New Actors Driving the Epithelial–Mesenchymal Transition in Cancer: The Role of Leptin

 , , ,
, , ,  ,
,  ,
, 
Abstract
:1. Introduction
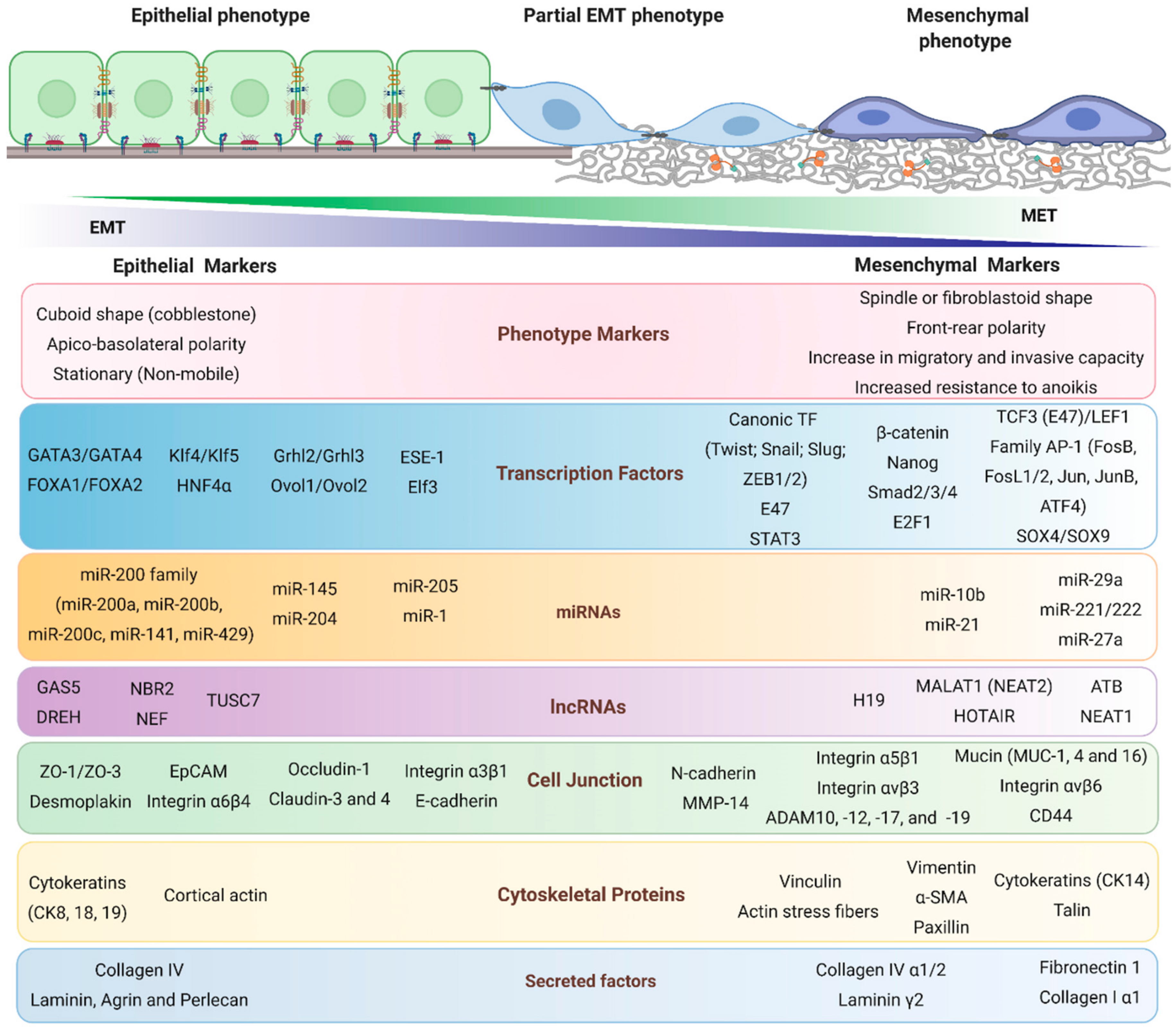
2. Epithelial–Mesenchymal Transition
2.1. Epithelial Markers
2.2. Mesenchymal Markers
3. Extracellular Signals Driving EMT
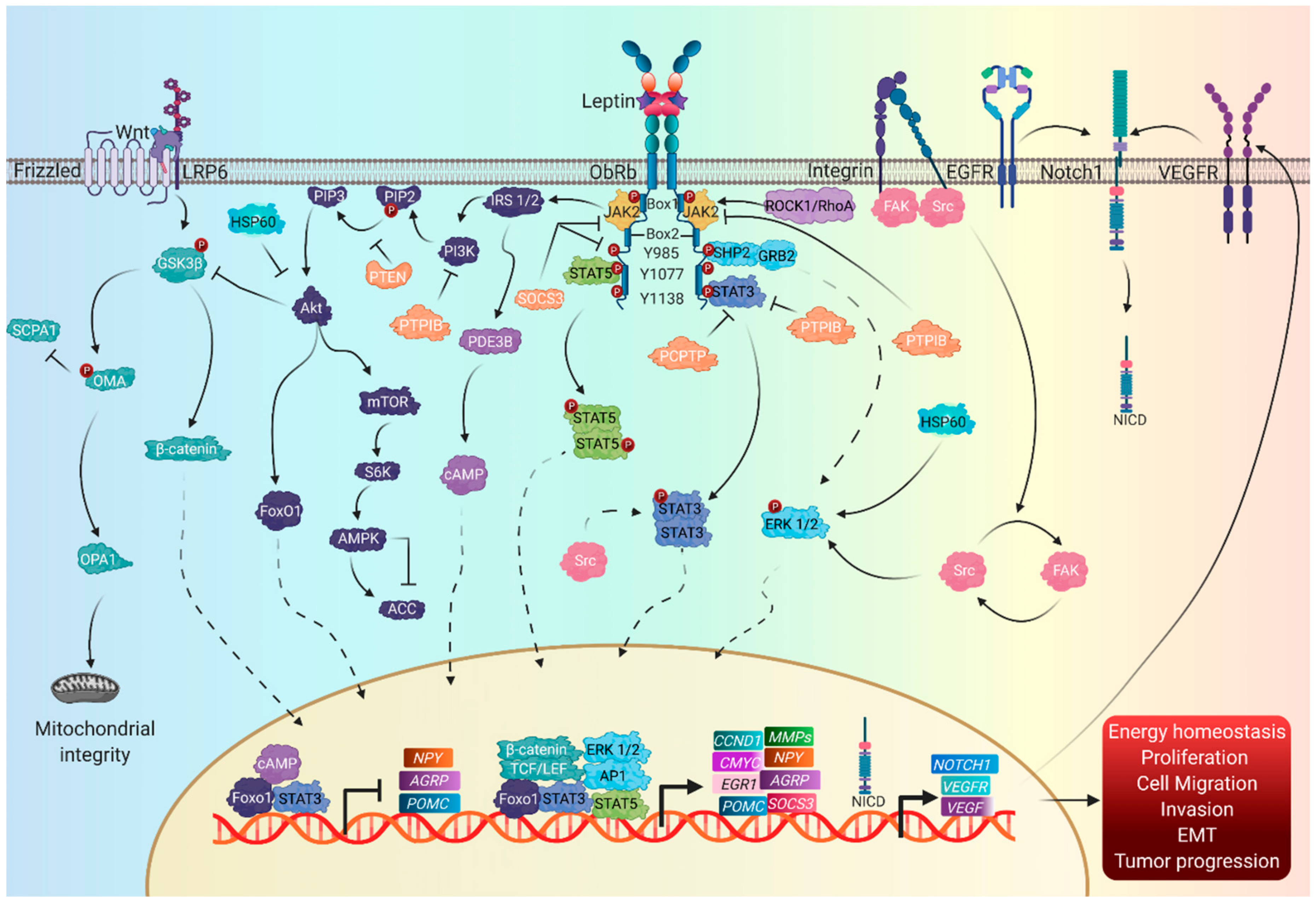
4. Leptin
4.1. Signaling Pathways
4.2. Canonical Leptin Pathways
4.3. Non-Canonical Leptin Signaling Pathways
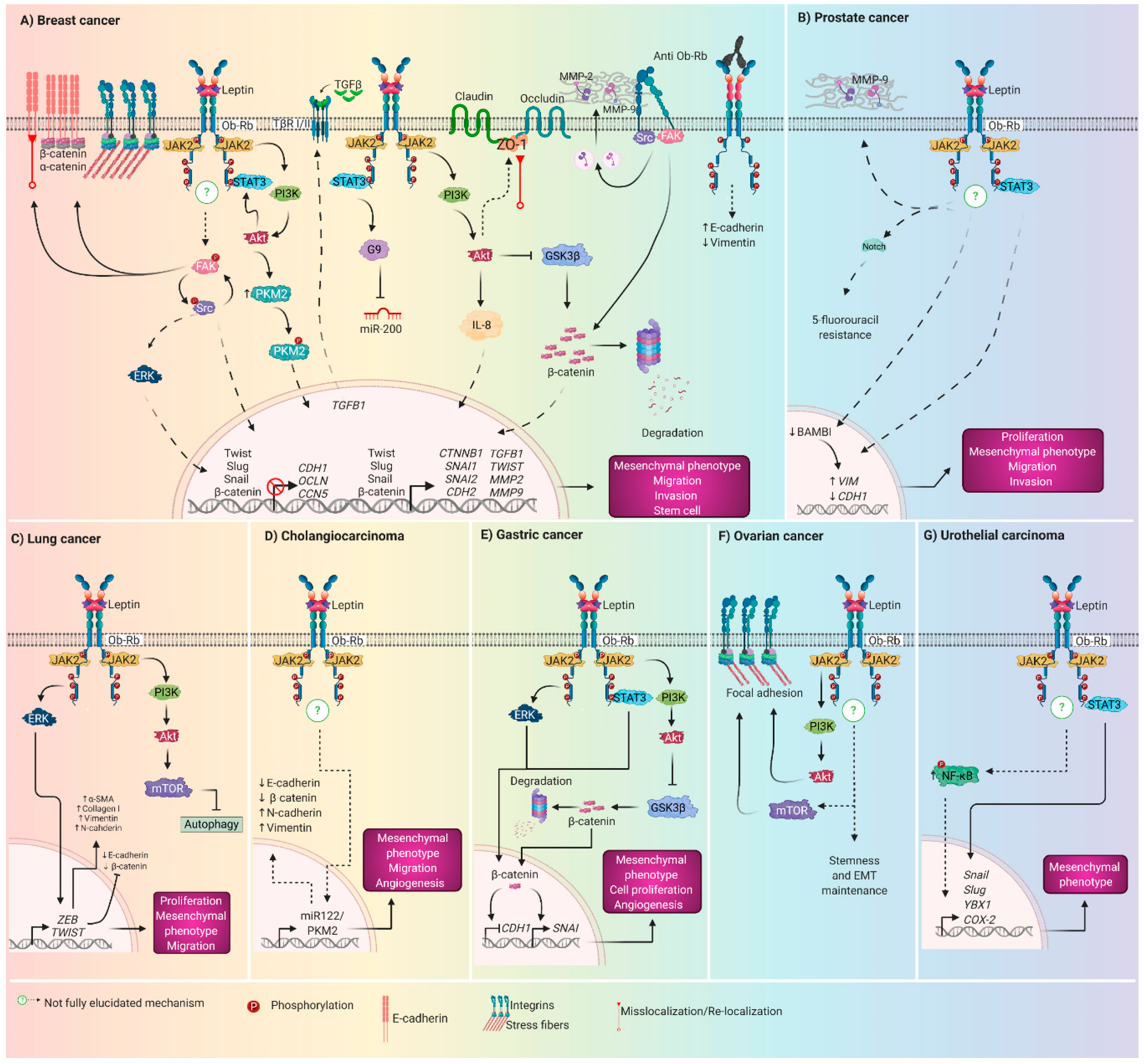
5. Role of Leptin Driving EMT in Breast cancer
5.1. In Vitro Assays
5.2. In Vivo Assays
5.3. Patient Samples
6. Role of Leptin Driving EMT in Gastric Cancer
6.1. In Vitro Assays
6.2. In Vivo Assays
6.3. Patients
7. Role of Leptin Driving EMT in Prostate Cancer
8. Role of Leptin Driving EMT in Lung Cancer
9. Other Cancers
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Cells Tissues Organs. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Kim, D.; Xing, T.; Yang, Z.; Dudek, R.; Lu, Q.; Chen, Y.-H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J. Clin. Med. 2017, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Rocha, M.R.; Barcellos-de-Souza, P.; Sousa-Squiavinato, A.C.M.; Fernandes, P.V.; de Oliveira, I.M.; Boroni, M.; Morgado-Diaz, J.A. Annexin A2 overexpression associates with colorectal cancer invasiveness and TGF-ß induced epithelial mesenchymal transition via Src/ANXA2/STAT3. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Song, L.; Zhang, Z.; Bai, X.X.; Cui, M.F.; Ma, L.J. MicroRNA-21 promotes TGF-β1-induced epithelial-mesenchymal transition in gastric cancer through up-regulating PTEN expression. Oncotarget 2016, 7, 66989–67003. [Google Scholar] [CrossRef] [Green Version]
- Brandi, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. Ikkα controls canonical TGFβ-SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in Panc1 cells. J. Cell Sci. 2013, 126, 2747. [Google Scholar] [CrossRef] [Green Version]
- Montorfano, I.; Becerra, A.; Cerro, R.; Echeverría, C.; Sáez, E.; Morales, M.G.; Fernández, R.; Cabello-Verrugio, C.; Simon, F. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-b1 and TGF-b2-dependent pathway. Lab. Investig. 2014, 94, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Gunaratne, A.; Chan, E.; El-Chabib, T.H.; Carter, D.; di Guglielmo, G.M. aPKC alters the TGFβ response in NSCLC cells through both Smad-dependent and Smad-independent pathways. J. Cell Sci. 2015, 128, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Grabowska, A.M.; Clarke, P.A.; Whelband, E.; Robinson, K.; Argent, R.H.; Tobias, A.; Kumari, R.; Atherton, J.C.; Watson, S.A. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 2010, 59, 1037–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, K.; Aoki, M.; Kannagi, R. Transcription factors c-Myc and CDX2 mediate E-selectin ligand expression in colon cancer cells undergoing EGF/bFGF-induced epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 7776–7781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.C.; Chen, X.H.; Song, H.X.; Wang, H.S.; Zhang, G.; Wang, H.; Chen, D.Y.; Fang, R.; Liu, H.; Cai, S.H.; et al. Snail regulated by PKC/GSK-3β pathway is crucial for EGF-induced epithelial-mesenchymal transition (EMT) of cancer cells. Cell Tissue Res. 2014, 358, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Buonato, J.M.; Lan, I.S.; Lazzara, M.J. EGF augments TGFβ-induced epithelial-mesenchymal transition by promoting SHP2 binding to GAB1. J. Cell Sci. 2015, 128, 3898–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Jiang, Y.; Steed, H.; Davidge, S.; Fu, Y.X. TGFβ and EGF synergistically induce a more invasive phenotype of epithelial ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 401, 376–381. [Google Scholar] [CrossRef]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 secreted from Cancer-Associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, K.; Inamurai, H.; Miyayamai, T.; Matsuokai, M. Involvement of Notch1 signaling in malignant progression of A549 cells subjected to prolonged cadmium exposure. J. Biol. Chem. 2017, 292, 7942–7953. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Shan, Z.; Shaikh, Z.A. Epithelial-mesenchymal transition in breast epithelial cells treated with cadmium and the role of Snail. Toxicol. Appl. Pharmacol. 2018, 344, 46–55. [Google Scholar] [CrossRef]
- Soto-Guzman, A.; Navarro-Tito, N.; Castro-Sanchez, L.; Martinez-Orozco, R.; Salazar, E.P. Oleic acid promotes MMP-9 secretion and invasion in breast cancer cells. Clin. Exp. Metastasis. 2010, 27, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Martinez-orozco, R.; Navarro-tito, N.; Soto-guzman, A.; Castro-sanchez, L. European Journal of Cell Biology Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Avtanski, D.; Garcia, A.; Caraballo, B.; Thangeswaran, P.; Marin, S.; Bianco, J.; Lavi, A.; Poretsky, L. Cytokine Resistin induces breast cancer cells epithelial to mesenchymal transition ( EMT ) and stemness through both adenylyl cyclase-associated protein 1 (CAP1) -dependent and CAP1-independent mechanisms. Cytokine 2019, 120, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Avtanski, D.; Garcia, A.; Caraballo, B.; Thangeswaran, P.; Marin, S.; Bianco, J.; Lavi, A.; Poretsky, L. Data in brief In vitro effects of resistin on epithelial to mesenchymal transition (EMT) in MCF-7 and MDA-MB-231 breast cancer cells e qRT-PCR and Westen blot analyses data. Data Br. 2019, 25, 104118. [Google Scholar] [CrossRef]
- Weingarten, C.; Jenudi, Y.; Tshuva, R.Y.; Moskovich, D.; Alfandari, A. The Interplay Between Epithelial-Mesenchymal Transition (EMT) and the Thyroid Hormones- α v β 3 Axis in Ovarian Cancer Horm. Cancer 2018, 9, 22–32. [Google Scholar]
- Aiwei, Y.B.; Behjatolah, M.K.; Hedges, R.A.; Rogers, L.J.; Kadlubar, S.A.; Thomas, K.E. Adipocyte hypoxia promotes epithelial-mesenchymal transition-related gene expression and estrogen receptor-negative phenotype in breast cancer cells. Oncol. Rep. 2015, 33, 2689–2694. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ding, G.; Liang, W.; Chen, C.; Yang, H. Role of LOX-1 and ROS in oxidized low-density lipoprotein induced epithelial-mesenchymal transition of NRK52E. Lipids Health Dis. 2010, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers (Basel) 2019, 11, 62. [Google Scholar] [CrossRef] [Green Version]
- Candelaria, P.V.; Rampoldi, A.; Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin signaling and cancer chemoresistance: Perspectives. World J. Clin. Oncol. 2017, 8, 106. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Auwerx, J.; Staels, B. Leptin. Lancet 1998, 351, 737–742. [Google Scholar] [CrossRef]
- Prolo, P.; Wong, M.L.; Licinio, J. Leptin. Int. J. Biochem. Cell Biol. 1998, 30, 1285–1290. [Google Scholar] [CrossRef]
- Naylor, C.; Petri, W.A. Leptin regulation of immune responses. Trends Mol. Med. 2016, 22, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Maymó, J.; Dueñas, J.L.; Varone, C.; Sánchez-Margalet, V. Role of leptin in female reproduction. Clin. Chem. Lab. Med. 2015, 53, 15–28. [Google Scholar] [CrossRef]
- Ray, A.; Cleary, M.P. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017, 38, 80–97. [Google Scholar] [CrossRef]
- Ghasemi, A.; Saeidi, J.; Azimi-Nejad, M.; Hashemy, S.I. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell. Oncol. 2019, 42, 243–260. [Google Scholar] [CrossRef]
- Kato, S.; Abarzua-catalan, L.; Trigo, C.; Delpiano, A.; García, K.; Ibañez, C.; Hormazábal, K.; Diaz, D.; Brañes, J.; Castellón, E.; et al. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: An explanation for poor outcomes in obese women. Oncotarget 2015, 6, 21100–21119. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Crean-Tate, K.K.; Reizes, O. Leptin Regulation of Cancer Stem Cells in Breast and Gynecologic Cancer. Endocrinology 2018, 159, 3069–3080. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Mendoza-Catalán, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling Pathways Induced by Leptin during Epithelial-Mesenchymal Transition in Breast Cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Meyer-schaller, N.; Cardner, M.; Diepenbruck, M.; Ivanek, R.; Beerenwinkel, N.; Christofori, G. Resource A Hierarchical Regulatory Landscape during the Multiple Stages of EMT A Hierarchical Regulatory Landscape during the Multiple Stages of EMT. Dev. Cell. 2019, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konge, J.; Leteurtre, F.; Goislard, M.; Biard, D.; Morel, S. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget 2018, 9, 23519–23531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Sun, H.; Zhang, W.; Zhang, M.; Yang, X.; Kuang, R. Meta-Analysis of EMT Datasets Reveals Different Types of EMT. PLoS ONE 2016, 1, e0156839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K.; Benson, J.A. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 2019, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, D.P.; Vanderhyden, B.C. Context specificity of the EMT transcriptional response. Nat. Commun. 2020, 11, 2142. [Google Scholar] [CrossRef]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription Factors OVOL1 and OVOL2 Induce the Mesenchymal to Epithelial Transition in Human Cancer. PLoS ONE 2013, 8, 1–20. [Google Scholar] [CrossRef]
- Somarelli, J.A.; Shetler, S.; Jolly, M.K.; Wang, X.; Dewitt, S.B.; Hish, A.J.; Gilja, S. Mesenchymal-Epithelial Transition in Sarcomas Is Controlled by the Combinatorial Expression of MicroRNA 200s and GRHL2. Mol. Cell. Biol. 2016, 36, 2503–2513. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.L.; Wang, W.J.; Qiu, Y.T.; Xie, X.F.; Bai, J.; Shi, Z.Z. miR-145-5p suppresses tumor cell migration, invasion and epithelial to mesenchymal transition by regulating the sp1/NF-kB signaling pathway in esophageal squamous cell carcinoma. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Sengez, B.; Aygün, I.; Shehwana, H.; Toyran, N.; Avci, S.T.; Konu, O.; Stemmler, M.P.; Alotaibi, H. The Transcription Factor Elf3 Is Essential for a Successful Mesenchymal to Epithelial Transition. Cells 2019, 8, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Q.; Zhou, D.; He, X.; Fan, J.; Tang, J.; Qiu, Z.; Zhang, Y.; Qiu, J.; Xu, Y.; Lai, G. The zinc finger protein GATA4 induces mesenchymal-to-epithelial transition and cellular senescence through the nuclear factor-κB pathway in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2019, 34, 2196–2205. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Chen, X.; Xie, C.; Chen, Z.; Liu, Y.; Ru, F.; He, Y. Leptin promotes epithelial-mesenchymal transition in benign prostatic hyperplasia through downregulation of BAMBI. Exp. Cell Res. 2020, 387, 111754. [Google Scholar] [CrossRef]
- Liu, Y.N.; Yin, J.J.; Abou-Kheir, W.; Hynes, P.G.; Casey, O.M.; Fang, L.; Yi, M.; Stephens, R.M.; Seng, V.; Sheppard-Tillman, H.; et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 2013, 32, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Long, J.; Du, R.; Ge, C.; Guo, K.; Xu, Y. miR-204 regulates the EMT by targeting snai1 to suppress the invasion and migration of gastric cancer. Tumor Biol. 2016, 37, 8327–8335. [Google Scholar] [CrossRef]
- Piperigkou, Z.; Franchi, M.; Götte, M.; Karamanos, N.K. Estrogen receptor beta as epigenetic mediator of miR-10b and miR-145 in mammary cancer. Matrix Biol. 2017, 64, 94–111. [Google Scholar] [CrossRef]
- Zhang, M.; Jabbari, N.; Satpathy, M.; Matyunina, L.V.; Wang, Y.; McDonald, L.D.E.; McDonald, J.F. Sequence diverse miRNAs converge to induce mesenchymal-to-epithelial transition in ovarian cancer cells through direct and indirect regulatory controls. Cancer Lett. 2019, 459, 168–175. [Google Scholar] [CrossRef]
- Cheng, J.T.; Wang, L.; Wang, H.; Tang, F.R.; Cai, W.Q.; Sethi, G.; Xin, H.W.; Ma, Z. Insights into Biological Role of LncRNAs in Epithelial-Mesenchymal Transition. Cells 2019, 8, 1178. [Google Scholar] [CrossRef] [Green Version]
- Gugnoni, M.; Ciarrocchi, A. Long noncoding RNA and epithelial mesenchymal transition in cancer. Int. J. Mol. Sci. 2019, 20, 1924. [Google Scholar] [CrossRef] [Green Version]
- Mierke, C.T.; Frey, B.; Fellner, M.; Herrmann, M.; Fabry, B. Integrin 5 1 facilitates cancer cell invasion through enhanced contractile forces. J. Cell Sci. 2011, 124, 369–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peixoto, P.; Etcheverry, A.; Aubry, M.; Missey, A.; Lachat, C.; Perrard, J.; Hendrick, E.; Delage-Mourroux, R.; Mosser, J.; Borg, C.; et al. EMT is associated with an epigenetic signature of ECM remodeling genes. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Lin, H.; Tang, M.; Wang, Y. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.N.; Ahn, D.H.; Kang, N.; Yeo, C.D.; Kim, Y.K.; Lee, K.Y.; Kim, T.-J.; Lee, S.H.; Park, M.S.; Yim, H.W.; et al. TGF-β induced EMT and stemness characteristics are associated with epigenetic regulation in lung cancer. Sci. Rep. 2020, 10, 10597. [Google Scholar] [CrossRef]
- Mekhdjian, A.H.; Kai, F.; Rubashkin, M.G.; Prahl, L.S.; Przybyla, L.M.; McGregor, A.L.; Bell, E.S.; Barnes, J.M.; DuFort, C.C.; Ou, G.; et al. Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol. Biol. Cell. 2017, 28, 1467–1488. [Google Scholar] [CrossRef]
- Galliher, A.J.; Schiemann, W.P. β3Integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [Green Version]
- Ramos, D.M.; Dang, D.; Sadler, S. The role of the integrin αvβ6 in regulating the epithelial to mesenchymal transition in oral cancer. Anticancer Res. 2009, 29, 125–130. [Google Scholar]
- Bianchi, A.; Gervasi, M.E.; Bakin, A.V. Role of β5-integrin in epithelial-mesenchymal transition in response to TGFβ. Cell Cycle 2010, 9, 1647–1659. [Google Scholar] [CrossRef] [Green Version]
- Conlon, G.A.; Murray, G.I. Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2019, 247, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.-X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int. J. Oncol. 2016, 48, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, M.P.; Lakshmanan, I.; Jain, M.; Das, S.; Chakraborty, S.; Dey, P.; Batra, S.K. MUC4 mucin-induced epithelial to mesenchymal transition: A novel mechanism for metastasis of human ovarian cancer cells. Oncogene 2010, 29, 5741–5754. [Google Scholar] [CrossRef] [Green Version]
- Roy, L.D.; Sahraei, M.; Subramani, D.B.; Besmer, D.; Nath, S.; Tinder, T.L.; Bajaj, E.; Shanmugam, K.; Lee, Y.Y. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 2011, 30, 1449–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef] [PubMed]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-Cadherin Promotes Metastasis via Multiple Downstream Transcriptional Pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alidadiani, N.; Ghaderi, S.; Dilaver, N.; Bakhshamin, S.; Bayat, M. Epithelial mesenchymal transition Transcription Factor (TF): The structure, function and microRNA feedback loop. Gene 2018, 674, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Avtanski, D.; Saxena, N.K.; Sharma, D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J. Biol. Chem. 2012, 287, 8598–8612. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, T.; Zhang, S.; Guo, K.; Liu, Y. Oct4 induces EMT through LEF1/β-catenin dependent WNT signaling pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 2599–2606. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-C.; Wu, M.-J.; Yang, J.-Y.; Camarillo, I.G.; Chang, C.-J. Leptin–STAT3–G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res. 2015, 75, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.M.; Marquardt, S.; Gupta, S.K.; Knoll, S.; Schmitz, U.; Spitschak, A.; Engelmann, D.; Vera, J.; Wolkenhauer, O.; Pützer, B.M. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Zhu, Y.; Gao, J.; Fu, J.; Liu, C.; Liu, Y.; Song, C.; Zhu, S.; Leng, Y.; Wang, G.; et al. MicroRNA-29a promotes colorectal cancer metastasis by regulating matrix metalloproteinase 2 and E-cadherin via KLF4. Br. J. Cancer 2014, 110, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Sun, J.; Wang, B.; Ren, J.C.; Su, W.; Zhang, T. MicroRNA-10b Triggers the Epithelial–Mesenchymal Transition (EMT) of Laryngeal Carcinoma Hep-2 Cells by Directly Targeting the E-cadherin. Appl. Biochem. Biotechnol. 2015, 176, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.F.; Lei, Y.; Liu, X.Q. MiR-29a promotes cell proliferation and EMT in breast cancer by targeting ten eleven translocation 1. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Liu, Z.; Shi, S.; Zhang, Z.; Zhang, J.; Lin, H.; Lu, H. MiR-221/222 activate the Wnt/β-catenin signaling to promote triple-negative breast cancer. J. Mol. Cell Biol. 2018, 10, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Chen, F.; Zheng, Y.; Zhang, D.; Qian, B.; Ji, H.; Long, F.; Cretoiu, D. MiR-21 regulates growth and EMT in lung cancer cells via PTEN/Akt/GSK3β signaling. Front. Biosci. Landmark 2019, 24, 1426–1439. [Google Scholar] [CrossRef] [Green Version]
- Salmerón-Bárcenas, E.; Mendoza-Catalán, M.A.; Illades-Aguiar, B.; Peralta-Arrieta, I.; Alquisiras-Burgos, I.; Ortiz-Ortiz, J.; Navarro-Tito, N.; Zacapala-Gómez, A.E. Long non-coding RNAs as new players in cervical carcinogenesis: An update. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8314–8328. [Google Scholar] [CrossRef]
- Fu, X.T.; Dai, Z.; Song, K.; Zhang, Z.J.; Zhou, Z.J.; Zhou, S.L.; Zhao, Y.M.; Xiao, Y.S.; Sun, Q.M.; Ding, Z.B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Shang, G.S.; Liu, L.; Qin, Y.W. IL-6 and TNF-α promote metastasis of lung cancer by inducing epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 4657–4660. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wen, H.; Zhou, C.; Su, Q.; Lin, Y.; Xie, Y.; Huang, Y.; Qiu, Q.; Lin, J.; Huang, X.; et al. TNF-α derived from M2 tumor-associated macrophages promotes epithelial-mesenchymal transition and cancer stemness through the Wnt/β-catenin pathway in SMMC-7721 hepatocellular carcinoma cells. Exp. Cell Res. 2019, 378, 41–50. [Google Scholar] [CrossRef]
- Wu, C.H.; Tang, S.C.; Wang, P.H.; Lee, H.; Ko, J.L. Nickel-induced epithelial-mesenchymal transition by reactive oxygen species generation and E-cadherin promoter hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef] [Green Version]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.O.Y.E.; Hwang, K.A.; Kim, C.H.O.W.O.N.; Jeung, E.U.I.B.A.E.; Choi, K.C. Altered expression of epithelial mesenchymal transition and pluripotent associated markers by sex steroid hormones in human embryonic stem cells. Mol. Med. Rep. 2017, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Singh, K.P. Nicotine-induced oxidative stress contributes to EMT and stemness during neoplastic transformation through epigenetic modi fi cations in human kidney epithelial cells. Toxicol. Appl. Pharmacol. 2019, 374, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.; Campfield, L.; Clark, F.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor. OB-R Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Masuzaki, H.; Ogawa, Y.; Isse, N.; Satoh, N.; Okazaki, T.; Shigemoto, M.; Mori, K.; Tamura, N.; Hosoda, K.; Yoshimasa, Y.; et al. Human obese gene expression: Adipocyte-specific expression and regional differences in the adipose tissue. Diabetes 1995, 44, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 292–295. [Google Scholar] [CrossRef]
- Maffei, M.; Halaas, J.; Ravussin, E.; Pratley, R.E.; Lee, G.H.; Zhang, Y.; Fei, H.; Kim, S.; Lallone, R.; Ranganathan, S.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1995, 1, 1155–1161. [Google Scholar] [CrossRef]
- Flier, J.S. What ’ s in a Name ? In Search of Leptin’s Physiologic Role. J. Clin. Endocrinol. Metab. 1998, 83, 1407–1413. [Google Scholar]
- Blüher, M. Obesity: Global epidemiology. Nat. Rev. 2011, 24, 205. [Google Scholar] [CrossRef]
- Farr, O.M.; Gavrieli, A.; Mantzoros, C.S. Leptin applications in 2015: What have we learned about leptin and obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2016, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tritos, N.A.; Mantzoros, C.S. Leptin: Its role in obesity and beyond. Diabetologia 2014. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Heist, K.; Depaoli, A.M.; Veldhuis, J.D.; Mantzoros, C.S. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J. Clin. Investig. 2003, 111, 1409–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J. The long road to leptin Find the latest version: The long road to leptin. J. Clin. Investig. 2016, 126, 4727–4734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkle, W.; Cordell, M.; Leibel, R.; Rosenbaum, M.; Hirsch, J. Effects of Reduced Weight Maintenance and Leptin Repletion on Functional Connectivity of the Hypothalamus in Obese Humans. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, A.; Clementi, L.; Alesse, E. Leptin in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 89–112. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Kwon, A.R.; Lee, Y.K.; Oh, S.W. Circulating adipokines and risk of obesity related cancers: A systematic review and meta-analysis. Obes. Res. Clin. Pract. 2019, 13, 329–339. [Google Scholar] [CrossRef]
- Giordano, C.; Barone, I.; Vircillo, V.; Panza, S.; Malivindi, R.; Gelsomino, L.; Pellegrino, M.; Rago, V.; Mauro, L.; Lanzino, M.; et al. Activated FXR Inhibits Leptin Signaling and Counteracts Tumor-promoting Activities of Cancer-Associated Fibroblasts in Breast Malignancy. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Cell. Oncol. 2013, 67, 537–547. [Google Scholar] [CrossRef]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441. [Google Scholar] [CrossRef] [Green Version]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S.; Ando, S. ScienceDirect Leptin, obesity and breast cancer: Progress to understanding the molecular connections. Pharmacology 2016, 83–89. [Google Scholar] [CrossRef]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.K.; Parish, C.R.; Wong, M.L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Park, S.H.; Leung, P.C.K.; Choi, K.C. Expression of leptin receptors and potential effects of leptin on the cell growth and activation of mitogen-activated protein kinases in ovarian cancer cells. J. Clin. Endocrinol. Metab. 2005, 90, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, S. Leptin: Regulation and clinical applications. Leptin Regul. Clin. Appl. 2015, 2, 1–287. [Google Scholar] [CrossRef]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef]
- Kielar, D.; Clark, J.S.C.; Ciechanowicz, A.; Kurzawski, G.; Sulikowski, T.; Naruszewicz, M. Leptin receptor isoforms expressed in human adipose tissue. Metabolism 1998, 47, 844–847. [Google Scholar] [CrossRef]
- Löllmann, B.; Grüninger, S.; Stricker-Krongrad, A.; Chiesi, M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem. Biophys. Res. Commun. 1997, 238, 648–652. [Google Scholar] [CrossRef]
- Moharana, K.; Zabeau, L.; Peelman, F.; Ringler, P.; Stahlberg, H.; Tavernier, J.; Savvides, S.N. Structural and mechanistic paradigm of Leptin receptor activation revealed by complexes with wild-type and antagonist Leptins. Structure 2014, 22, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Mancour, L.V.; Daghestani, H.N.; Dutta, S.; Westfield, G.H.; Schilling, J.; Oleskie, A.N.; Herbstman, J.F.; Chou, S.Z.; Skiniotis, G. Ligand-Induced Architecture of the Leptin Receptor Signaling Complex. Mol. Cell. 2012, 48, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Wauman., J. Leptin receptor signaling: Pathways to leptin resistance. Front. Biosci. 2011, 16, 2771–2793. [Google Scholar] [CrossRef] [Green Version]
- Devos, R.; Guisez, Y.; van der Heyden, J.; White, D.W.; Kalai, M.; Fountoulakis, M.; Plaetinck, G. Ligand-independent dimerization of the extracellular domain of the leptin receptor and determination of the stoichiometry of leptin binding. J. Biol. Chem. 1997, 272, 18304–18310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yang, X.; Yu, S.; Zheng, R. The leptin signaling. Adv. Exp. Med. Biol. 2018, 1090, 123–144. [Google Scholar] [CrossRef]
- Bates, S.H.; Stearns, W.H.; Dundon, T.A.; Schubert, M.; Tso, A.W.K.; Wang, Y.; Banks, A.S.; Lavery, H.J.; Haq, A.K.; Maratos-Flier, E.; et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 2003, 421, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Ishida-Takahashi, R.; Villanueva, E.C.; Fingar, D.C.; Münzberg, H.; Myers, M.G. The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J. Biol. Chem. 2007, 282, 31019–31027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, J.; Zerbinatti, C.; Zhan, Y.; Kolber, B.J.; Herz, J.; Muglia, L.J.; Bu, G. Lipoprotein receptor LRP1 regulates leptin signaling and energy homeostasis in the adult central nervous system. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M.; Yan, D.; Avtanski, D.; Saxena, N.K.; Sharma, D.; et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature 2016, 8, 1–21. [Google Scholar] [CrossRef]
- Mütze, J.; Roth, J.; Gerstberger, R.; Hübschle, T. Nuclear translocation of the transcription factor STAT5 in the rat brain after systemic leptin administration. Neurosci. Lett. 2007, 417, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Wauman, J.; Zabeau, L.; Tavernier, J. The leptin receptor complex: Heavier than expected? Front. Endocrinol. (Lausanne) 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, N.K.; Sharma, D.; Ding, X.; Lin, S.; Marra, F.; Merlin, D.; Anania, F.A. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef] [Green Version]
- Ren, D.; Li, M.; Duan, C.; Rui, L. Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab. 2005, 2, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Ma, X.; Hampel, B.; Balthasar, N.; Coppari, R.; Münzberg, H.; Shanabrough, M.; Burdakov, D.; Rother, E.; Janoschek, R.; et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Investig. 2006, 116, 1886–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metabolism 2013, 6, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.J.; Shin, B.S.; An, G.; Morris, M.E. Biochanin A Inhibits Breast Cancer Tumor Growth in A Murine Xenograft Model. Pharm. Res. 2008, 25, 2158–2163. [Google Scholar] [CrossRef]
- Li, C.; Friedman, J.M. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc. Natl. Acad. Sci. USA 1999, 96, 9677–9682. [Google Scholar] [CrossRef] [Green Version]
- Park, H.K.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef]
- Yang, G.; Lim, C.Y.; Li, C.; Radda, G.K.; Li, C.; Cao, X.; Han, W. FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J. Biol. Chem. 2009, 284, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 Kinase Phosphorylates AMPK on Serine 491 to Mediate Leptin’s Effect on Food Intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Matter, E.K.; Woods, S.C.; Seeley, R.J. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J. Neurosci. 2008, 28, 7202–7208. [Google Scholar] [CrossRef] [Green Version]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Märker, T.; Sell, H.; Zilleßen, P.; Glöde, A.; Kriebel, J.; Ouwens, D.M.; Pattyn, P.; Ruige, J.; Famulla, S.; Roden, M.; et al. Heat shock protein 60 as a mediator of adipose tissue inflammation and insulin resistance. Diabetes 2012, 61, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Li, Z.; Rui, L. Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J. Biol. Chem. 2008, 283, 28066–28073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaías-Tizapa, R.; Acosta, E.; Tacuba-Saavedra, A.; Mendoza-Catalán, M.; Navarro-Tito, N. Leptina promueve la expresión de Hic-5 y la formación de puntos de actina de manera dependiente de las cinasas FAK y Src en células epiteliales mamarias MCF10A. Biomédica 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Villanueva-Duque, A.; Zuniga-Eulogio, M.D.; Dena-Beltran, J.; Castaneda-Saucedo, E.; Calixto-Galvez, M.; Mendoza-Catalán, M.A.; Ortuno-Pineda, C.; Navarro-Tito, N. Leptin induces partial epithelial-mesenchymal transition in a FAK-ERK dependent pathway in MCF10A mammary non-tumorigenic cells. Int. J. Clin. Exp. Pathol. 2017, 10, 10334–10342. [Google Scholar]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-Catalán, M.A.; Ortuño-Pineda, C.; Moreno-Godinez, M.E.; Villegas-Comonfort, S.; Padilla-Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src- dependent manner in breast cancer cells. Endocr. Connect. 2019. [Google Scholar] [CrossRef] [Green Version]
- Caro, J.F.; Kolaczynski, J.W.; Nyce, M.R.; Ohannesian, J.P.; Opentanova, I.; Goldman, W.H.; Lynn, R.B.; Zhang, P.L.; Sinha, M.K.; Considine, R.V. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: A possible mechanism for leptin resistance. Lancet 1996, 348, 159–161. [Google Scholar] [CrossRef]
- Cui, H.; Cai, F.; Belsham, D.D. Anorexigenic hormones leptin, insulin, and α-melanocyte-stimulating hormone directly induce neurotensin (NT) gene expression in novel NT-expressing cell models. J. Neurosci. 2005, 25, 9497–9506. [Google Scholar] [CrossRef] [Green Version]
- Benomar, Y.; Roy, A.F.; Aubourg, A.; Djiane, J.; Taouis, M. Cross down-regulation of leptin and insulin receptor expression and signalling in a human neuronal cell line. Biochem. J. 2005, 388, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Olea-Flores, M.; Zuñiga-Eulogio, M.; Tacuba-Saavedra, A.; Bueno-Salgado, M.; Sánchez-Carvajal, A.; Vargas-Santiago, Y.; Mendoza-Catalán, M.A.; Salazar, E.P.; García-Hernández, A.; Padilla-Benavides, T.; et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced ER-α-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, L.W.; Rossi, E.L.; Mcdonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; Linda, A.; Hursting, S.D. Leptin Signaling Mediates Obesity-associated CSC Enrichment and EMT in Preclinical TNBC Models. Mol. Cancer Res. 2019, 16, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Tenvooren, I.; Jenks, M.Z.; Rashid, H.; Cook, K.L.; Joëlle, K.; Sistrunk, C.; Holmes, J.; Wang, K.; Bonin, K.; Lo, H.; et al. Elevated leptin disrupts epithelial polarity and promotes premalignant alterations in the mammary gland. Oncogene 2019, 38, 3855–3870. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, Q.; Su, M.; Ji, F.; Wang, N.; Zhong, C.; Jiang, Y.; Liu, Y.; Zhang, Z.; Yang, J.; et al. Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell. Oncol. 2017, 40, 537–547. [Google Scholar] [CrossRef]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A.; et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr. Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef]
- Laforest, S.; Ennour-Idrissi, K.; Ouellette, G.; Gauthier, M.F.; Michaud, A.; Durocher, F.; Tchernof, A.; Diorio, C. Associations between markers of mammary adipose tissue dysfunction and breast cancer prognostic factors. Int. J. Obes. 2020. [Google Scholar] [CrossRef]
- Chen, X.; Zha, X.; Chen, W.; Zhu, T.; Qiu, J.; Røe, O.D.; Li, J.; Wang, Z.; Yin, Y. Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Biomed. Pharmacother. 2013, 67, 22–30. [Google Scholar] [CrossRef]
- Lee, K.N.; Choi, H.S.; Yang, S.Y.; Park, H.K.; Lee, Y.Y.; Lee, O.Y.; Yoon, B.C.; Hahm, J.S.; Paik, S.S. The role of leptin in gastric cancer: Clinicopathologic features and molecular mechanisms. Biochem. Biophys. Res. Commun. 2014, 446, 822–829. [Google Scholar] [CrossRef]
- Pai, R.; Lin, C.; Tran, T.; Tarnawski, A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochem. Biophys. Res. Commun. 2005, 331, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Kitayama, J.; Mori, K.; Watanabe, T.; Nagawa, H. Transactivation of epidermal growth factor receptor is involved in leptin-induced activation of Janus-activated kinase 2 and extracellular signal-regulated kinase 1/2 in human gastric cancer cells. Cancer Res. 2005, 65, 9159–9163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki-Ohara, K.; Mayuzumi, H.; Kato, S.; Minokoshi, Y.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Matsuzaki, G.; Yoshimura, A. Enhancement of leptin receptor signaling by SOCS3 deficiency induces development of gastric tumors in mice. Oncogene 2014, 33, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Judd, L.M.; Bredin, K.; Kalantzis, A.; Jenkins, B.J.; Ernst, M.; Giraud, A.S. STAT3 Activation Regulates Growth, Inflammation, and Vascularization in a Mouse Model of Gastric Tumorigenesis. Gastroenterology 2006, 131, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Arita, S.; Kinoshita, Y.; Ushida, K.; Enomoto, A.; Inagaki-Ohara, K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch. Biochem. Biophys. 2016, 610, 16–24. [Google Scholar] [CrossRef]
- Inagaki-ohara, K.; Okamoto, S.; Takagi, K.; Saito, K.; Arita, S.; Tang, L.; Hori, T.; Kataoka, H.; Matsumoto, S.; Minokoshi, Y. Leptin receptor signaling is required for high-fat diet-induced atrophic gastritis in mice. Nutr. Metab. (Lond.) 2016, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, S.; Ogawa, T.; Murakami, Y.; Kinoshita, Y.; Okazaki, M. Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice. Nutrients 2019, 11, 2127. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Expression pattern of leptin and leptin receptor (OB-R) in human gastric cancer. World J. Gastroenterol. 2006, 12, 5517–5522. [Google Scholar] [CrossRef]
- Capelle, L.G.; de Vries, A.C.; Haringsma, J.; Steyerberg, E.W.; Looman, C.W.N.; Nagtzaam, N.M.A.; van Dekken, H.; Borg, F.T.; de Vries, R.A.; Kuipers, E.J. Serum levels of leptin as marker for patients at high risk of gastric cancer. Helicobacter 2009, 14, 596–604. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, M.; Zhou, J.; Luo, G.; Chen, X.; Zhang, L.; Liang, C. Circulating levels of adiponectin and leptin in patients with prostate cancer. Int. J. Clin. Exp. Med. 2018, 11, 5784–5792. [Google Scholar]
- Kim, J.H.; Lee, S.Y.; Myung, S.C.; Kim, Y.S.; Kim, T.H.; Kim, M.K. Clinical significance of the leptin and leptin receptor expressions in prostate tissues. Asian J. Androl. 2008, 10, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Fontana, C.M.L.; Maselli, M.E.; Elizalde, R.F.P.; Di, N.A.; Mónaco, M.; Recupero, A.L.U.; Laur, J.D.L. Leptin increases prostate cancer aggressiveness. J. Physiol. Biochem. 2011, 531–538. [Google Scholar] [CrossRef]
- Feng, H.; Liu, Q.; Zhang, N.; Zheng, L.; Sang, M.; Feng, J. Leptin Promotes Metastasis by Inducing an Epithelial–Mesenchymal Transition in A549 Lung Cancer Cells. Logy Res. 2017, 21, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Price, R.S.; Cavazos, D.A.; de Angel, R.E.; Hursting, S.D. Obesity-related systemic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer Prostatic Dis. 2012, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrab, A.; Pagano, A.; Ayed, K.; Chebil, M.; Derouiche, A.; Kovacic, H.; Gati, A. Leptin Promotes Prostate Cancer Proliferation and Migration by Stimulating STAT3 Pathway Leptin Promotes Prostate Cancer Proliferation and Migration by Stimulating. Nutr. Cancer 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Harbuzariu, A.; Gonzalez-perez, R.R. Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 2018, 9, 18239–18253. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.J.; Dong, L.L.; Kang, X.L.; Li, Z.M.; Zhang, H.Y. Leptin promotes proliferation and inhibits apoptosis of prostate cancer cells by regulating ERK1/2 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8341–8348. [Google Scholar] [CrossRef]
- Fava, G.; Alpini, G.; Rychlicki, C.; Saccomanno, S.; Demorrow, S.; Trozzi, L.; Candelaresi, C.; Venter, J.; Di, A.; Marzioni, M.; et al. Leptin Enhances Cholangiocarcinoma Cell Growth Sharon. Cancer Res. 2009, 68, 6752–6761. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Yu, H.S.; Lai, T.Y.; Yeh, Y.L.; Su, C.C.; Hsu, H.H.; Tsai, F.J.; Tsai, C.H.; Wu, H.C.; Tang, C.H. Leptin increases motility and integrin up-regulation in human prostate cancer cells. J. Cell. Physiol. 2011, 226, 1274–1282. [Google Scholar] [CrossRef]
- Noda, T.; Kikugawa, T.; Tanji, N.; Miura, N.; Asai, S.; Higashiyama, S.; Yokoyama, M. Long-term exposure to leptin enhances the growth of prostate cancer cells. Int. J. Oncol. 2015, 46, 1535–1542. [Google Scholar] [CrossRef]
- Ribeiro, R.; Araújo, A.P.; Coelho, A.; Catarino, R.; Pinto, D.; Araújo, A.; Calçada, C.; Lopes, C.; Medeiros, R. A functional polymorphism in the promoter region of leptin gene increases susceptibility for non-small cell lung cancer. Eur. J. Cancer 2006, 42, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Carpagnano, G.E.; Spanevello, A.; Curci, C.; Salerno, F.; Palladino, G.P.; Resta, O.; di Gioia, G.; Carpagnano, F.; Barbaro, M.P.F. IL-2, TNF-α, and leptin: Local versus systemic concentrations in NSCLC patients. Oncol. Res. 2007, 16, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.I.N.; Cao, F.; Li, N.; Gao, X.I.N.; Su, X.; Jiang, X. Leptin induces epithelial-to-mesenchymal transition via activation of the ERK signaling pathway in lung cancer cells. Oncol. Lett. 2018, 4782–4788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zhao, S.; Guo, T.; Li, J.; Gu, C. The Nutritional Cytokine Leptin Promotes NSCLC by Activating the PI3K/AKT and MAPK/ERK Pathways in NSCLC Cells in a Paracrine Manner. Biomed. Res. Int. 2019, 2019, 2585743. [Google Scholar] [CrossRef] [Green Version]
- Gui, X.; Chen, H.; Cai, H.; Sun, L.; Gu, L. Leptin promotes pulmonary fi brosis development by inhibiting autophagy via PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 498, 660–666. [Google Scholar] [CrossRef]
- Song, C.H.; Liao, J.; Deng, Z.H.; Zhang, J.Y.; Xue, H.; Li, Y.M.; Liang, C.; Han, M.; Zhang, K.; Yan, G.T. Is leptin a predictive factor in patients with lung cancer? Clin. Biochem. 2014, 47, 230–232. [Google Scholar] [CrossRef]
- Peng, C.; Sun, Z.; Li, O.U.; Guo, C.; Yi, W.; Tan, Z.; Jiang, B.O. Leptin stimulates the epithelial-mesenchymal transition and pro-angiogenic capability of cholangiocarcinoma cells through the miR-122/PKM2 axis. Int. J. Oncol. 2019, 298–308. [Google Scholar] [CrossRef]
- Chen, C.; Chang, Y.C.; Lan, M.S.; Breslin, M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways. Int. J. Oncol. 2013, 42, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, A.; Hashemy, S.I.; Aghaei, M.; Panjehpour, M. Leptin induces matrix metalloproteinase 7 expression to promote ovarian cancer cell invasion by activating ERK and JNK pathways. J. Cell. Biochem. 2018, 119, 2333–2344. [Google Scholar] [CrossRef]
- Kumar, J.; Fang, H.; Mcculloch, D.R.; Crowley, T. Leptin receptor signaling via Janus kinase 2/Signal transducer and activator of transcription 3 impacts on ovarian cancer cell phenotypes. Oncotarget 2017, 8, 93530–93540. [Google Scholar] [CrossRef]
- Méndez-López, L.F.; Dávila-Rodríguez, M.I.; Zavala-Pompa, A.; Torres-López, E.; González-Martínez, B.E.; López-Cabanillas-Lomelí, M. Expression of leptin receptor in endometrial biopsies of endometrial and ovarian cancer patients. Biomed. Rep. 2013, 1, 659–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, F.; Zhang, H.; Yao, L.; Jiang, S.; Lu, H.; Xing, X.; Zhang, C.; Jiang, P.; Zhang, R. Leptin contributes to the taxol chemoresistance in epithelial ovarian cancer. Oncol. Lett. 2019, 18, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.S.F.; Chung, Y.F.; Chen, H.W.; Tsai, K.B.; Chang, H.L.; Huang, C.H.; Su, J.H. Aberrant expression and possible involvement of the leptin receptor in bladder cancer. Urology 2004, 63, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Abe, T.; Kinoshita, F.; Ushijima, M.; Masaoka, H.; Shiota, M. The role of adipocytokines and their receptors in bladder cancer: Expression of adiponectin or leptin is an independent prognosticator. Am. J. Transl. Res. 2020, 12, 3033–3045. [Google Scholar] [CrossRef]
- Somasundar, P.; Riggs, D.; Jackson, B.; Vona-Davis, L.; McFadden, D.W. Leptin stimulates esophageal adenocarcinoma growth by nonapoptotic mechanisms. Am. J. Surg. 2003, 186, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.; Mutungi, G.; Beales, I.L.P. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology 2006, 147, 4505–4516. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L.P. Leptin stimulates the proliferation of human oesophageal adenocarcinoma cells via HB-EGF- and TGFα-mediated transactivation of the epidermal growth factor receptor. Br. J. Biomed. Sci. 2008, 65, 121–127. [Google Scholar] [CrossRef]
- Trevellin, E.; Scarpa, M.; Carraro, A.; Lunardi, F.; Kotsafti, A.; Porzionato, A.; Saadeh, L.; Cagol, M.; Alfieri, R.; Tedeschi, U.; et al. Esophageal adenocarcinoma and obesity: Peritumoral adipose tissue plays a role in lymph node invasion. Oncotarget 2015, 6, 11203–11215. [Google Scholar] [CrossRef]
- Spyridopoulos, T.N.; Petridou, E.T.; Dessypris, N.; Terzidis, A.; Skalkidou, A.; Deliveliotis, C.; Chrousos, G.P.; Giannopoulos, A.; Alivizatos, G.; Vassilakis, G.; et al. Inverse association of leptin levels with renal cell carcinoma: Results from a case-control study. Hormones 2009, 8, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Perumal, K.; Mun, K.S.; Yap, N.Y.; Hassan, A.; Razack, A.; Gobe, G.C.; Ong, T.A.; Kuppusamy, S.; Rajandram, R. Research Article A Study on the Immunohistochemical Expressions of Leptin and Leptin Receptor in Clear Cell Renal Cell Carcinoma. Biomed. Res. Int. 2020, 2020, 3682086. [Google Scholar] [CrossRef]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Leptin Promotes Invasiveness of Murine Renal Cancer Cells Via Extracellular Signal-Regulated Kinases and Rho Dependent Pathway. J. Urol. 2006, 176, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Sumitomo, M.; Asakuma, J.; Asano, T.; Zheng, R.; Asano, T.; Nanus, D.M.; Hayakawa, M. Increased Serum Leptin Levels and Over Expression of Leptin Receptors are Associated With the Invasion and Progression of Renal Cell Carcinoma. J. Urol. 2006, 176, 1631–1635. [Google Scholar] [CrossRef] [PubMed]
- Cymbaluk, A.; Chudecka-Głaz, A.; Rzepka-Górska, I. Leptin levels in serum depending on Body Mass Index in patients with endometrial hyperplasia and cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 136, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.T.; Wu, Q.J.; Wang, Y.L.; Ma, X.X. Circulating adiponectin, leptin and adiponectin-leptin ratio and endometrial cancer risk: Evidence from a meta-analysis of epidemiologic studies. Int. J. Cancer 2015, 137, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Carino, C.; Olawaiye, A.B.; Cherfils, S.; Serikawa, T.; Lynch, M.P.; Rueda, B.R.; Gonzalez, R.R. Leptin regulation of pro-angiogenic molecules in benign and cancer endometrial cells. Int. J. Cancer 2012, 23, 1–7. [Google Scholar] [CrossRef]
- Sharma, D.; Saxena, N.K.; Vertino, P.M.; Anania, F.A. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal- transduction pathways. Endocr. Relat. Cancer 2006, 23, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Gao, Y.; Zhang, L.L.; He, D.L. Concomitant activation of the JAK/STAT3 and ERK1/2 signaling is involved in leptin-mediated proliferation of renal cell carcinoma Caki-2 cells. Cancer Biol. Ther. 2008, 7, 1787–1792. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Choi, Y.S.; Choi, J.H. Leptin promotes human endometriotic cell migration and invasion by up-regulating MMP-2 through the JAK2/STAT3 signaling pathway. Mol. Hum. Reprod. 2015, 21, 792–802. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.S.; Lee, G.S.R.; Nam, S.Y.; Kim, S.J. Progesterone inhibits leptin-induced invasiveness of BeWo cells. Int. J. Med. Sci. 2015, 12, 773–779. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Inductor | Cell Model | The Effect in the EMT | References | |
|---|---|---|---|---|
| Growth factor | TGF-β | HT-29 human colorectal cancer cells | ↓ E-cadherin ↓ Cell-Cell contacts ↑ Vimentin ↑ Elongated phenotype | [7] |
| SGC-7901 and KATO-III human gastric cancer cells | ↓ E-cadherin ↑ Vimentin ↑ N-cadherin ↑ β-catenin ↑ miR-21 | [8] | ||
| A549 human lung adenocarcinoma cells H1299 human non-small cell lung carcinoma cells | ↑ MMP-2 ↑ MMP-9 | [11] | ||
| Endothelial cells | ↑ α-SMA ↑ Fibronectin | [10] | ||
| Panc1 human pancreatic cancer cells MDA-MB-231 human breast cancer cells | ↑ SNAIL ↑ SLUG | [9] | ||
| EGF | AGS, MGLVA1, ST16 human gastric epithelial cells | ↑ SNAIL ↑ SLUG ↑ MMP-7 | [12] | |
| HT29 and DLD-1 colorectal adenocarcinoma cells | ↓ E-cadherin ↑ SNAIL ↑ ZEB ↑ Vimentin | [13] | ||
| H322, PC9 human non-small cell lung carcinoma CAPAN-2 human pancreatic cancer cells | ↓ E-cadherin ↑ Vimentin | [15] | ||
| OVCA429 Human ovarian cancer cells | ↓ E-cadherin ↑ Slug ↑ Snail ↑ MMP-2 | [16] | ||
| PC3 human prostate cancer cells A549 human lung adenocarcinoma cells | ↑ Typical fibroblast-like morphology ↑ N-cadherin ↑ Fibronectin ↑ Snail ↑ Twist | [14] | ||
| Cytokines | IL-6 | Cancer gastric patients’ tissue | ↑ α-SMA | [17] |
| SGC-7901 human gastric cancer cells | ↓ E-cadherin ↑ Vimentin ↑ N-cadherin ↑ ZEB | [17] | ||
| A549 and NCIH358 human lung adenocarcinoma cells | ↓ E-cadherin ↑ Vimentin ↑ α-SMA | [18] | ||
| IL-8 | HCC hepatocellular carcinoma cells | ↓ E-cadherin ↑ Spindle-shaped ↑ Snail ↑ N-cadherin | [87] | |
| Patients with NSCLC | ↑ Vimentin ↑ N-cadherin | [88] | ||
| TNF-α | SMMC-7721 hepatocellular carcinoma cells | ↓ E-cadherin ↑ Spindle-shaped morphology ↑ Vimentin ↑ N-cadherin ↑ β-catenin | [89] | |
| Chemical | Cadmium | A549 human lung adenocarcinoma cells | ↓ E-cadherin ↑ Spindle-shaped morphology ↑ Formation of stress fibers ↑ N-cadherin ↑ Vimentin ↑ Snail ↑Slug | [19] |
| MCF10A nontumorigenic epithelial cells MDA-MB-231, HCC 1937, and HCC 38 human breast cancer cells | ↓ Intercellular adhesion ↓ E-cadherin ↑ Spindle-like morphology ↑ N-cadherin ↑ ZEB-1 ↑ Snail | [20] | ||
| Sodium arsenite, Nickel (II) chloride, and Nickel (II) sulfate | BEAS-2B human bronchial epithelial cell line | ↓ E-cadherin ↑ Fibronectin | [90] | |
| Fatty acids | Palmitate | HepG2 human hepatocarcinoma cells | ↑ SNAIL1 ↑ ZEB2 ↑ TWIST1 | [91] |
| Free fatty acids | Patients with primary liver cancer and hepatocellular carcinoma | ↑ SNAIL1 ↑ VIM ↑ Wnt and TGF-β signaling | [91] | |
| Oleic acid | MCF10A nontumorigenic epithelial cells MCF7, and MDA-MB-31 human breast cancer cells | ↑ MMP-9 ↑ PKC ↑ Src ↑ EGFR | [21] | |
| Arachidonic acid | MCF10A nontumorigenic epithelial cell line | ↑ Vimentin ↑ N-cadherin ↑ Cytokeratin 5/8 ↑ MMP-9 secretion | [22] | |
| Oxidative stress and Hypoxia | ROS | NRK-52E normal rat kidney cells | ↓ E-cadherin ↑ α-SMA | [27] |
| STC2 | SKOV3 human ovarian cancer cells | ↓ E-cadherin ↑ Snail ↑ N-cadherin ↑ Vimentin ↑ MMP-2 ↑ MMP-9 | [94] | |
| Hormones | Estradiol and Progesterone | Embryonic stem cells | ↓ E-cadherin ↑ N-cadherin ↑ Snail ↑ Slug | [92] |
| L-thyroxine (T4) 3,5,3′-triiodo-L-thyronine (T3) | OVCAR-3, SKOV-3 and A2780 human ovarian adenocarcinoma cells | ↑ ZEB-1 ↑ β-catenin ↑ Slug ↑ Vimentin | [25] | |
| Resistin | MCF-7, MDA-MB-231 human breast cancer cells MCF10A nontumorigenic epithelial cell line | ↑ SNAI1 ↑ SNAI2 ↑ TCF8 ↑ TWIST1 ↑ Snail ↑ Vimentin ↑ Fibronectin ↓ E-cadherin ↓ Claudin-1 | [23,24] | |
| Others | Nicotine | HK-2 human renal proximal tubular epithelial cells | ↑ Vimentin ↑ E-cadherin | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olea-Flores, M.; Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Acosta, E.; García-Rodríguez, E.; Zacapala-Gomez, A.E.; Mendoza-Catalán, M.A.; Ortiz-Ortiz, J.; Ortuño-Pineda, C.; Navarro-Tito, N. New Actors Driving the Epithelial–Mesenchymal Transition in Cancer: The Role of Leptin. Biomolecules 2020, 10, 1676. https://doi.org/10.3390/biom10121676
Olea-Flores M, Juárez-Cruz JC, Zuñiga-Eulogio MD, Acosta E, García-Rodríguez E, Zacapala-Gomez AE, Mendoza-Catalán MA, Ortiz-Ortiz J, Ortuño-Pineda C, Navarro-Tito N. New Actors Driving the Epithelial–Mesenchymal Transition in Cancer: The Role of Leptin. Biomolecules. 2020; 10(12):1676. https://doi.org/10.3390/biom10121676
Chicago/Turabian StyleOlea-Flores, Monserrat, Juan C. Juárez-Cruz, Miriam D. Zuñiga-Eulogio, Erika Acosta, Eduardo García-Rodríguez, Ana E. Zacapala-Gomez, Miguel A. Mendoza-Catalán, Julio Ortiz-Ortiz, Carlos Ortuño-Pineda, and Napoleón Navarro-Tito. 2020. "New Actors Driving the Epithelial–Mesenchymal Transition in Cancer: The Role of Leptin" Biomolecules 10, no. 12: 1676. https://doi.org/10.3390/biom10121676