
Methods for Differentiating Prion Types in Food-Producing Animals

Abstract
:1. Introduction
1.1. Transmissible Spongiform Encephalopathies, and the Concept of Prions as an Infectious Agent
1.2. Examples of Prion Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prion types * | Host Species |
|---|---|
| Classical BSE (bovine spongiform encephalopathy) | Cattle, Goats # |
| Atypical BSE: H-BSE, L-BSE/bovine amyloidotic spongiform encephalopathy (BASE) | Cattle |
| Classical Scrapie ^ | Sheep, Goats |
| CH1641 Scrapie + | Sheep |
| Atypical/Nor98 Scrapie | Sheep, Goats |
| CWD (chronic wasting disease) | White-tailed deer, Elk, Mule Deer, Moose |
1.3. TSE Diagnosis
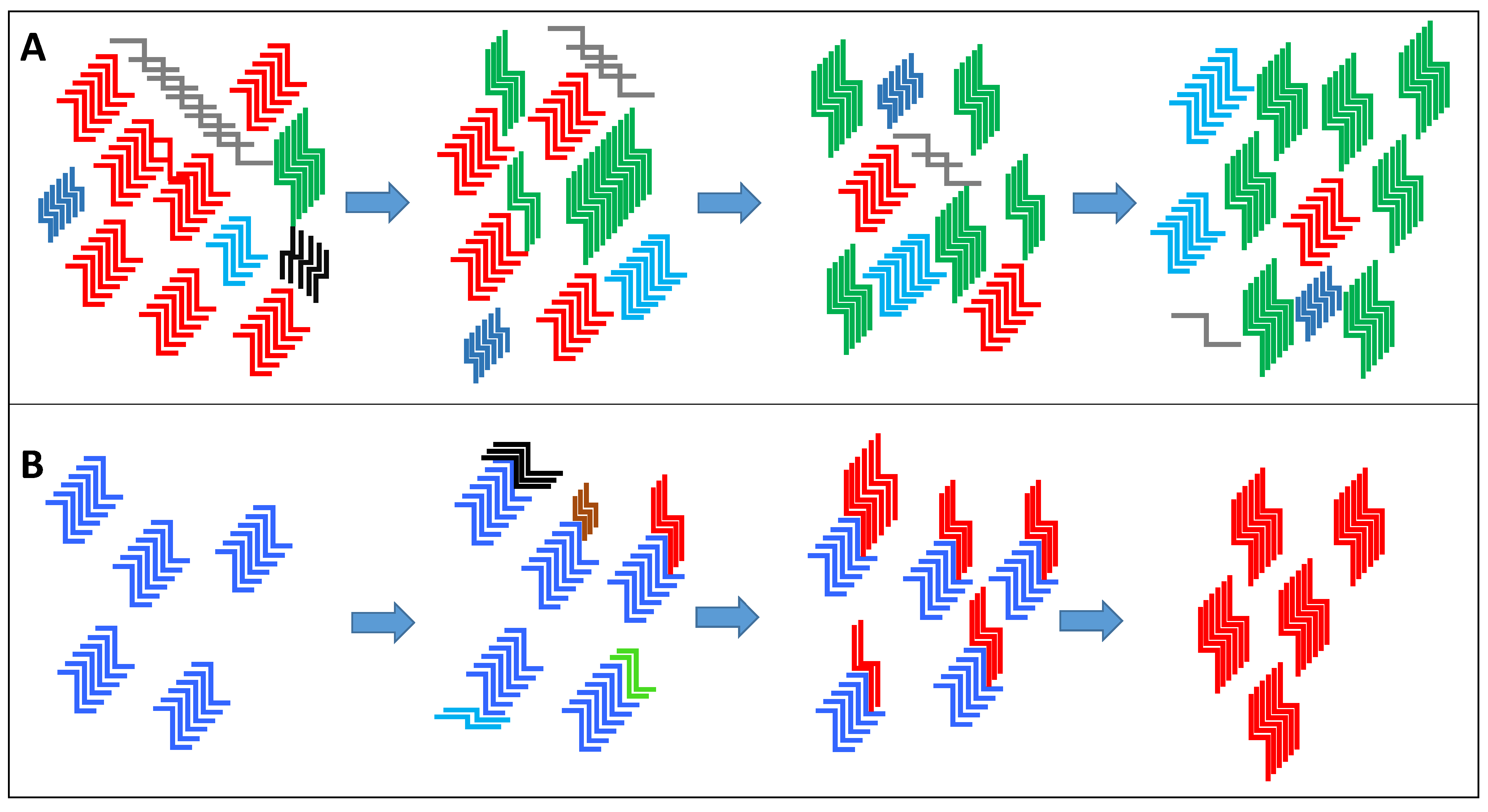
1.4. The Concept of Prion Strains and Species Barrier
1.5. The Emergence of “New” Prion Strains
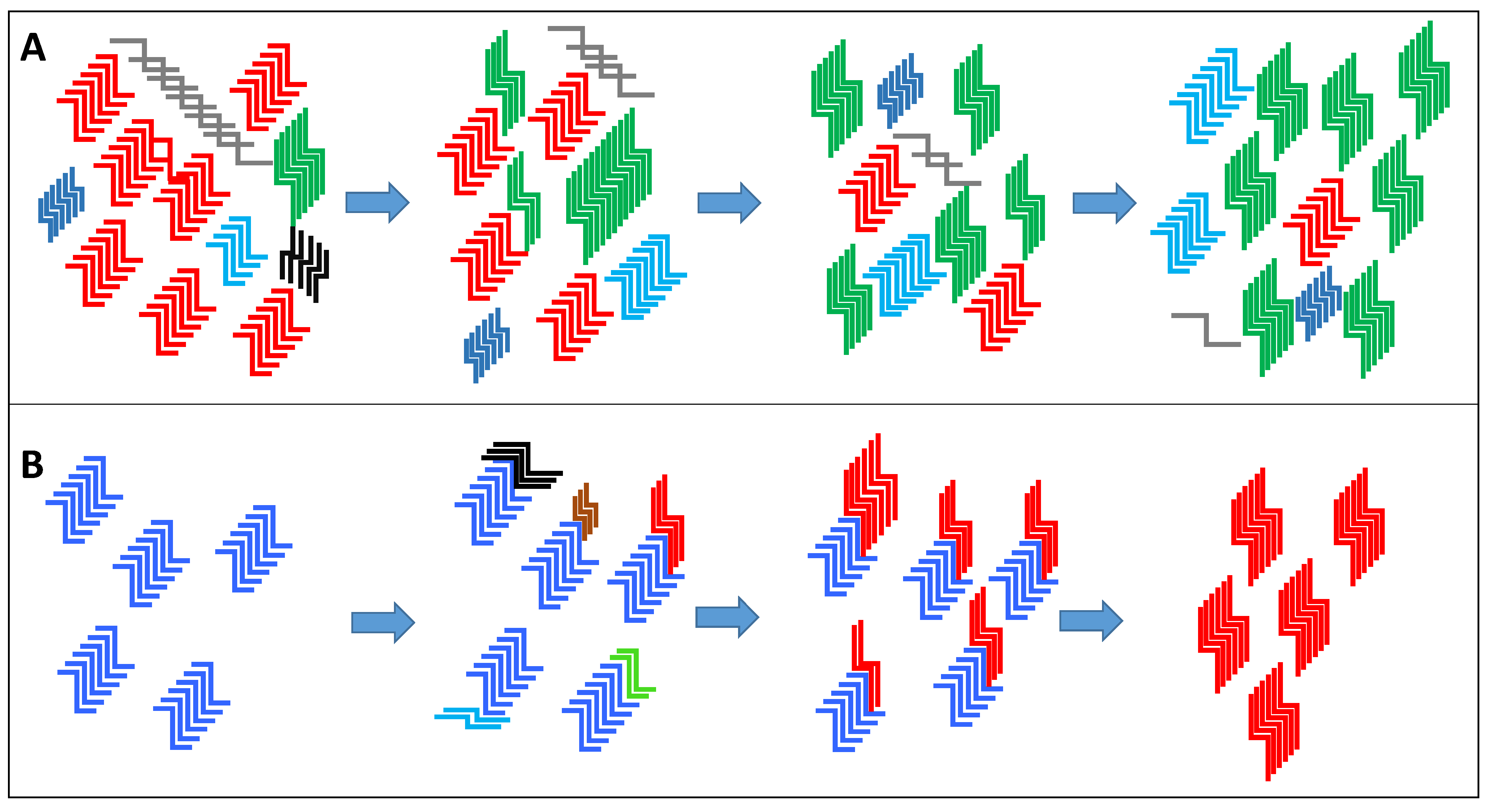
1.6. The Significance of New Strain Emergence
1.7. Prion Diseases that Affect Food-Producing Animals
1.7.1. BSE
1.7.2. Scrapie
1.7.3. CWD
2. Typing of TSEs in Food-Producing Animals
2.1. Rodent Bioassay: Wild-Type Mouse Lines
2.2. Rodent Bioassay: Transgenic Mouse Lines
2.3. Bioassay in Other Model Species
3. Histological and Immunohistochemical Analysis of Tissue from Natural Hosts
3.1. Histology
3.2. Immunohistochemistry
3.3. Diagnostic Applications of Histology and Immunohistochemistry
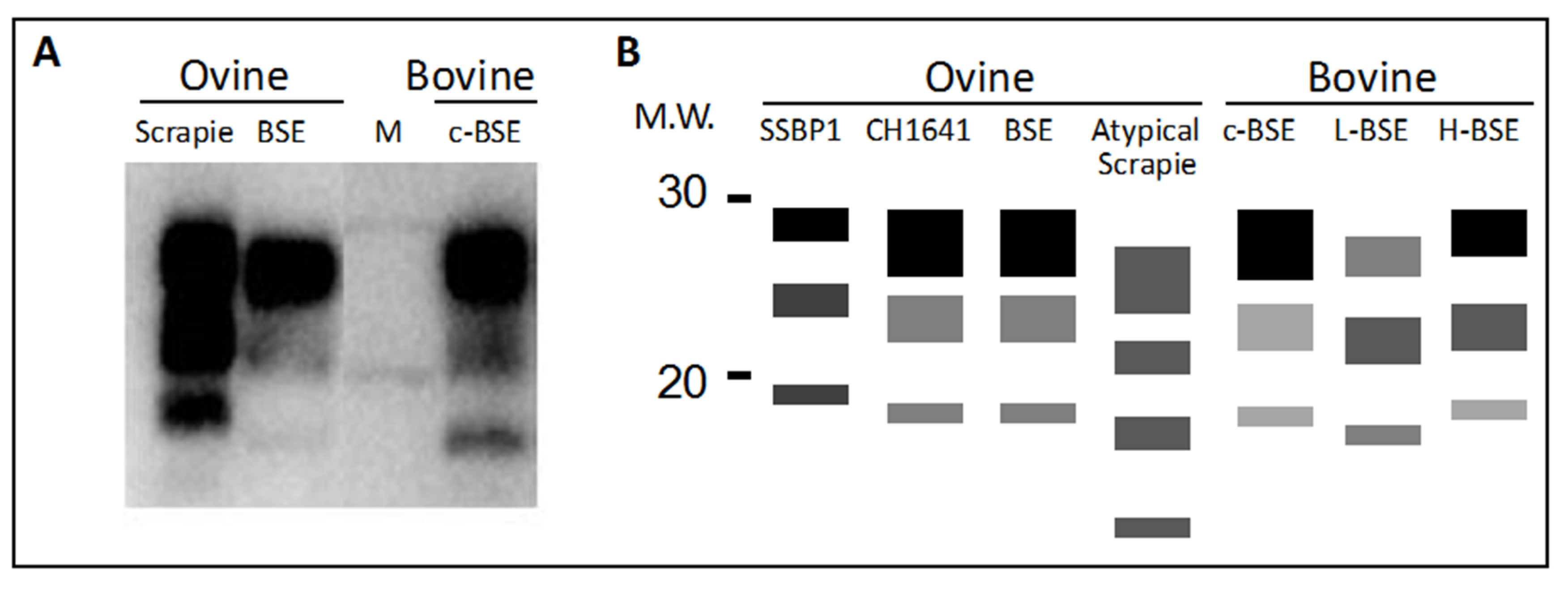
4. Protease Fragmentation of PrPSc and Glycoform Ratios
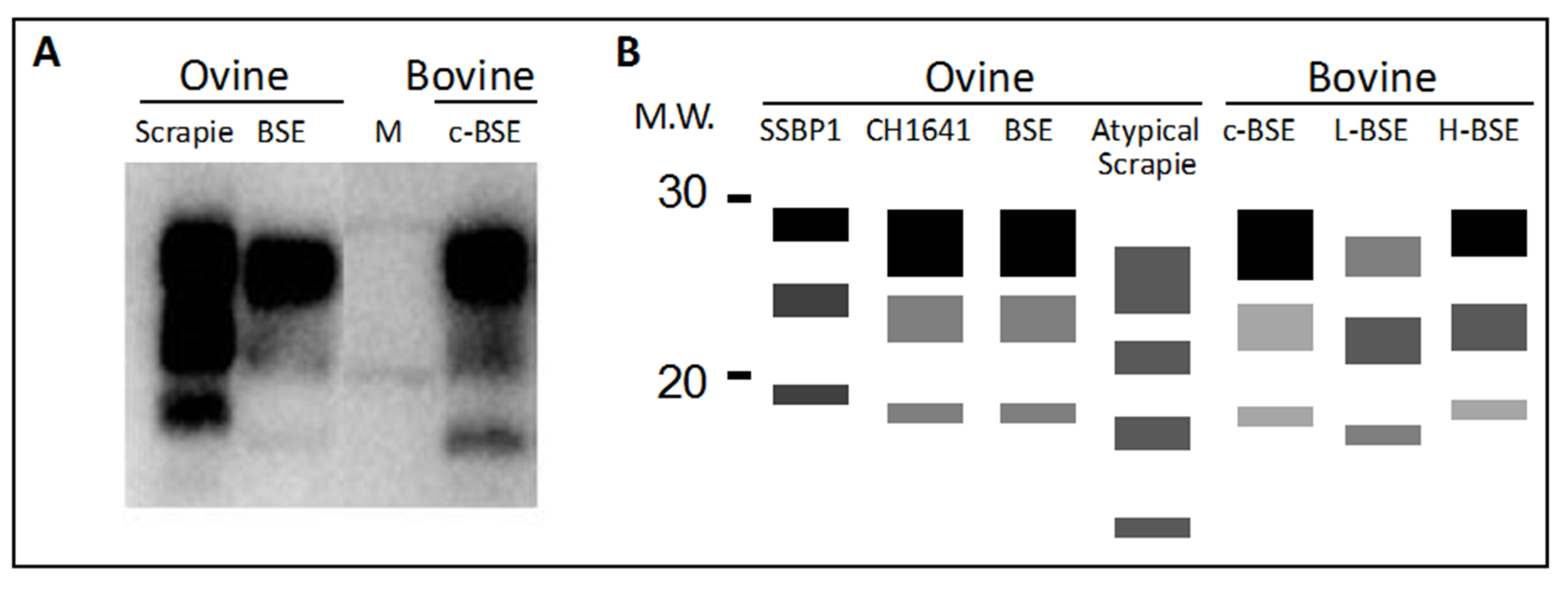
4.1. Differentiating Prion Isolates in Small Ruminants
4.2. Differentiating Cattle BSE Types
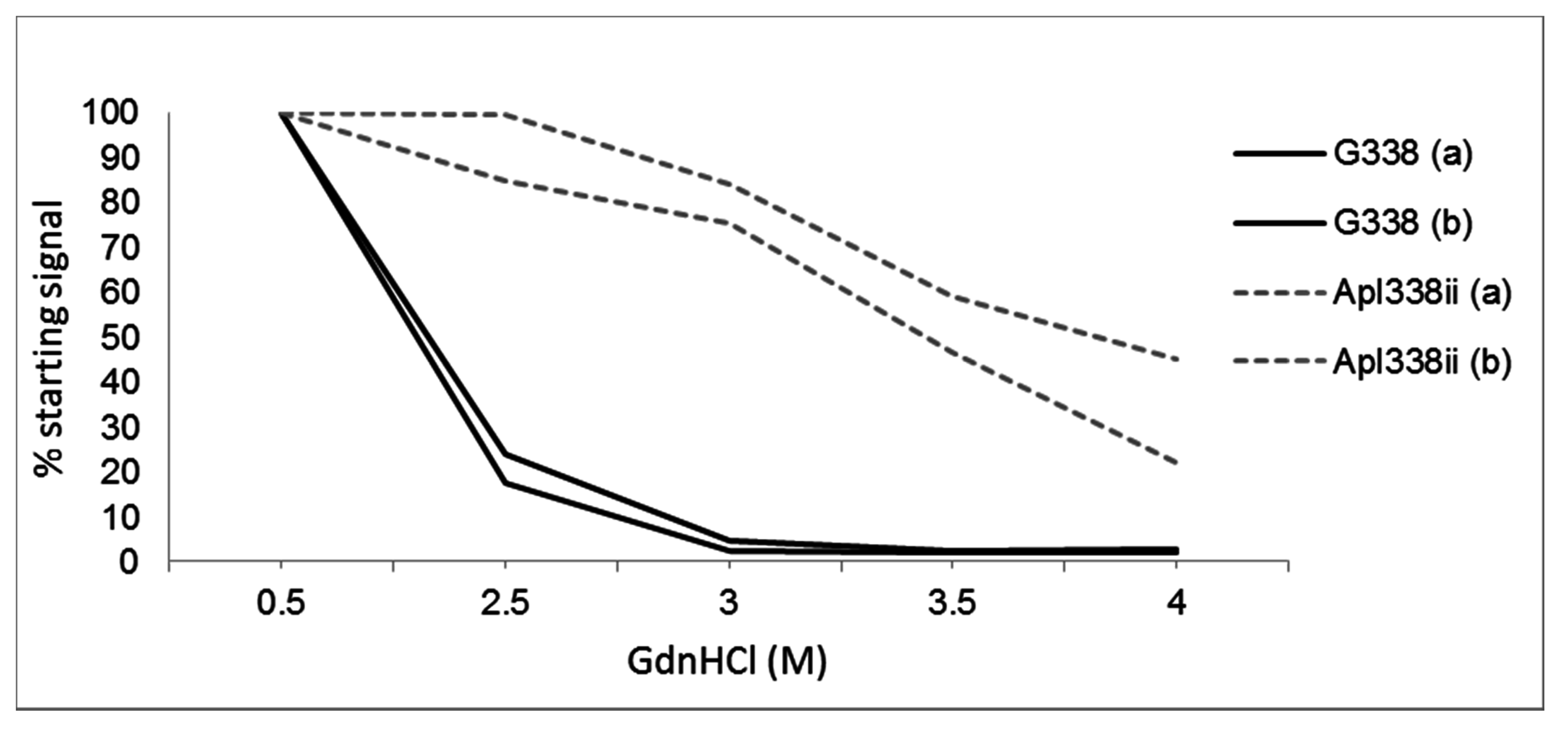
5. Differential Stability of Prion Types
6. Cell Culture Methods
7. In Vitro Replication of Prion Types
8. Conclusions and Future Perspectives
Author Contributions
Conflicts of Interest
Abbreviations
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.K.; McKinley, M.P.; Bowman, K.A.; Braunfeld, M.B.; Barry, R.A.; Prusiner, S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Morignat, E.; Oussaid, N.; Gay, C.; Abrial, D.; Ducrot, C.; Calavas, D. Individual factors associated with l- and h-type bovine spongiform encephalopathy in france. BMC Vet. Res. 2009, 8, e74. [Google Scholar] [CrossRef] [PubMed]
- Biacabe, A.; Morignat, E.; Vulin, J.; Calavas, D.; Baron, T. Atypical bovine spongiform encephalopathies, France, 2001–2007. Emerg. Infect. Dis. 2008, 14, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Simmons, H.A.; Simmons, M.M.; Spencer, Y.I.; Chaplin, M.J.; Povey, G.; Davis, A.; Ortiz-Pelaez, A.; Hunter, N.; Matthews, D.; Wrathall, A.E. Atypical scrapie in sheep from a uk research flock which is free from classical scrapie. BMC Vet. Res. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Fediaevsky, A.; Maurella, C.; Noremark, M.; Ingravalle, F.; Thordeirsdottir, S.; Orge, L.; Poizat, R.; Hautaniemi, M.; Liam, B.; Calavas, D.; et al. The prevalence of atypical scrapie in sheep from positive flocks is not higher than in the general sheep population in 11 European countries. BMC Vet. Res. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.G.; Sauer, M.; van Keulen, L.J.M.; Tang, Y.; Bossers, A.; Langeveld, J.P.M. Differentiation of ruminant transmissible spongiform encephalopathy isolate types, including bovine spongiform encephalopathy and ch1641 scrapie. J. Gen. Virol. 2011, 92, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Langeveld, J.P.; Erkens, J.H.; Rammel, I.; Jacobs, J.G.; Davidse, A.; van Zijderveld, F.G.; Bossers, A.; Schildorfer, H. Four independent molecular prion protein parameters for discriminating new cases of C, L, and h bovine spongiform encephalopathy in cattle. J. Clin. Microbiol. 2011, 49, 3026–3028. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E. Tse strain variation. Br. Med. Bull. 2003, 66, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.; Jeffrey, M.; Gubbins, S.; Grimmer, S.; Gonzalez, L.; Martin, S.; Chaplin, M.; Webb, P.; Simmons, M.; Spencer, Y.; et al. Monitoring for bovine spongiform encephalopathy in sheep in great britain, 1998–2004. J. Gen. Virol. 2006, 87, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itrri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have prpsc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.R.; Peretz, D.; Nguyen, H.O.B.; DeArmond, S.J.; Prusiner, S.B. Transmission barriers for bovine ovine, and human prions in transgenic mice. J. Virol. 2005, 79, 5259–5271. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Hope, J.; Fraser, H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet. Rec. 1993, 133, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.K.; Cunningham, A.A. Epidemiologic observations on spongiform encephalopathies in captive wild animals in the british-isles. Vet. Rec. 1994, 135, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A.; Fraser, H. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J. Gen. Virol. 1989, 70, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Baskakov, I.V. The evolution of transmissible prions: The role of deformed templating. PLoS Pathog. 2013, 9, e1003759. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Wain, R.; Baskakov, I.V.; Legname, G.; Palmer, C.G.; Nguyen, H.-O.B.; Lemus, A.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Protease-sensitive synthetic prions. PLoS Pathog. 2010, 6, e1000736. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Lee, Y.J.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Changes in prion replication environment cause prion strain mutation. FASEB J. 2013, 27, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Ahn, M.; Lessard, P.; Giles, K.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009, 5, e1000673. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Maddison, B.C.; Spiropoulos, J.; Vickery, C.M.; Lockey, R.; Owen, J.P.; Bishop, K.; Baker, C.A.; Gough, K.C. Incubation of ovine scrapie with environmental matris results in biological and biochemical changes of prpsc over time. Vet. Res. 2015, 46. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.H.; Scott, A.C.; Johnson, C.T.; Gunning, R.F.; Hancock, R.D.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.G.; Langeveld, J.P.M.; Biacabe, A.G.; Acutis, P.L.; Polak, M.P.; Gavier-Widen, D.; Buschmann, A.; Caramelli, M.; Casalone, C.; Mazza, M.; et al. Molecular discrimination of atypical bovine spongiform encephalopathy strains from a geographical region spanning a wide area in europe. J. Clin. Microbiol. 2007, 45, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, A.; Gretzschel, A.; Biacabe, A.G.; Schiebel, K.; Corona, C.; Hoffmann, C.; Eiden, M.; Baron, T.; Casalone, C.; Groschup, M.H. Atypical BSE in Germany—Proof of transmissibility and biochemical characterization. Vet. Microbiol. 2006, 117, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Casalone, C.; Zanusso, G.; Acutis, P.; Ferrari, S.; Capucci, L.; Tagliavini, F.; Monaco, S.; Caramelli, M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic creutzfeldt-jakob disease. Proc. Natl. Acad. Sci. USA 2004, 101, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Biacabe, A.G.; Laplanche, J.L.; Ryder, S.; Baron, T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004, 5, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Eloit, M.; Adjou, K.; Coulpier, M.; Fontaine, J.J.; Hamel, R.; Lilin, T.; Messiaen, S.; Andreoletti, O.; Baron, T.; Bencsik, A.; et al. BSE agent signatures in a goat. Vet. Rec. 2005, 156, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; Gonzalez, L.; Foster, J.; Langeveld, J.P.M.; van Zijderveld, F.G.; Grassi, J.; Hunter, N. Immunohistochemical features of PrP d accumulation in natural and experimental goat transmissible spongiform encephalopathies. J. Comp. Pathol. 2006, 134, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Sarradin, P.; Thu, B.; Schonheit, J.; Tranulis, M.A.; Bratberg, B. Cases of scrapie with unusual features in norway and designation of a new type, nor98. Vet. Rec. 2003, 153, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Everest, S.J.; Thorne, L.; Barnicle, D.A.; Edwards, J.C.; Elliott, H.; Jackman, R.; Hope, J. Atypical prion protein in sheep brain collected during the british scrapie-surveillance programme. J. Gen. Virol. 2006, 87, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Arsac, J.N.; Goldmann, W.; Noremark, M. Atypical/nor98 scrapie: Properties of the agent, genetics, and epidemiology. Vet. Res. 2008, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seuberlich, T.; Botteron, C.; Benestad, S.L.; Brunisholz, H.; Wyss, R.; Kihm, U.; Schwermer, H.; Friess, M.; Nicolier, A.; Heim, D.; et al. Atypical scrapie in a swiss goat and implications for transmissible spongiform encephalopathy surveillance. J. Vet. Diagn. Investig. 2007, 19, 2–8. [Google Scholar] [CrossRef]
- Mazza, M.; Iulini, B.; Vaccari, G.; Acutis, P.L.; Martucci, F.; Esposito, E.; Peletto, S.; Barocci, S.; Chiappini, B.; Corona, C.; et al. Co-existence of classical scrapie and nor98 in a sheep from an italian outbreak. Res. Vet. Sci. 2010, 88, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Baeten, L.A.; Powers, B.E.; Jewell, J.E.; Spraker, T.R.; Miller, M.W. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). J. Wildl. Dis. 2007, 43, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; Miller, M.W.; Williams, E.S.; Getzy, D.M.; Adrian, W.J.; Schoonveld, G.G.; Spowart, R.A.; Orourke, K.I.; Miller, J.M.; Merz, P.A. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and rocky mountain elk (Cervus elaphus nelsoni) in northcentral colorado. J. Wildl. Dis. 1997, 33, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.K.; Cunningham, A.A.; Wells, G.A.; Wilesmith, J.W.; Barnett, J.E. Spongiform encephalopathy in a herd of greater kudu (Tragelaphus strepsiceros): Epidemiological observations. Vet. Rec. 1993, 133, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Bons, N.; Mestre-Frances, N.; Belli, P.; Cathala, F.; Gajdusek, D.C.; Brown, P. Natural and experimental oral infection of nonhuman primates by bovine spongiform encephalopathy agents. Proc. Natl. Acad. Sci. USA 1999, 96, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; McConnell, I.; Fraser, H.; Dickinson, A.G. The disease characteristics of different strains of scrapie in sinc congenic mouse lines—Implications for the nature of the agent and host control of pathogenesis. J. Gen. Virol. 1991, 72, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. Distribution of experimentally induced scrapie lesions in the brain. Nature 1967, 216, 1310–1311. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; McBride, P.A.; Farquhar, C.F. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci. Lett. 1989, 102, 1–6. [Google Scholar] [CrossRef]
- Thackray, A.M.; Hopkins, L.; Spiropoulos, J.; Bujdoso, R. Molecular and transmission characteristics of primary-passaged ovine scrapie isolates in conventional and ovine PrP transgenic mice. J. Virol. 2008, 82, 11197–11207. [Google Scholar] [CrossRef] [PubMed]
- Le Dur, A.; Beringue, V.; Andreoletti, O.; Reine, F.; Lai, T.L.; Baron, T.; Bratberg, B.; Vilotte, J.L.; Sarradin, P.; Benestad, S.L.; et al. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc. Natl. Acad. Sci. USA 2005, 102, 16031–16036. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; Hopkins, L.; Lockey, R.; Spiropoulos, J.; Bujdoso, R. Propagation of ovine prions from “poor” transmitter scrapie isolates in ovine PrP transgenic mice. Exp. Mol. Pathol. 2012, 92, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Masujin, K.; Shu, Y.J.; Yamakawa, Y.; Hagiwara, K.; Sata, T.; Matsuura, Y.; Iwamaru, Y.; Imamura, M.; Okada, H.; Mohri, S.; et al. Biological and biochemical characterization of l-type-like bovine spongiform encephalopathy (BSE) detected in japanese black beef cattle. Prion 2008, 2, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Masujin, K.; Iwamaru, Y.; Imamura, M.; Matsuura, Y.; Mohri, S.; Czub, S.; Yokoyama, T. Experimental transmission of h-type bovine spongiform encephalopathy to bovinized transgenic mice. Vet. Pathol. 2011, 48, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Hart, P.; Piccardo, P.; Hunter, N.; Casalone, C.; Baron, T.; Barron, R.M. Bovine PrP expression levels in transgenic mice influence transmission characteristics of atypical bovine spongiform encephalopathy. J. Gen. Virol. 2012, 93, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Mason, G.L.; Seward, T.; Green, M.; Eliason, G.A.J.; Mathiason, C.; Miller, M.W.; Williams, E.S.; Hoover, E.; Telling, G.C. Transmission of prions from mule deer and elk with chronic wasting disease to transgenic mice expressing cervid PrP. J. Virol. 2004, 78, 13345–13350. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.A.; Nonno, R.; Castilla, J.; D’Agostino, C.; Pirisinu, L.; Riccardi, G.; Conte, M.; Richt, J.; Kunkle, R.; Langeveld, J.; et al. Chronic wasting disease in bank voles: Characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013, 9, e1003219. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, M.A.; Chianini, F.; Vaccari, G.; Esposito, E.; Conte, M.; Eaton, S.L.; Hamilton, S.; Finlayson, J.; Steele, P.J.; Dagleish, M.P.; et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J. Gen. Virol. 2008, 89, 2975–2985. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.J.; Raymond, L.D.; Meade-White, K.D.; Hughson, A.G.; Favara, C.; Gardner, D.; Williams, E.S.; Miller, M.W.; Race, R.E.; Caughey, B. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: Evidence for strains. J. Virol. 2007, 81, 4305–4314. [Google Scholar] [CrossRef] [PubMed]
- Perrott, M.R.; Sigurdson, C.J.; Mason, G.L.; Hoover, E.A. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 2012, 93, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.; Goldmann, W.; Parnham, D.; Chong, A.; Hunter, N. Partial dissociation of prpsc deposition and vacuolation in the brains of scrapie and BSE experimentally affected goats. J. Gen. Virol. 2001, 82, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Begara-McGorum, I.; Gonzalez, L.; Simmons, M.; Hunter, N.; Houston, F.; Jeffrey, M. Vacuolar lesion profile in sheep scrapie: Factors influencing its variation and relationship to disease-specific PrP accumulation. J. Comp. Pathol. 2002, 127, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Gavier-Widen, D.; Noremark, M.; Benestad, S.; Simmons, M.; Renstrom, L.; Bratberg, B.; Elvander, M.; af Segerstad, C.H. Recognition of the nor98 variant of scrapie in the swedish sheep population. J. Vet. Diagn. Investig. 2004, 16, 562–567. [Google Scholar] [CrossRef]
- Ligios, C.; Dexter, G.; Spiropoulos, J.; Maestrale, C.; Carta, A.; Simmons, M.M. Distribution of vascular amyloid in scrapie-affected sheep with different genotypes. J. Comp. Pathol. 2004, 131, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.H.; Wilesmith, J.W. The neuropathology and epidemiology of bovine spongiform encephalopathy. Brain Pathol. 1995, 5, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Gavier-Widen, D.; Wells, G.A.H.; Simmons, M.M.; Wilesmith, J.W.W.; Ryan, J. Histological observations on the brains of symptomless 7-year-old cattle. J. Comp. Pathol. 2001, 124, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Gavier-Widen, D.; Stack, M.J.; Baron, T.; Balachandran, A.; Simmons, M. Diagnosis of transmissible spongiform encephalopathies in animals: A review. J. Vet. Diagn. Investig. 2005, 17, 509–527. [Google Scholar] [CrossRef]
- Stack, M.J.; Moore, S.J.; Davis, A.; Webb, P.R.; Bradshaw, J.M.; Lee, Y.H.; Chaplin, M.; Focosi-Snyman, R.; Thurston, L.; Spencer, Y.I.; et al. Bovine spongiform encephalopathy: Investigation of phenotypic variation among passive surveillance cases. J. Comp. Pathol. 2011, 144, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Siso, S.; Doherr, M.G.; Botteron, C.; Fatzer, R.; Zurbriggen, A.; Vandevelde, M.; Seuberlich, T. Neuropathological and molecular comparison between clinical and asymptomatic bovine spongiform encephalopathy cases. Acta Neuropathol. 2007, 114, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Gonzalez, L.; Chong, A.; Foster, J.; Goldmann, W.; Hunter, N.; Martin, S. Ovine infection with the agents of scrapie (CH1641 isolate) and bovine spongiform encephalopathy: Immunochemical similarities can be resolved by immunohistochemistry. J. Comp. Pathol. 2006, 134, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.; McKelvey, W.; Fraser, H.; Chong, A.; Ross, A.; Parnham, D.; Goldmann, W.; Hunter, N. Experimentally induced bovine spongiform encephalopathy did not transmit via goat embryos. J. Gen. Virol. 1999, 80, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Gonzalez, L. Classical sheep transmissible spongiform encephalopathies: Pathogenesis, pathological phenotypes and clinical disease. Neuropathol. Appl. Neurobiol. 2007, 33, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Siso, S.; Monleon, E.; Casalone, C.; van Keulen, L.J.M.; Balkema-Buschmann, A.; Ortiz-Pelaez, A.; Iulini, B.; Langeveld, J.P.M.; Hoffmann, C.; et al. Variability in disease phenotypes within a single PRNP genotype suggests the existence of multiple natural sheep scrapie strains within europe. J. Gen. Virol. 2010, 91, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, S.; Seuberlich, T.; Oevermann, A.; Baron, T.; Bencsik, A. Comparison of brain PrPd distribution in ovine BSE and scrapie. Vet. Pathol. 2011, 48, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Martin, S.; Jeffrey, M. Distinct profiles of PrPd immunoreactivity in the brain of scrapie- and BSE-infected sheep: Implications for differential cell targeting and PrP processing. J. Gen. Virol. 2003, 84, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Martin, S.; Houston, F.E.; Hunter, N.; Reid, H.W.; Bellworthy, S.J.; Jeffrey, M. Phenotype of disease-associated PrP accumulation in the brain of bovine spongiform encephalopathy experimentally infected sheep. J. Gen. Virol. 2005, 86, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulos, J.; Casalone, C.; Caramelli, M.; Simmons, M.M. Immunohistochemistry for PrPSc in natural scrapie reveals patterns which are associated with the PrP genotype. Neuropathol. Appl. Neurobiol. 2007, 33, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; Gonzalez, L.; Ryder, S.J.; Bellworthy, S.J.; Jackman, R. Differential diagnosis of infections with the bovine spongiform encephalopathy (BSE) and scrapie agents in sheep. J. Comp. Pathol. 2001, 125, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Martin, S.; Gonzalez, L. Cell-associated variants of disease-specific prion protein immunolabelling are found in different sources of sheep transmissible spongiform encephalopathy. J. Gen. Virol. 2003, 84, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, S.; Martin, S.; Simon, S.; Comoy, E.; Bencsik, A.; Deslys, J.P.; Grassi, J.; Jeffrey, M.; Baron, T. Comparative molecular analysis of the abnormal prion protein in field scrapie cases and experimental bovine spongiform encephalopathy in sheep by use of western blotting and immunohistochemical methods. J. Virol. 2004, 78, 3654–3662. [Google Scholar] [CrossRef] [PubMed]
- Siso, S.; Jeffrey, M.; Martin, S.; Chianini, F.; Dagleish, M.P.; Gonzalez, L. Characterization of strains of ovine transmissible spongiform encephalopathy with a short PrPd profiling method. J. Comp. Pathol. 2010, 142, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Barron, R.M.; Campbell, S.L.; King, D.; Bellon, A.; Chapman, K.E.; Williamson, R.A.; Manson, J.C. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of prpsc in vivo. J. Biol. Chem. 2007, 282, 35878–35886. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Sidle, K.C.L.; Joiner, S.; Keyes, P.; Martin, T.C.; Dawson, M.; Collinge, J. Molecular screening of sheep for bovine spongiform encephalopathy. Neurosci. Lett. 1998, 255, 159–162. [Google Scholar] [CrossRef]
- Wadsworth, J.D.F.; Hill, A.F.; Joiner, S.; Jackson, G.S.; Clarke, A.R.; Collinge, J. Strain-specific prion-protein conformation determined by metal ions. Nat. Cell Biol. 1999, 1, 55–59. [Google Scholar] [PubMed]
- Notari, S.; Capellari, S.; Giese, A.; Westner, I.; Baruzzi, A.; Ghetti, B.; Gambetti, P.; Kretzschmar, H.A.; Parchi, P. Effects of different experimental conditions on the prpsc core generated by protease digestion—Implications for strain typing and molecular classification of CJD. J. Biol. Chem. 2004, 279, 16797–16804. [Google Scholar] [CrossRef] [PubMed]
- Hope, J.; Wood, S.C.E.R.; Birkett, C.R.; Chong, A.; Bruce, M.E.; Cairns, D.; Goldmann, W.; Hunter, N.; Bostock, C.J. Molecular analysis of ovine prion protein identifies similarities between BSE and an experimental isolate of natural scrapie, ch1641. J. Gen. Virol. 1999, 80, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.; Kuczius, T.; McElroy, M.; Parada, M.G.; Groschup, M.H. Molecular analysis of irish sheep scrapie cases. J. Gen. Virol. 2000, 81, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Stack, M.J.; Chaplin, M.J.; Clark, J. Differentiation of prion protein glycoforms from naturally occurring sheep scrapie, sheep-passaged scrapie strains (CH1641 and SSBP1), bovine spongiform encephalopathy (BSE) cases and romney and cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathol. 2002, 104, 279–286. [Google Scholar] [PubMed]
- Somerville, R.A. Host and transmissible spongiform encephalopathy agent strain control glycosylation of PrP. J. Gen. Virol. 1999, 80, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.A.; Hamilton, S.; Fernie, K. Transmissible spongiform encephalopathy strain, PrP genotype and brain region all affect the degree of glycosylation of prpsc. J. Gen. Virol. 2005, 86, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Madec, J.Y.; Groschup, M.H.; Calavas, D.; Junghans, F.; Baron, T. Protease-resistant prion protein in brain and lymphoid organs of sheep within a naturally scrapie-infected flock. Microbiol. Pathog. 2000, 28, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kuczius, T.; Haist, I.; Groschup, M.H. Molecular analysis of bovine spongiform encephalopathy and scrapie strain variation. J. Infect. Dis. 1998, 178, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Nonno, R.; Esposito, E.; Vaccari, G.; Conte, M.; Marcon, S.; di Bari, M.; Ligios, C.; di Guardo, G.; Agrimi, U. Molecular analysis of cases of italian sheep scrapie and comparison with cases of bovine spongiform encephalopathy (BSE) and experimental BSE in sheep. J. Clin. Microbiol. 2003, 41, 4127–4133. [Google Scholar] [CrossRef] [PubMed]
- Race, R.E.; Raines, A.; Baron, T.G.M.; Miller, M.W.; Jenny, A.; Williams, E.S. Comparison of abnormal prion protein glycoform patterns from transmissible spongiform encephalopathy agent-infected deer, elk, sheep, and cattle. J. Virol. 2002, 76, 12365–12368. [Google Scholar] [CrossRef] [PubMed]
- Gretzschel, A.; Buschmann, A.; Eiden, M.; Ziegler, U.; Luhken, G.; Erhardt, G.; Groschup, M.H. Strain typing of german transmissible spongiform encephalopathies field cases in small ruminants by biochemical methods. J. Vet. Med. Ser. B 2005, 52, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.P.; Rees, H.C.; Maddison, B.C.; Terry, L.A.; Thorne, L.; Jackman, R.; Whitelam, G.C.; Gough, K.C. Molecular profiling of ovine prion diseases by using thermolysin-resistant PrPSc and endogenous C2 PrP fragments. J. Virol. 2007, 81, 10532–10539. [Google Scholar] [CrossRef] [PubMed]
- Pirisinu, L.; Migliore, S.; di Bari, M.A.; Esposito, E.; Baron, T.; D’Agostino, C.; de Grossi, L.; Vaccari, G.; Agrimi, U.; Nonno, R. Molecular discrimination of sheep bovine spongiform encephalopathy from scrapie. Emerg. Infect. Dis. 2011, 17, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gielbert, A.; Jacobs, J.G.; Baron, T.; Andreoletti, O.; Yokoyama, T.; Langeveld, J.P.M.; Sauer, M.J. All major prion types recognised by a multiplex immunofluorometric assay for disease screening and confirmation in sheep. J. Immunol. Meth. 2012, 380, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Vulin, J.; Biacabe, A.G.; Cazeau, G.; Calavas, D.; Baron, T. Molecular typing of protease-resistant prion protein in transmissible spongiform encephalopathies of small ruminants, france, 2002–2009. Emerg. Infect. Dis. 2011, 17, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Gretzschel, A.; Buschmann, A.; Langeveld, J.; Groschup, M.H. Immunological characterization of abnormal prion protein from atypical scrapie cases in sheep using a panel of monoclonal antibodies. J. Gen. Virol. 2006, 87, 3715–3722. [Google Scholar] [CrossRef] [PubMed]
- Polak, M.P.; Zmudzinski, J.F.; Jacobs, J.G.; Langeveld, J.P. Atypical status of bovine spongiform encephalopathy in Poland: A molecular typing study. Arch. Virol. 2008, 153, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Terry, L.A.; Jenkins, R.; Thorne, L.; Everest, S.J.; Chaplin, M.J.; Davis, L.A.; Stack, M.J. First case of h-type bovine spongiform encephalopathy identified in great britain. Vet. Rec. 2007, 160, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Pirisinu, L.; Di Bari, M.; Marcon, S.; Vaccari, G.; D’Agostino, C.; Fazzi, P.; Esposito, E.; Galeno, R.; Langeveld, J.; Agrimi, U.; et al. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrPSc. PLoS ONE 2010, 5, e12723. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Scott, M.R.; Groth, D.; Williamson, R.A.; Burton, D.R.; Cohen, F.E.; Prusiner, S.B. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001, 10, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D.; Andreoletti, O.; Archer, F.; Madelaine, M.F.; Vilotte, J.L.; Lehmann, S.; Laude, H. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 4055–4059. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.J.; Olsen, E.A.; Lee, K.S.; Raymond, L.D.; Bryant, P.K.; Baron, G.S.; Caughey, W.S.; Kocisko, D.A.; McHolland, L.E.; Favara, C.; et al. Inhibition of protease-resistant prion protein formation in a transformed deer cell line infected with chronic wasting disease. J. Virol. 2006, 80, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Neale, M.H.; Mountjoy, S.J.; Edwards, J.C.; Vilette, D.; Laude, H.; Windl, O.; Saunders, G.C. Infection of cell lines with experimental and natural ovine scrapie agents. J. Virol. 2010, 84, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D. Cell models of prion infection. Vet. Res. 2008, 39, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Baker, C.A.; Demczyk, C.A.; Smith, E.W.; Julius, C.; Weissmann, C. Prion strain discrimination in cell culture: The cell panel assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20908–20913. [Google Scholar] [CrossRef] [PubMed]
- Eiden, M.; Palm, G.J.; Hinrichs, W.; Matthey, U.; Zahn, R.; Groschup, M.H. Synergistic and strain-specific effects of bovine spongiform encephalopathy and scrapie prions in the cell-free conversion of recombinant prion protein. J. Gen. Virol. 2006, 87, 3753–3761. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saa, P.; Soto, C. Detection of prions in blood. Nat. Med. 2005, 11, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Relationship between conformational stability and amplification efficiency of prions. Biochemistry 2011, 50, 7933–7940. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Baskakov, I.V. Assessment of strain-specific prpsc elongation rates revealed a transformation of prpsc properties during protein misfolding cyclic amplification. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Saa, P.; Sferrazza, G.F.; Ottenberg, G.; Oelschlegel, A.M.; Dorsey, K.; Lasmezas, C.I. Strain-specific role of RNAs in prion replication. J. Virol. 2012, 86, 10494–10504. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Baskakov, I.V. Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J. Biol. Chem. 2013, 288, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Taema, M.M.; Maddison, B.C.; Thorne, L.; Bishop, K.; Owen, J.; Hunter, N.; Baker, C.A.; Terry, L.A.; Gough, K.C. Differentiating ovine BSE from CH1641 scrapie by serial protein misfolding cyclic amplification. Mol. Biotechnol. 2012, 51, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Gough, K.C.; Bishop, K.; Maddison, B.C. Highly sensitive detection of small ruminant bovine spongiform encephalopathy within transmissible spongiform encephalopathy mixes by serial protein misfolding cyclic. J. Clin. Microbiol. 2014, 52, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.M.; Chaplin, M.J.; Vickery, C.M.; Simon, S.; Davis, L.; Denyer, M.; Lockey, R.; Stack, M.J.; O’Connor, M.J.; Bishop, K.; et al. Does the presence of scrapie affect the ability of current statutory discriminatory tests to detect the presence of bovine spongiform encephalopathy? J. Clin. Microbiol. 2015, 53, 2593–2604. [Google Scholar] [PubMed]
- Thorne, L.; Holder, T.; Ramsay, A.; Edwards, J.; Taema, M.M.; Windl, O.; Maddison, B.C.; Gough, K.C.; Terry, L.A. In vitro amplification of ovine prions from scrapie-infected sheep from great britain reveals distinct patterns of propagation. BMC Vet. Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Orru, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.Q.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank vole prion protein as an apparently universal substrate for RT-QUIC-based detection and discrimination of prion strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gough, K.C.; Rees, H.C.; Ives, S.E.; Maddison, B.C. Methods for Differentiating Prion Types in Food-Producing Animals. Biology 2015, 4, 785-813. https://doi.org/10.3390/biology4040785
Gough KC, Rees HC, Ives SE, Maddison BC. Methods for Differentiating Prion Types in Food-Producing Animals. Biology. 2015; 4(4):785-813. https://doi.org/10.3390/biology4040785
Chicago/Turabian StyleGough, Kevin C., Helen C. Rees, Sarah E. Ives, and Ben C. Maddison. 2015. "Methods for Differentiating Prion Types in Food-Producing Animals" Biology 4, no. 4: 785-813. https://doi.org/10.3390/biology4040785