Front. Plant Sci. 2015, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.-M.; Hu, C.-Y.; Pang, Y.; Kastaniotis, A.J.; Hiltunen, J.K.; Zhu, Y. Saturated very-long-chain fatty acids promote cotton fiber and Arabidopsis cell elongation by activating ethylene biosynthesis. Plant Cell 2007, 19, 3692–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, C.-Y.; Wang, H.; Pang, Y.; Xu, C.; Jiao, Y.; Qin, Y.-M.; Western, T.L.; Yu, S.; Zhu, Y. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol. Cell. Proteom. 2010, 9, 2019–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Wei, H.; Sun, H.; Hao, P.; Wang, H.; Su, J.; Yu, S. Proteomic analysis of differences in fiber development between wild and cultivated Gossypium hirsutum L. J. Proteome Res. 2017, 16, 2811–2824. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hu, W.; Li, B.; Yang, Y.; Zhang, Y.; Thow, K.; Fan, L.; Qu, Y. Proteomic profiling of cotton fiber developmental transition from cell elongation to secondary wall deposition. Acta Biochim. Biophys. Sin. (Shanghai) 2019, 51, 1168–1177. [Google Scholar] [CrossRef]
- Tan, J.; Tu, L.; Deng, F.; Hu, H.; Nie, Y.; Zhang, X. A genetic and metabolic analysis revealed that cotton fiber cell development was retarded by flavonoid naringenin. Plant Physiol. 2013, 162, 86–95. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Liu, C.; Liu, Y.; Luo, M. Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics. Biomolecules 2020, 10, 1258. https://doi.org/10.3390/biom10091258
Wang L, Liu C, Liu Y, Luo M. Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics. Biomolecules. 2020; 10(9):1258. https://doi.org/10.3390/biom10091258
Chicago/Turabian StyleWang, Li, Chen Liu, Yujie Liu, and Ming Luo. 2020. "Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics" Biomolecules 10, no. 9: 1258. https://doi.org/10.3390/biom10091258
APA StyleWang, L., Liu, C., Liu, Y., & Luo, M. (2020). Fumonisin B1-Induced Changes in Cotton Fiber Elongation Revealed by Sphingolipidomics and Proteomics. Biomolecules, 10(9), 1258. https://doi.org/10.3390/biom10091258