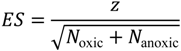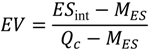2.1. Metabolic Network Expansion Does Not Conclude When Taking Into Account Phylogenetic Relationships
Raymond and Segré [
17] investigated the increased number of metabolic reactions (or enzymes) in 44 organisms after including oxygen, and used known oxic reactions and anoxic reactions as a parameter. They found that network expansion due to oxygen use was highest in eukaryotes and aerobic prokaryotes. This result supports the hypothesis that oxygen contributes to metabolic network expansion.
We re-evaluated this hypothesis using a larger dataset of metabolic networks, compared with the previous study [
17]. In particular we focused on 174 organisms (see
Section 3.1 and
Table S1 in supplementary). This dataset covers most (~90%) of 44 organisms, which were investigated in the previous study, indicating that it includes ~4 times as many organisms as, compared with the dataset in the previous study. Furthermore, 174 organisms in our dataset can be classified into 105 genera, which have ~2.5 times the number of (44) genera, investigated in the previous study. Although phylogenetic heterogeneity in our dataset is slightly higher than that in the previous study, we concluded that a more comprehensive analysis is possible using this phylogenetic tree. In addition, this difference in phylogenetic heterogeneity did not pose a significant problem in this study because we obtained a similar conclusion using our dataset when performing traditional statistical tests, as in the previous study. In particular, we investigated the increase in enzyme and metabolite development. As in the previous study, the increased rate in oxic
versus anoxic network size was calculated based on oxic and anoxic reactions (see
Section 3.3), and was defined as the ratio of the total number of enzymes (metabolites) to anoxic enzymes (metabolites) (
i.e., metabolic space, which can be available without oxygen).
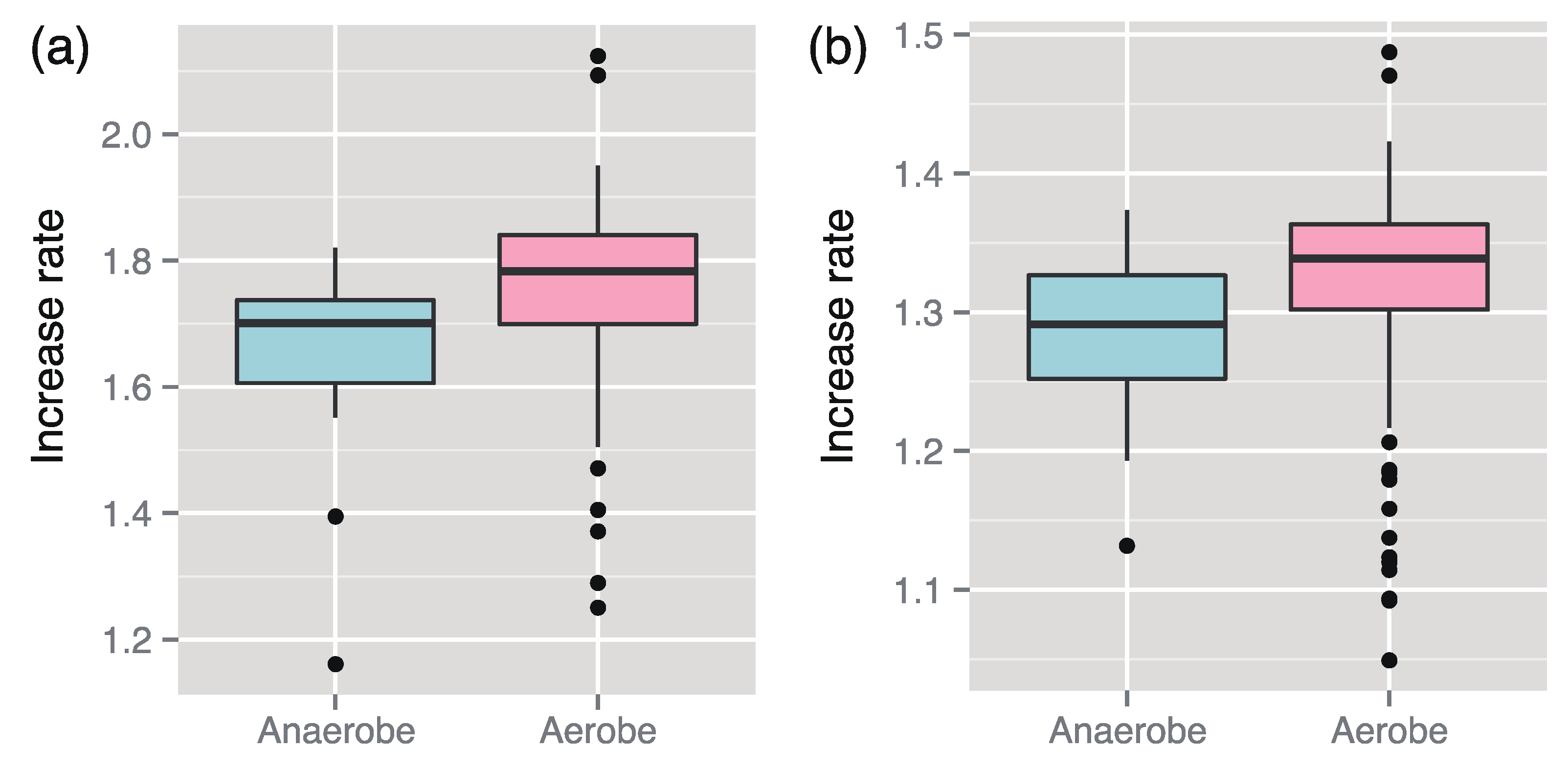
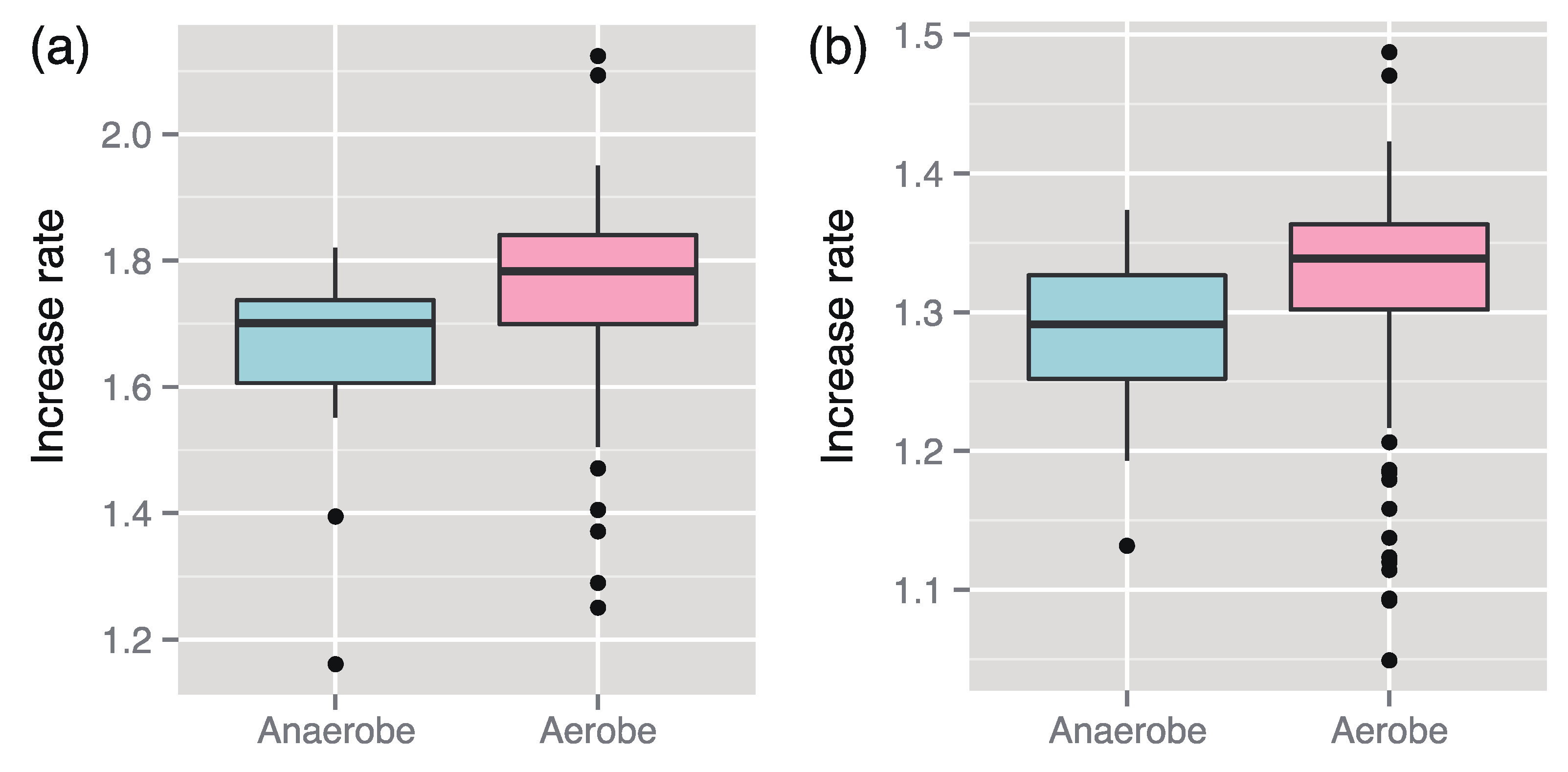
We found that the increased rates of 145 aerobes (including facultative aerobes) were larger than those of 29 anaerobes (
Figure 1). A similar result could also be derived through statistical analysis using the linear model (
Table 1b), which was performed using the lm function in a statistical software R version 3.0.0 [
31].
Figure 1.
Median comparison of the increase rates between aerobes and anaerobes. The medians can be concluded to be significantly different between them in both case of the enzyme-based increase rates (a) (p-value p = 4.6 × 10−5 using the Wilcoxon–Mann–Whitney (WMW) test) and the metabolite-based increase rate (b) (p = 5.2 × 10−5 using the WMW test).
Table 1.
Statistical tests for the impact of species oxygen requirements on the increase rate, calculated based on the number of enzymes and the number of metabolites, using linear models with (a) and without (b) consideration of a phylogenetic relationship. Estimates are shown for the categorical variable corresponding to species oxygen requirements (i.e., aerobic or not aerobic). In particular, in this study, we considered that the p-value of less than 0.01 indicates a non-zero estimate. A positive estimate means that the increase rate of aerobes is higher than that of anaerobes.
| Phylogeny | Enzyme context | Metabolite context |
|---|
| Estimate ± SE | t-value | p-value | Estimate ± SE | t-value | p-value |
|---|
| (a) Considered | 0.04 ± 0.05 | 0.74 | 0.47 | 0.02 ± 0.02 | 0.83 | 0.42 |
| (b) Not considered | 0.10 ± 0.03 | 3.8 | 2.2 × 10−4 | 0.04 ± 0.01 | 3.0 | 3.5 × 10−3 |
Statistical analyses indicated that oxygen expands metabolic networks, as reported in a previous study; however, more careful examination is required as traditional statistical methods assume independence of all observations (or star phylogeny; see [
26,
29] for details). In particular, this traditional statistical method assumes that anaerobes are ancestral. As explained in
Section 1, such an assumption may be not satisfied because species traits such as increased rates in metabolic networks and oxygen requirements depend upon the phylogenetic relationship (
i.e., evolutionary history). In fact, anaerobes are discretely distributed in the phylogenetic tree (
Figure S1 in supplementary), suggesting that it remains possible that an evolutionary history (
i.e., gain and loss of oxygen requirements) causes the observed association between the network expansion and oxygen requirements. To control for phylogenetic relationships among species, therefore, we performed phylogenetic comparative analysis, which can test an association between traits with consideration of such an evolutionary history (see also
Section 3.1), using the brunch algorithm [
28], which calculates phylogenetically independent contrasts for linear models including binary categorical variables. Particularly, we used the statistical software R version 3.0.0 with its function brunch, which is available in the R package caper (Comparative Analyses of Phylogenetics and Evolution in R) version 0.5. A phylogenetic relationship, which is required to perform phylogenetic comparative analysis, was generated based on a highly resolved tree of life [
32] (see Figure S1 and
Section 3.2 for details). As a result, we could not draw conclusions regarding the effect of oxygen on metabolic network expansion when considering phylogenetic relationships (
Table 1a). This finding suggests that increased metabolic space due to oxygen utilization is taxonomically constrained (e.g., Figure 7 in Reference [
26]). That is, a phylogenetic signal causes an association between the increased space and oxygen requirements. This means that an association between oxygen requirements and metabolic network expansion is not likely to exist.
However, note that this conclusion may be debatable, because phylogenetic comparative analyses have several practical and theoretical limitations [
29]. Specifically, such analyses assume a simple evolutionary model, which deems random, Brownian-motion-like traits to be change on a phylogenetic tree with accurate branch lengths.
First, analysis of species, which can be investigated using phylogenetic comparative analyses, depends on the availability of phylogenetic trees. In this study, only ~20% (174) of organisms were investigated because a high-quality phylogenetic tree was used, although we collected metadata (
i.e., oxygen requirements) for 930 organisms (see
Section 3.1 for details). More comprehensive analysis is required to evaluate the association between oxygen and metabolic network expansion in greater detail.
More comprehensive phylogenetic trees (i.e., trees including more species) can be generated using 16S ribosomal RNA sequences or protein sequences; however, quality of the phylogenetic tree (e.g., branch lengths) affects the results of phylogenetic comparative analyses. To avoid this limitation, we used a highly resolved tree of life in this study. Moreover, if the assumption of Brownian motion is invalid, as when evolutionary trends have occurred, estimates may not be accurate.
2.2. Overestimated or Underestimated Differences of Chemical Properties between Oxic Metabolites and Anoxic Metabolites
Despite these limitations in data analysis, our findings cast doubt on whether oxygen availability increases chemical diversity in metabolic networks. Therefore, whether oxygen has led to the development of novel (
i.e., oxic) metabolites with chemical properties different from anoxic metabolites [
19] merely due to its presence remains unknown. Detailed examination is required as this conclusion has been derived through strongly biased evaluation, as described in
Section 1.
To determine whether previous observations are overestimated, underestimated, or accurate, we investigated the degrees of differences in the chemical properties of oxic metabolites and anoxic metabolites in individual metabolic networks, and calculated the likelihood that the degree computed from the integral network was obtained from the set of degrees calculated from individual metabolic networks.
In this study, we considered the effect size of the Wilcoxon–Mann–Whitney (WMW) test, a nonparametric test, as an indication of the degree of differences between the chemical properties of oxic metabolites and anoxic metabolites. Note that the
p-value is unsuitable for the degree of differences between two groups because sample size influences the
p-value. Thus, we examined effect sizes. Effect sizes are normalized measures for statistical analyses that are not influenced by sample sizes. Effect size (
ES) of the WMW test was obtained as follows:
where
z is the
z-score calculated from the
p-value obtained using the one-tailed WMW test and
Noxic and
Nanoxic are the number of oxic metabolites and the number of anoxic metabolites, respectively. Particularly,
ES > 0 or
ES < 0 indicates that a chemical property’s median of oxic metabolites is larger or smaller, respectively, than that of anoxic metabolites examined in this study.
The likelihood that the effect size (
ESint) of differences in chemical properties calculated from the integral networks was obtained from the set of effect sizes of the differences computed from individual metabolic networks was evaluated based on quantiles, which describe various subdivisions of a frequency distribution as they are distributed into equal proportions, as well as the evaluation value (
EV) [
33,
34], which is defined as:
where
MES is the median (
i.e., 50% quantile) of the effect sizes obtained from individual metabolic networks.
Qc denotes the effect size at 2.5 or 97.5% quantiles, depending on whether
ESint is lower or higher than
MES.
Using EV, we evaluated whether an observed value (i.e., ESint) was significantly lower or higher than MES. An EV larger than 1 indicates that ESint is not within the most likely 95% of effect sizes from individual metabolic networks, suggesting an overestimation or underestimation of ESint in individual metabolic networks.
Similarly, the Z-test can be used; however, this test assumes a normal distribution of variables. On the other hand, an evaluation using EV does not require assumptions regarding the distribution shape of the effect sizes obtained from individual metabolic networks; thus, it is more accurate than the Z-test.
We evaluated the differences (
i.e.,
Eint) in 84 chemical properties between oxic metabolites and anoxic metabolite, as were calculated in the previous study by Jiang
et al. [
19].
Jiang
et al. found that hydrophobicity (e.g., measured as AlogP98, the logarithm of a water/octanol partition coefficient) of oxic metabolites is a discriminative chemical property (
i.e., oxic metabolites are more hydrophobic than anoxic metabolites). As in the previous study, our analysis supported this conclusion when focusing on integral metabolic networks; however, we identified several overestimated and underestimated differences in the discriminative chemical properties of oxic metabolites, reported in the previous study, when considering the individual metabolic networks (
Table 2) of non-redundant species belonging to the same domain to reduce phylogenetic effects as much as possible (see
Section 3.1 and Tables S2 and S3 for details). In particular, the difference (
i.e.,
Eint) in hydrophobicity (e.g., AlogP98) between oxic metabolites and anoxic metabolites in the previous study were overestimated for aerobic unicellular organisms; however, this overestimation is not observed for multicellular eukaryotes (see also
Figure 2). Similar conclusions can be derived from the context of molecular solubility and fraction of charged atoms distributed on the molecular surface area. Additionally, the toxicity of oxic metabolites was overestimated in aerobic unicellular organisms, although some descriptors reflecting hydrophobicity (e.g., SOL and FPSA) and toxicity (
i.e., pLC50) were not highly overestimated in unicellular eukaryotes.
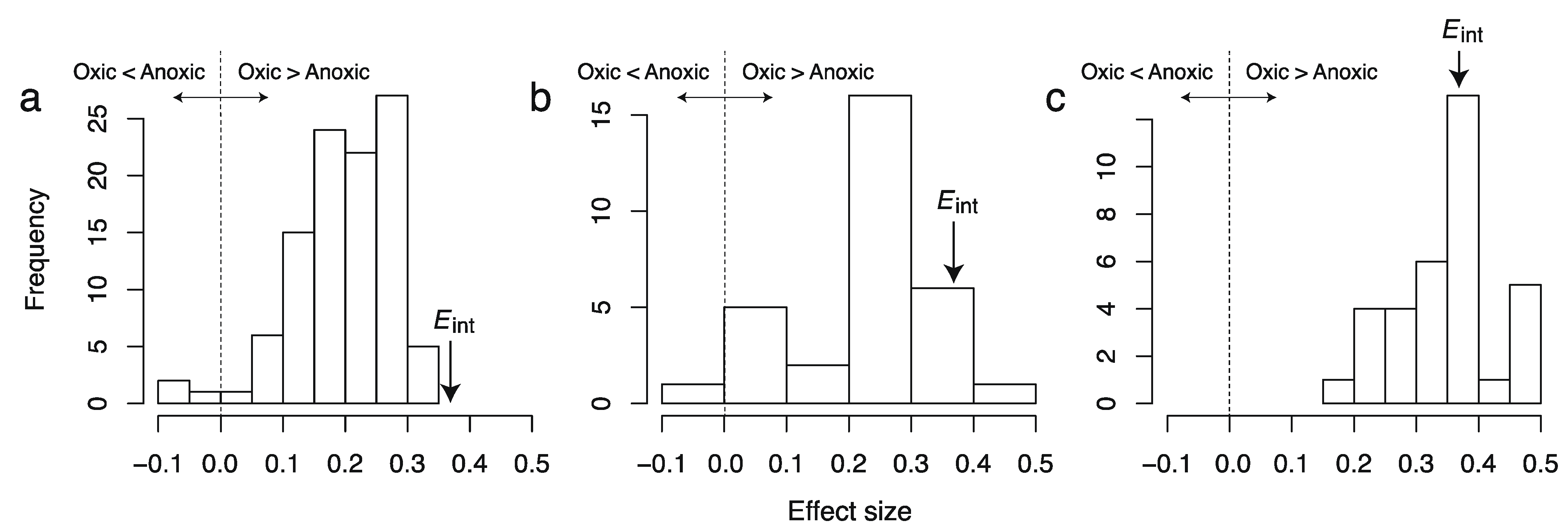
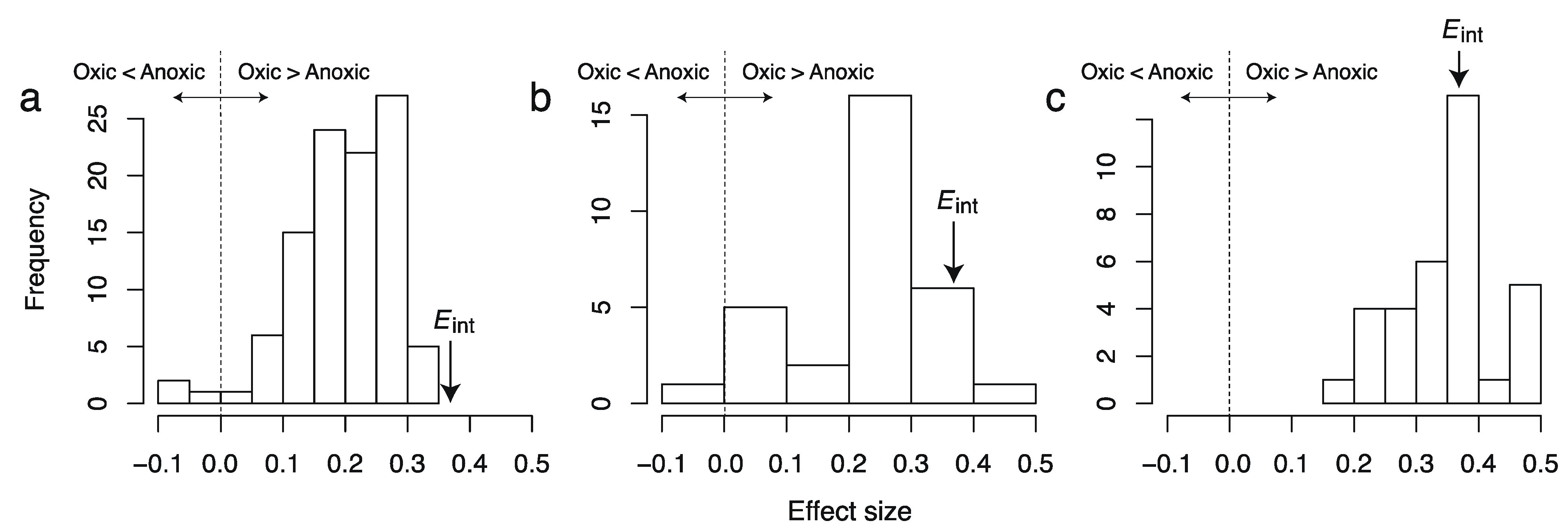
Figure 2.
Distributions of effect sizes of the difference in AlogP98 between oxic and anoxic metabolites in individual metabolic networks of (a) aerobic bacteria, (b) unicellular eukaryotes, and (c) multicellular eukaryotes. Eint is the effect size obtained from the integral network.
This result indicates that differences in hydrophobicity and toxicity are lower than previously thought, suggesting a lower impact of oxygen on increased chemical differences in this context. For example, alkaloids, which are a type of secondary metabolite in plant species and discriminative oxic metabolites, are strongly hydrophobic and toxic. It has been reported that other discriminative oxic metabolites are important for transmembrane export and import (steroids), signal transfer (steroids, diterpenoids, and polyphenols), defense against biotic factors (macrocyclic lactones in addition to alkaloids), and organism protection from oxidation (polyphenols) [
19]; however, these metabolites should be observed characteristically in higher organisms such plants. When using the integral metabolic network to investigate differences between oxic and anoxic metabolites, these metabolites were weighted regardless of whether they are less conserved among species. In fact, differences in these chemical properties are lower in aerobic unicellular organisms because these organisms synthesize only small amounts of such metabolites. In contrast, neither overestimation nor underestimation was observed for multicellular eukaryotes, as expected.
However, observed overestimations and underestimations do not completely reject the contribution of oxygen to metabolic evolution, as differences in chemical properties are partially concluded. For example, 70% of aerobic bacteria and 80% of unicellular eukaryotes showed significant difference in AlogP98 between oxic and anoxic metabolites (
p-value < 0.01, WMW test). Additionally, the rigidity of rotatable bonds (
i.e., RotBonds in
Table 2), another discriminative chemical property of oxic metabolites examined in the previous study [
19], was not overestimated for individual metabolic networks. This is because oxidoreductases, which are enriched in oxic enzymes [
19], contribute to increased rigidity because they convert single bonds and double bonds between two atoms.
Table 2.
Likelihood (evaluation value EV) that the effect size (Eint) of differences in chemical properties between oxic metabolites and anoxic metabolites calculated from the integral networks was obtained from the set of effect sizes of the differences computed from individual metabolic networks. A positive and negative effect size indicates that the value for a chemical property of oxic metabolites was larger and smaller, respectively, than that of anoxic metabolites. An EV value of larger than 1 indicates overestimation or underestimation of (Eint) in an individual network. P corresponds to the negative logarithmic p-value (i.e., –log10(p-value)) obtained from the Wilcoxon–Mann–Whitney (WMW) test. MES and MP denote the median of Ps and ESs in individual networks. 6 representative chemical properties between oxic metabolites and anoxic metabolites in a previous study [19] are shown. The descriptors are as follows: the logarithm of partition coefficient, atom-type value, using latest parameters (AlogP98), molecular solubility (SOL), ratio of atomic charge weighted partial negative surface area on total molecular surface area (FNSA), ratio of atomic charge weighted partial positive surface area on total molecular surface area (FPSA), negative log of lethal concentration 50% (pLC50), and rotatable bond count (RotBonds).
| Descriptor | Integral | Aerobic bacteria | Unicellular eukaryotes | Multicellular eukaryotes |
|---|
| Eint | P | MES | MP | EV | MES | MP | EV | MES | MP | EV |
|---|
| AlogP98 | 0.37 | 34.9 | 0.21 | 3.76 | 1.56 | 0.26 | 5.72 | 1.27 | 0.36 | 11.2 | 0.08 |
| SOL | −0.34 | 30.1 | −0.17 | 2.58 | 2.03 | −0.29 | 6.92 | 0.67 | −0.33 | 10.1 | 0.07 |
| FNSA | 0.32 | 26.4 | 0.13 | 1.78 | 1.08 | 0.16 | 2.43 | 1.00 | 0.26 | 7.06 | 0.30 |
| FPSA | −0.23 | 13.8 | −0.06 | 0.58 | 1.90 | −0.14 | 2.01 | 0.60 | −0.22 | 4.36 | 0.10 |
| pLC50 | 0.27 | 19.7 | 0.09 | 1.00 | 1.70 | 0.21 | 3.93 | 0.86 | 0.27 | 6.75 | 0.01 |
| RotBonds | −0.23 | 13.8 | −0.27 | 5.78 | 0.38 | −0.27 | 4.59 | 0.24 | −0.28 | 7.60 | 0.49 |
2.3. Does Oxygen Contribute to Metabolic Evolution?
The observation that network expansion was not conclusive when considering a phylogenetic relationship, and that overestimations or underestimations of the difference in chemical properties between oxic metabolites and anoxic metabolites were observed in aerobic unicellular organisms (particularly in aerobic bacteria) but not in multicellular organisms, suggests a limited association between oxygen requirements and chemical differences. Rather, phylogenetic signals and multicellularity (i.e., cell-cell communication) increase chemical diversity in metabolic networks.
Many studies state that oxygen accelerates evolution, such the emergence of multicellularity (e.g., reviewed in [
11,
12]), because most multicellular organisms showing great diversity (in the context of metabolites in this study) are obligately aerobic. This result suggests an association between oxygen and evolution; however, obligately aerobic unicellular organisms are less complex. For example, differences in chemical properties between oxic and anoxic metabolites are less significant than previously thought in obligately aerobic bacteria (
Table 2 and
Figure 2). Thus, its remains possible that the contribution of oxygen to metabolic evolution is an artifact due to strongly biased observations of (aerobic) multicellular organisms, as discussed in
Section 2.2.
To more effectively identify the presence of an association between oxygen and metabolic evolution, investigation of anaerobic multicellular organisms is required. For example, Danovaro
et al. [
35] demonstrated that a representative from the animal phylum Loricifera lives in a deep, permanently anaerobic, and hypersaline water basin in the Mediterranean Sea. This organism may be useful in investigating whether oxygen contributes to molecular diversity, although its genomic and metabolic data are not available. If oxygen accelerates the increase in molecular diversity, then such an increase in diversity should not be observed for anaerobic multicellular organisms.
An opposite hypothesis is that the contribution of oxygen to metabolic evolution is limited and can be examined by investigating anaerobic multicellular organisms. Our hypothesis assumes that an association between oxygen and metabolic evolution is a side effect of the relationship between oxygen requirements and multicellularity. In particular, the acquirement of multicellularity may be a strategy for protecting against oxidative stress. Oxygen is useful for energy production; however, it also plays a role as a biomolecular denaturant. Unicellular organisms are quite susceptible to oxidative stress, but they can avoid such stress by aggregating to reduce their exposed surface areas. In this case, cell-cell communication is required. Thus, unicellular organisms acquired more hydrophobic compounds for signal transfer (
i.e., communications between membranes), and finally evolved into multicellular organisms. This hypothesis may be supported that fact that the difference in chemical properties between oxic metabolites and anoxic metabolites are significant in multicellular organisms, but not in unicellular organisms (aerobic bacteria, in particular) (
Table 2). This indicates that oxygen is just one possible reason for the emergence of multicellularity (
i.e., cell-cell communication), suggesting that increased chemical diversity was influenced by cell-cell communication rather than oxygen. Therefore, if our hypothesis is more fitting, anaerobic multicellular organisms show increased chemical diversity, such as network expansion and overestimated differences in the chemical properties between oxic and anoxic metabolites, which contradicts the classical hypothesis.
Our conclusion that oxygen has a weak effect on evolution is limited to the context of chemical diversity in metabolic networks. As described in
Section 1, many previous studies have provided evidence that oxygen contributes to species and morphological diversity in higher organisms such as fish and insects. Such macroscopic evolution after the emergence of multicellular organisms may be explained in a different context.
Our analysis (in
Section 2.1 and
Section 2.2) has general limitations, as do many other works on metabolic network analyses, including limited knowledge of metabolic reactions (
i.e., missing links), reconstruction of metabolic networks based on genomic information. That is, it remains possible that our conclusion is affected by the difference in the percentage of functionally-unknown proteins in a species genome between aerobes and anaerobes. Thereby, we evaluated the fraction of functionally-unknown proteins in genome for each species (see
Section 3.5).
When using the dataset (see Table S1), for example, we cannot conclude any difference in the fraction of functionally-unknown proteins among aerobes, facultative aerobes, and anaerobes (Figure S2). (p = 0.33 using the Kruskal-Wallis test, whose alternative hypothesis is that at least one group is different from the other groups). Thus, the difference in the fraction of functionally-unknown proteins might not seriously affect our conclusions.
Nevertheless, metabolic networks have not been fully understood; thus, there is a need for a more careful examination in data analysis in the future. For example, enzyme promiscuity [
36], which implies that enzymes can catalyze multiple reactions, act on more than one substrate, or exert a range of suppressions [
37], in which an enzymatic function is suppressed by over-expressing enzymes showing originally different functions, suggests the existence of many hidden metabolic reactions. Consideration of these hidden metabolic reactions is important for designing metabolic pathways and understanding metabolic evolution.
Although data analysis has these limitations, our finding encourages a more careful examination of an association between oxygen and evolution in both macroscopic and microscopic contexts.



{kind=link}
{kind=link}