Intracellular Metabolite Pool Changes in Response to Nutrient Depletion Induced Metabolic Switching in Streptomyces coelicolor

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
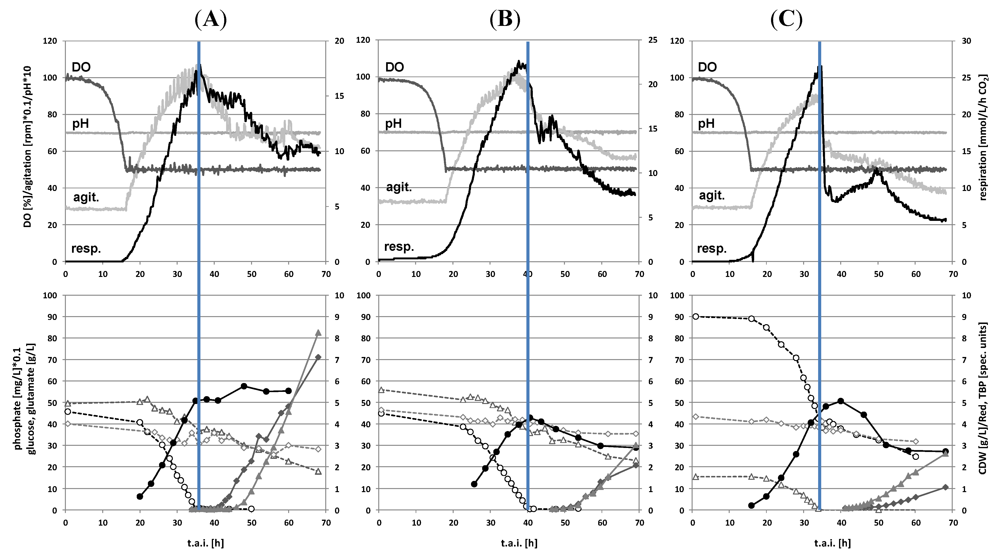
2.1. Batch Cultivations with Time Series Metabolite Sampling
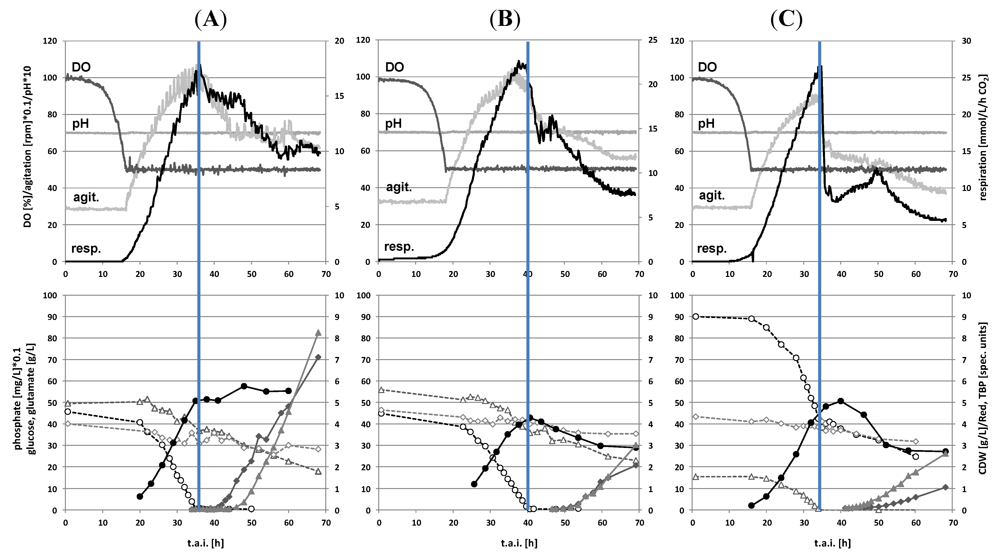
2.2. Metabolite Profiling of the Transition Phase

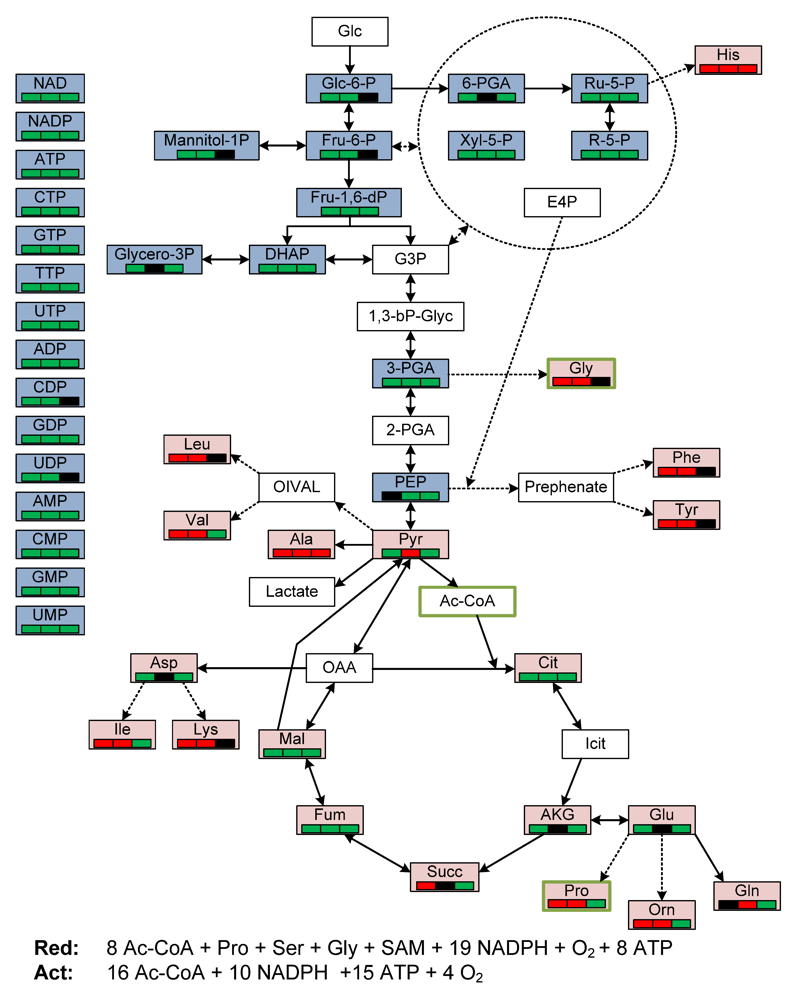
2.3. General Discussion
3. Experimental
3.1. Strain and General Cultivation Parameters
3.2. Sampling and Quenching
3.3. Metabolite Extraction
3.4. Metabolite Derivatization with Methyl Chloroformate (MCF) and GC-MS Analysis
3.5. LC-MS/MS Analysis
3.6. Data Processing and Visualization
4. Conclusions
Acknowledgments
References and Notes
- Chater, K.F. Streptomyces inside-out: A new perspective on the bacteria that provide us with antibiotics. Philos. Trans. R. Soc. B-Biol. Sci. 2006, 361, 761–768. [Google Scholar]
- Hesketh, A.; Bucca, G.; Laing, E.; Flett, F.; Hotchkiss, G.; Smith, C.P.; Chater, K.F. New pleiotropic effects of eliminating a rare tRNA from Streptomyces coelicolor, revealed by combined proteomic and transcriptomic analysis of liquid cultures. BMC Genomics 2007, 8, 261. [Google Scholar]
- Lian, W.; Jayapal, K.P.; Charaniya, S.; Mehra, S.; Glod, F.; Kyung, Y.S.; Sherman, D.H.; Hu, W.S. Genome-wide transcriptome analysis reveals that a pleiotropic antibiotic regulator, AfsS, modulates nutritional stress response in Streptomyces coelicolor A3(2). BMC Genomics 2008, 9, 56. [Google Scholar]
- Zhou, Z.; Gu, J.Y.; Du, Y.L.; Li, Y.Q.; Wang, Y.F. The -omics era- toward a systems-level understanding of Streptomyces. Curr. Genomics 2011, 12, 404–416. [Google Scholar]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar]
- Nieselt, K.; Battke, F.; Herbig, A.; Bruheim, P.; Wentzel, A.; Jakobsen, O.M.; Sletta, H.; Alam, M.T.; Merlo, M.E.; Moore, J.; et al. The dynamic architecture of the metabolic switch in Streptomyces coelicolor. BMC Genomics 2010, 11, 10. [Google Scholar]
- Alam, M.T.; Merlo, M.E.; Hodgson, D.A.; Wellington, E.M.; Takano, E.; Breitling, R. Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genomics 2010, 11, 202. [Google Scholar]
- Waldvogel, E.; Herbig, A.; Battke, F.; Amin, R.; Nentwich, M.; Nieselt, K.; Ellingsen, T.; Wentzel, A.; Hodgson, D.; Wohlleben, W.; et al. The Pii protein GlnK is a pleiotropic regulator for morphological differentiation and secondary metabolism in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 2011, 92, 1219–1236. [Google Scholar]
- Thomas, L.; Hodgson, D.A.; Wentzel, A.; Nieselt, K.; Ellingsen, T.E.; Moore, J.; Morrissey, E.R.; Legaie, R.; the STREAM Consortium; Wohlleben, W.; et al. Metabolic switches and adaptations deduced from the proteomes of Streptomyces coelicolor wild type and phoP mutant grown in batch culture. Mol. Cell. Proteomics 2011. [Google Scholar]
- Battke, F.; Herbig, A.; Wentzel, A.; Jakobsen, O.M.; Bonin, M.; Hodgson, D.A.; Wohlleben, W.; Ellingsen, T.E.; Nieselt, K. A technical platform for generating reproducible expression data from Streptomyces coelicolor batch cultivations. Adv. Exp. Med. Biol. 2011, 696, 3–15. [Google Scholar]
- Villas-Boas, S.G.; Bruheim, P. The potential of metabolomics tools in bioremediation studies. OMICS 2007, 11, 305–313. [Google Scholar] [CrossRef]
- Nielsen, J.; Oliver, S. The next wave in metabolome analysis. Trends Biotechnol. 2005, 23, 544–546. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Overkamp, K.M.; Muilwijk, B.; Coulier, L.; Hankemeier, T. Microbial metabolomics: Toward a platform with full metabolome coverage. Anal. Biochem. 2007, 370, 17–25. [Google Scholar]
- Kvitvang, H.F.N.; Andreassen, T.; Adam, T.; Villas-Boas, S.G.; Bruheim, P. Highly sensitive GC/MS/MS method for quantitation of amino and nonamino organic acids. Anal. Chem. 2011, 83, 2705–2711. [Google Scholar]
- Geng, P.; Meng, X.S.; Bai, G.; Luo, G.A. Profiling of acarviostatin family secondary metabolites secreted by Streptomyces coelicoflavus ZG0656 using ultraperformance liquid chromatography coupled with electrospray ionization mass spectrometry. Anal. Chem. 2008, 80, 7554–7561. [Google Scholar]
- Brautaset, T.; Bruheim, P.; Sletta, H.; Hagen, L.; Ellingsen, T.E.; Strom, A.R.; Valla, S.; Zotchev, S.B. Hexaene derivatives of nystatin produced as a result of an induced rearrangement within the nysC polyketide synthase gene in S. noursei ATCC 11455. Chem. Biol. 2002, 9, 367–373. [Google Scholar] [CrossRef]
- Bruheim, P.; Borgos, S.E.F.; Tsan, P.; Sletta, H.; Ellingsen, T.E.; Lancelin, J.M.; Zotchev, S.B. Chemical diversity of polyene macrolides produced by Streptomyces noursei ATCC 11455 and recombinant strain ERD44 with genetically altered polyketide synthase NysC. Antimicrob. Agents Chemother. 2004, 48, 4120–4129. [Google Scholar]
- Jankevics, A.; Merlo, M.E.; de Vries, M.; Vonk, R.J.; Takano, E.; Breitling, R. Metabolomic analysis of a synthetic metabolic switch in Streptomyces coelicolor A3(2). Proteomics 2011, 11, 4622–4631. [Google Scholar]
- Rodriguez-Garcia, A.; Barreiro, C.; Santos-Beneit, F.; Sola-Landa, A.; Martin, J.F. Genome-wide transcriptomic and proteomic analysis of the primary response to phosphate limitation in Streptomyces coelicolor M145 and in a deltaphoP mutant. Proteomics 2007, 7, 2410–2429. [Google Scholar]
- Santos-Beneit, F.; Rodriguez-Garcia, A.; Sola-Landa, A.; Martin, J.F. Cross-talk between two global regulators in streptomyces: PhoP and AfsR interact in the control of afsS, pstS and phoRP transcription. Mol. Microbiol. 2009, 72, 53–68. [Google Scholar]
- Winder, C.L.; Dunn, W.B.; Schuler, S.; Broadhurst, D.; Jarvis, R.; Stephens, G.M.; Goodacre, R. Global metabolic profiling of Escherichia coli cultures: An evaluation of methods for quenching and extraction of intracellular metabolites. Analy. Chem. 2008, 80, 2939–2948. [Google Scholar]
- Kirschner, M.W. The meaning of systems biology. Cell 2005, 121, 503–504. [Google Scholar] [CrossRef]
- Manteca, A.; Fernandez, M.; Sanchez, J. Cytological and biochemical evidence for an early cell dismantling event in surface cultures of Streptomyces antibioticus. Res. Microbiol. 2006, 157, 143–152. [Google Scholar]
- Yague, P.; Manteca, A.; Simon, A.; Diaz-Garcia, M.E.; Sanchez, J. New method for monitoring programmed cell death and differentiation in submerged Streptomyces cultures. Appl. Environ. Microbiol. 2010, 76, 3401–3404. [Google Scholar]
- Dolezal, J.; Kapralek, F. Physiological characteristics of chemostatically grown Citrobacter freundii as a function of the specific growth rate and type of nutrient limitation. Folia Microbiol. (Praha) 1976, 21, 168–177. [Google Scholar]
- O'Sullivan, E.; Condon, S. Relationship between acid tolerance, cytoplasmic pH, and ATP and H+-ATPase levels in chemostat cultures of Lactococcus lactis. Appl. Environ. Microbiol. 1999, 65, 2287–2293. [Google Scholar]
- Rane, K.D.; Sims, K.A. Oxygen uptake and citric acid production by Candida lipolytica Y 1095. Biotechnol. Bioeng. 1994, 43, 131–137. [Google Scholar] [CrossRef]
- Liras, P.; Villanueva, J.R.; Martin, J.F. Sequential expression of macromolecule biosynthesis and candicidin formation in Streptomyces griseus. J. Gen. Microbiol. 1977, 102, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Vu-Trong, K.; Bhuwapathanapun, S.; Gray, P.P. Metabolic regulation in tylosin-producing Streptomyces fradiae: Regulatory role of adenylate nucleotide pool and enzymes involved in biosynthesis of tylonolide precursors. Antimicrob. Agents Chemother. 1980, 17, 519–525. [Google Scholar]
- Bascaran, V.; Sanchez, L.; Hardisson, C.; Brana, A.F. Stringent response and initiation of secondary metabolism in Streptomyces clavuligerus. J. Gen. Microbiol. 1991, 137, 1625–1634. [Google Scholar] [CrossRef]
- van der Werf, M.J.; Overkamp, K.M.; Muilwijk, B.; Koek, M.M.; van der Werff-van der Vat, B.J.; Jellema, R.H.; Coulier, L.; Hankemeier, T. Comprehensive analysis of the metabolome of Pseudomonas putida S12 grown on different carbon sources. Mol. Biosyst. 2008, 4, 315–327. [Google Scholar] [CrossRef]
- Barrette, W.C., Jr.; Hannum, D.M.; Wheeler, W.D.; Hurst, J.K. Viability and metabolic capability are maintained by Escherichia coli, Pseudomonas aeruginosa, and Streptococcus lactis at very low adenylate energy charge. J. Bacteriol. 1988, 170, 3655–3659. [Google Scholar]
- Battke, F.; Symons, S.; Nieselt, K. Mayday—Integrative analytics for expression data. BMC Bioinformatics 2010, 11, 121. [Google Scholar] [CrossRef]
- Gunnarsson, N.; Bruheim, P.; Nielsen, J. Glucose metabolism in the antibiotic producing actinomycete Nonomuraea sp ATCC 39727. Biotechnol. Bioeng. 2004, 88, 652–663. [Google Scholar] [CrossRef]
- Bruheim, P.; Butler, M.; Ellingsen, T.E. A theoretical analysis of the biosynthesis of actinorhodin in a hyper-producing Streptomyces lividans strain cultivated on various carbon sources. Appl. Microbiol. Biotechnol. 2002, 58, 735–742. [Google Scholar] [CrossRef]
- Butler, M.J.; Takano, E.; Bruheim, P.; Jovetic, S.; Marinelli, F.; Bibb, M.J. Deletion of scbA enhances antibiotic production in Streptomyces lividans. Appl. Microbiol. Biotechnol. 2003, 61, 512–516. [Google Scholar]
- Becker, J.; Zelder, O.; Hafner, S.; Schroder, H.; Wittmann, C. From zero to hero—Design-based systems metabolic engineering of Corynebacterium glutamicum for L-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef]
- Ikeda, M.; Ohnishi, J.; Hayashi, M.; Mitsuhashi, S. A genome-based approach to create a minimally mutated Corynebacterium glutamicum strain for efficient L-lysine production. J. Ind. Microbiol. Biotechnol. 2006, 33, 610–615. [Google Scholar]
- Claessen, D.; Rink, R.; de Jong, W.; Siebring, J.; de Vreugd, P.; Boersma, F.G.; Dijkhuizen, L.; Wosten, H.A. A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes Dev. 2003, 17, 1714–1726. [Google Scholar]
- Villas-Boas, S.G.; Delicado, D.G.; Akesson, M.; Nielsen, J. Simultaneous analysis of amino and nonamino organic acids as methyl chloroformate derivatives using gas chromatography-mass spectrometry. Anal. Biochem. 2003, 322, 134–138. [Google Scholar]
- Luo, B.; Groenke, K.; Takors, R.; Wandrey, C.; Oldiges, M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J. Chromatogr. A 2007, 1147, 153–164. [Google Scholar]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wentzel, A.; Sletta, H.; Consortium, S.; Ellingsen, T.E.; Bruheim, P. Intracellular Metabolite Pool Changes in Response to Nutrient Depletion Induced Metabolic Switching in Streptomyces coelicolor. Metabolites 2012, 2, 178-194. https://doi.org/10.3390/metabo2010178
Wentzel A, Sletta H, Consortium S, Ellingsen TE, Bruheim P. Intracellular Metabolite Pool Changes in Response to Nutrient Depletion Induced Metabolic Switching in Streptomyces coelicolor. Metabolites. 2012; 2(1):178-194. https://doi.org/10.3390/metabo2010178
Chicago/Turabian StyleWentzel, Alexander, Havard Sletta, Stream Consortium, Trond E. Ellingsen, and Per Bruheim. 2012. "Intracellular Metabolite Pool Changes in Response to Nutrient Depletion Induced Metabolic Switching in Streptomyces coelicolor" Metabolites 2, no. 1: 178-194. https://doi.org/10.3390/metabo2010178