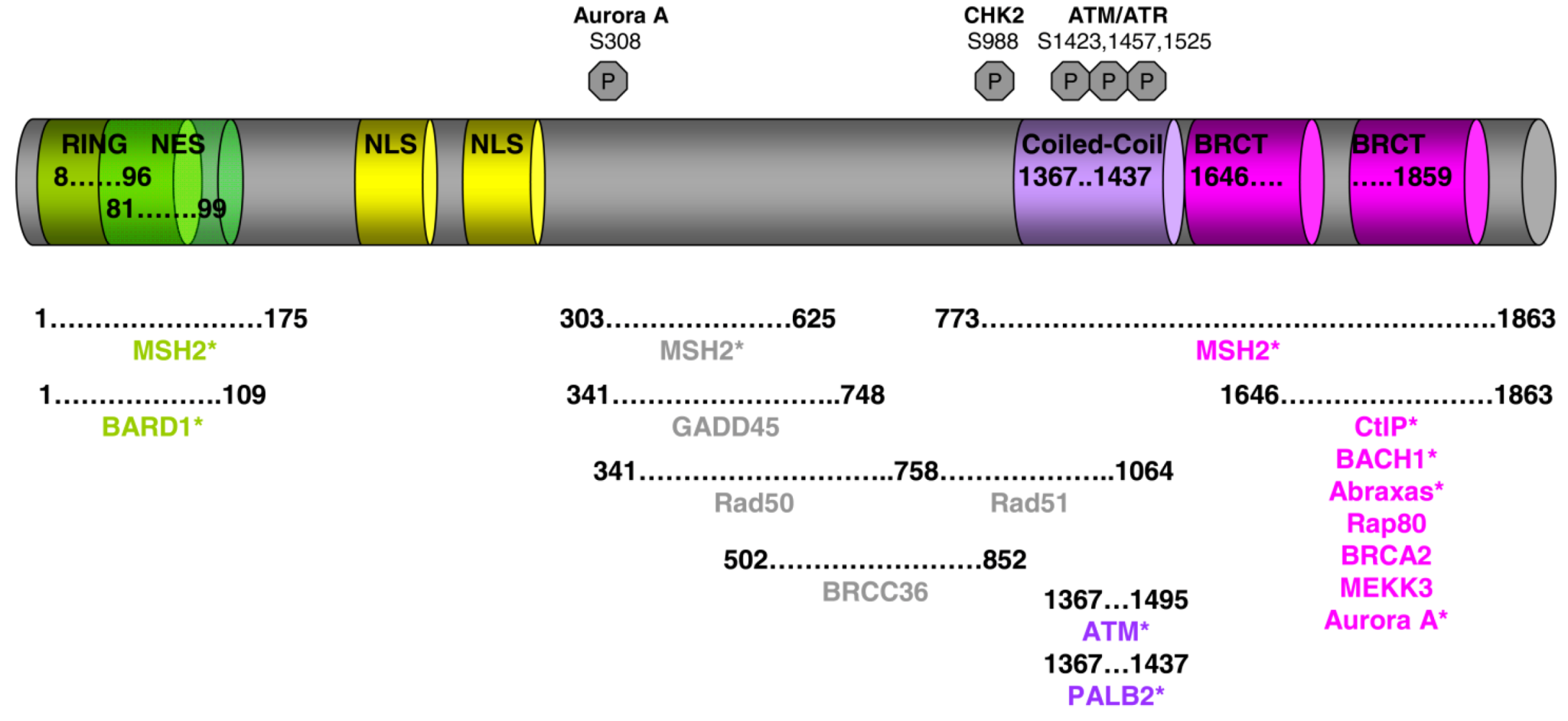
BRCA1 and Its Network of Interacting Partners

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
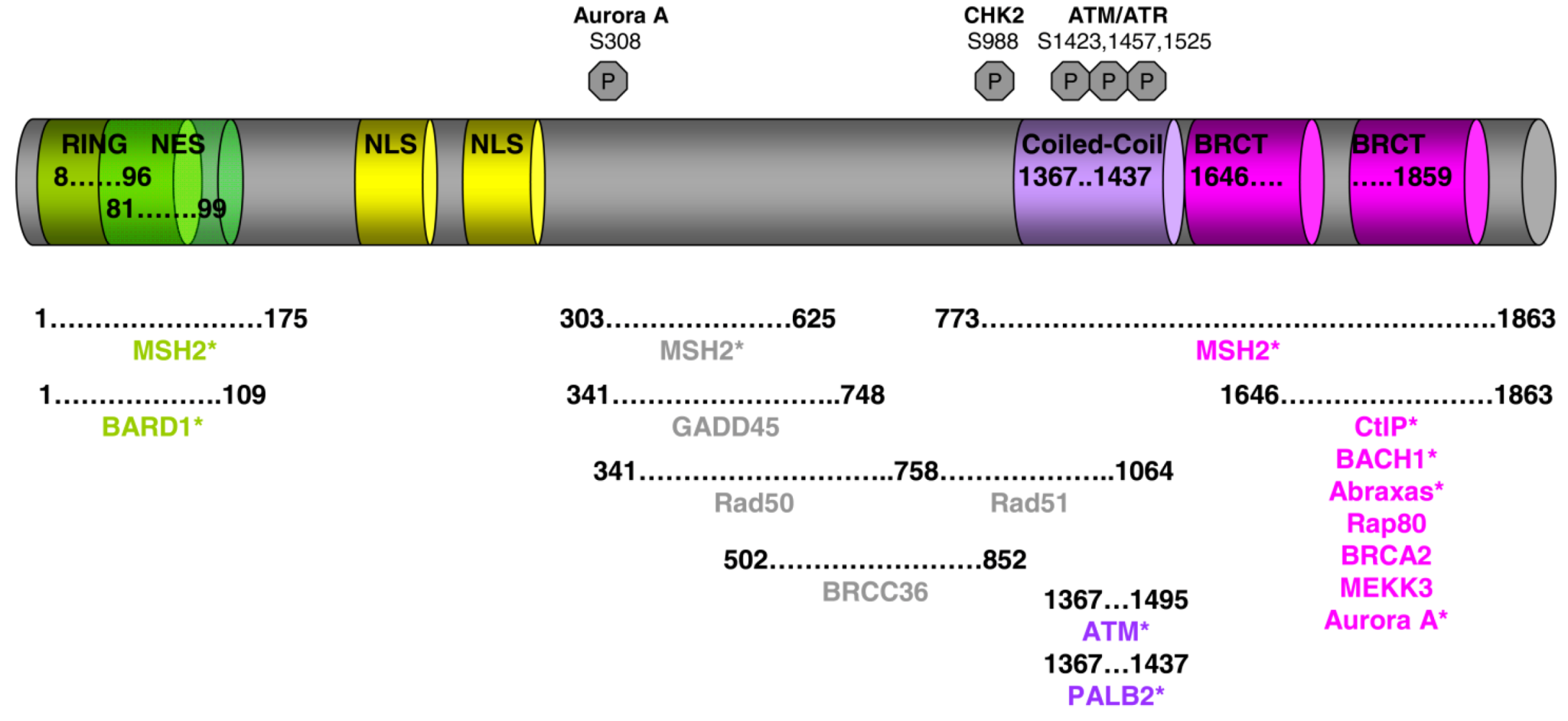
2. BRCA1 Interaction with the DNA Recombinase RAD51
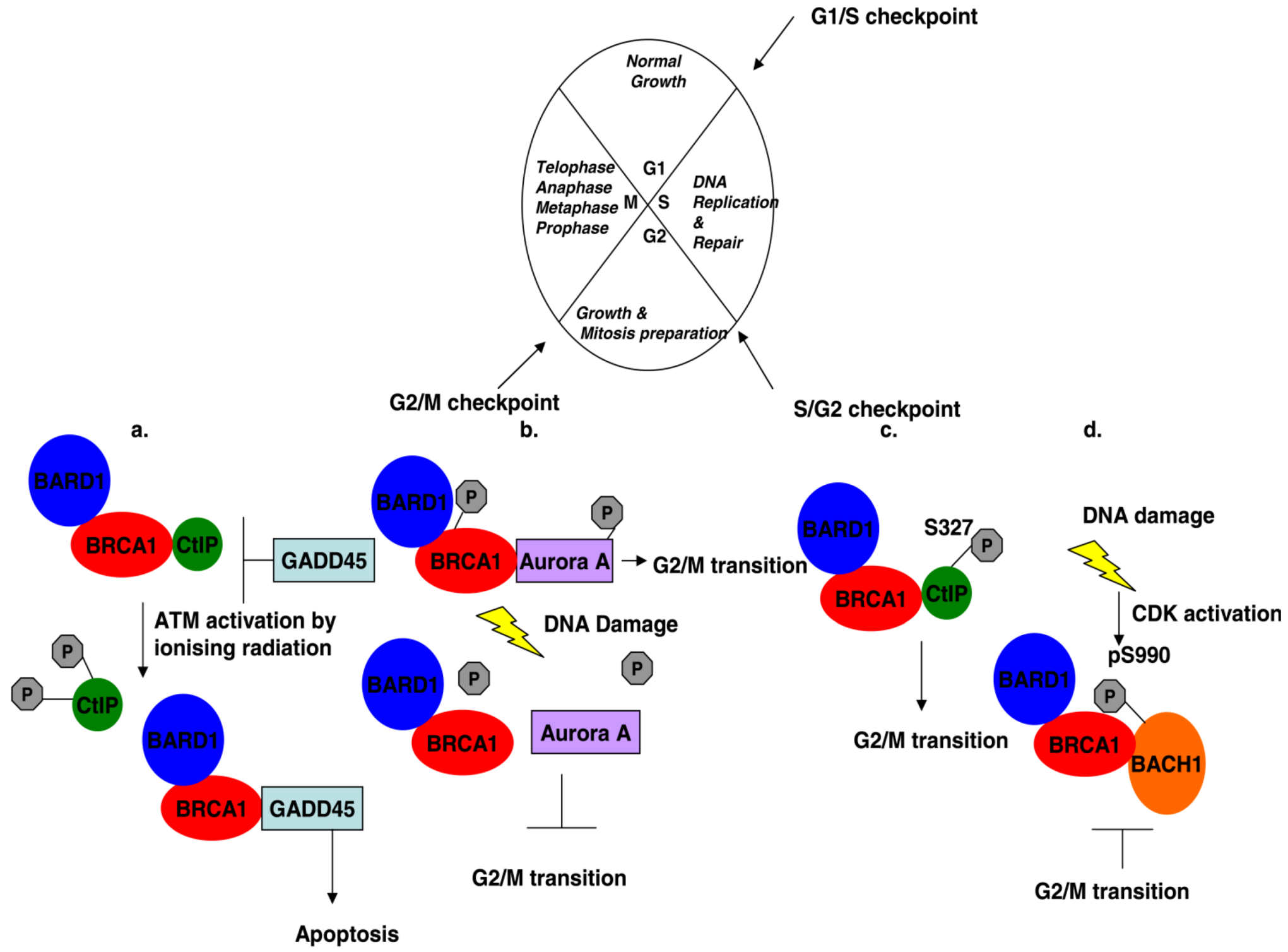
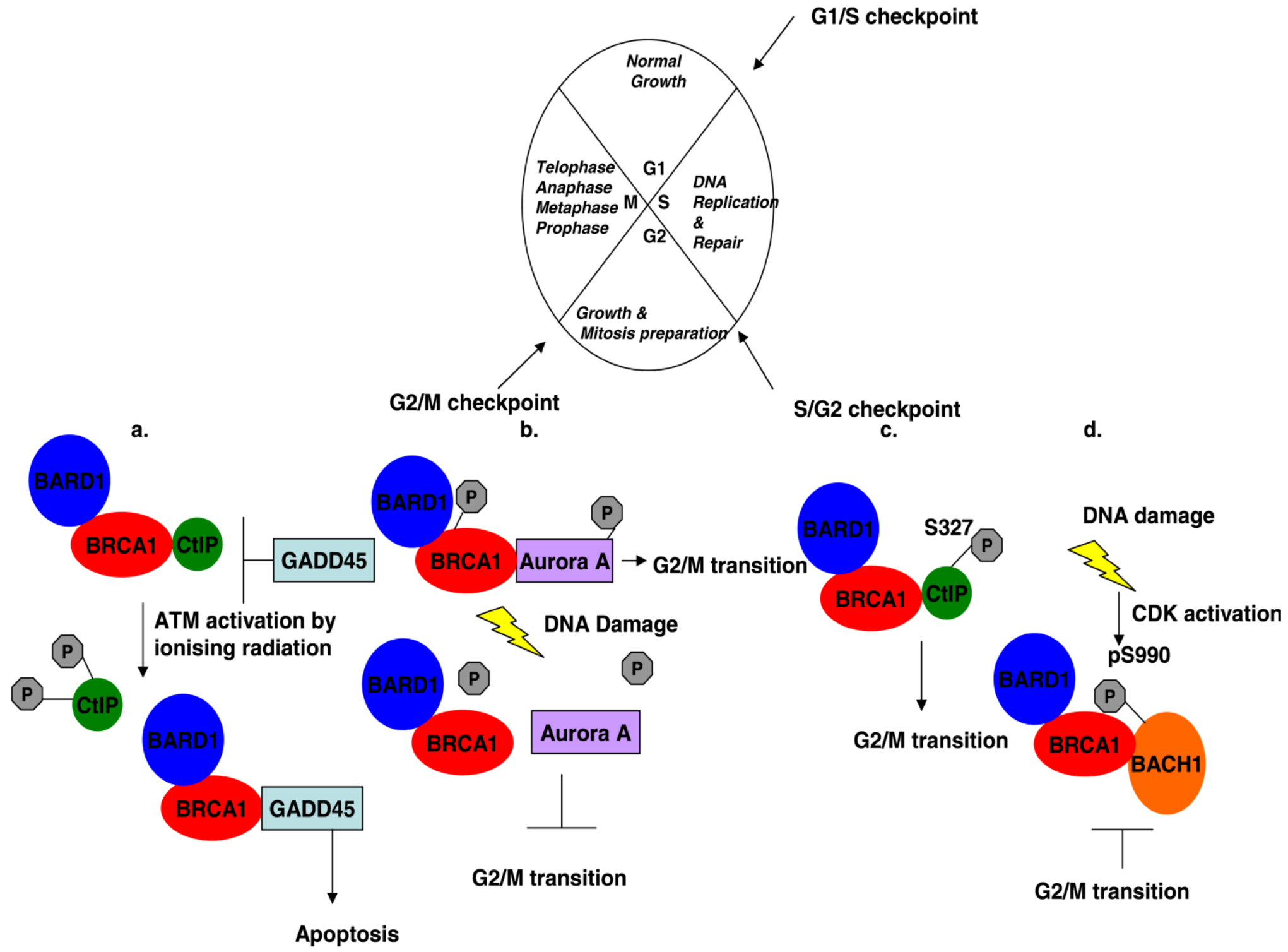
3. Phosphorylated BRCA1 Mediated Interactions and Their Function Cell Cycle Regulation
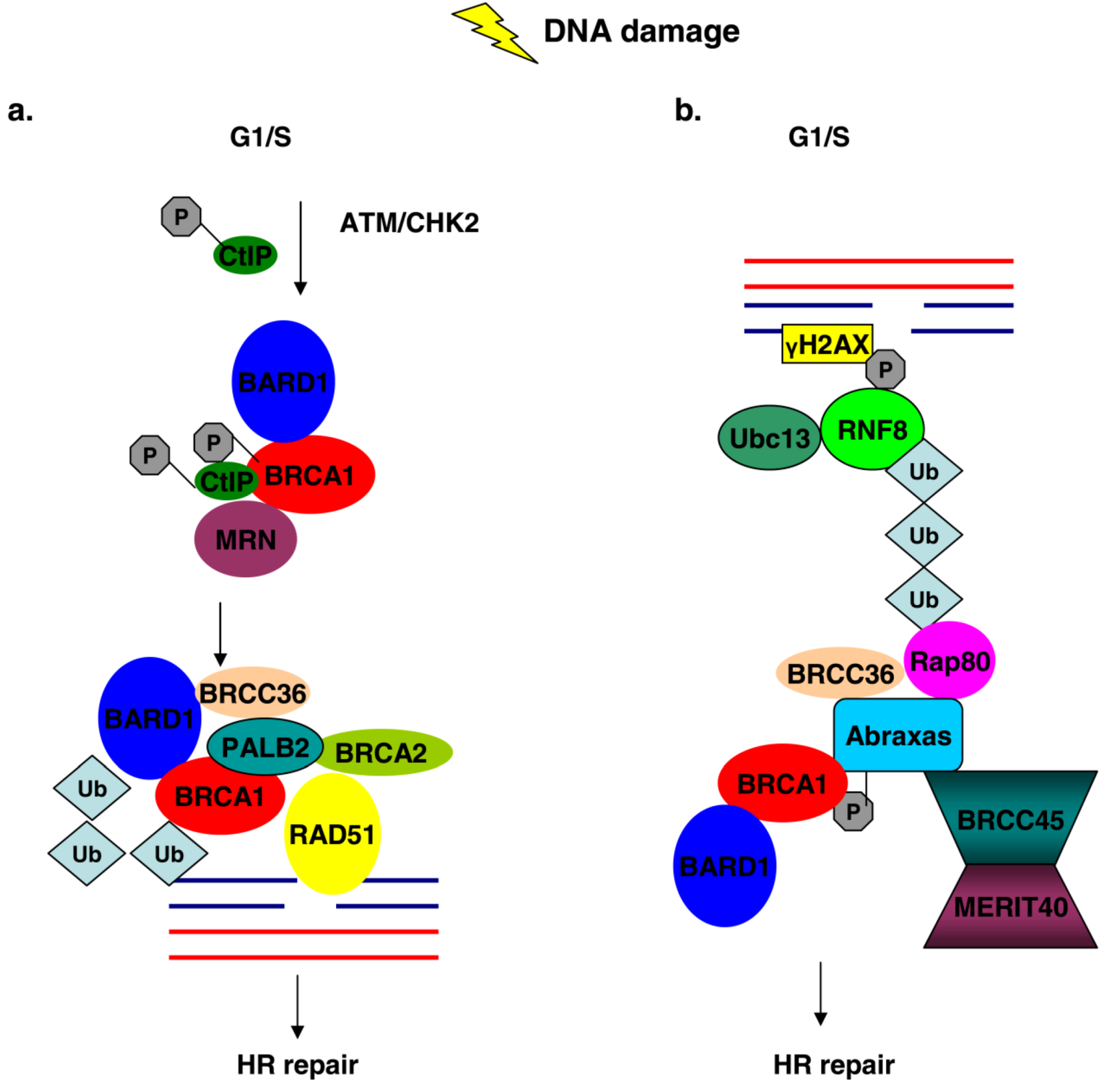
4. The BRCC Complex of Proteins
5. PALB2 Acts as Bridge between BRCA1 and BRCA2
6. BRCA1 Associates with the Mrn Complex during the Late S/G2 Phase of the Cell Cycle
7. The Interaction of BRCA1 with CtIP at Different Stages of the Cell Cycle Has Specific Functions

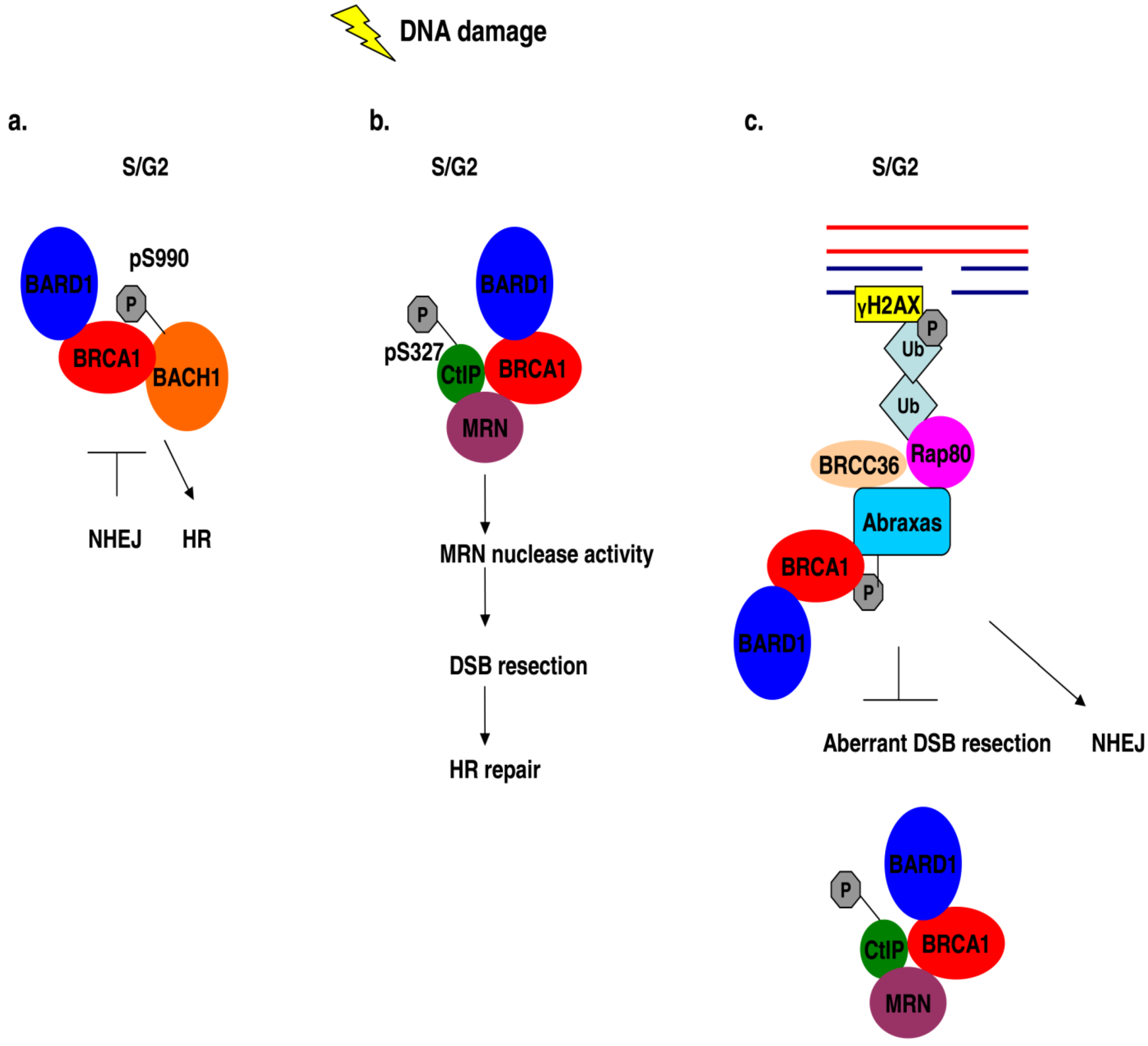
8. BRCA1 Forms a Complex with Phosphorylated BACH1 (BRIP1)
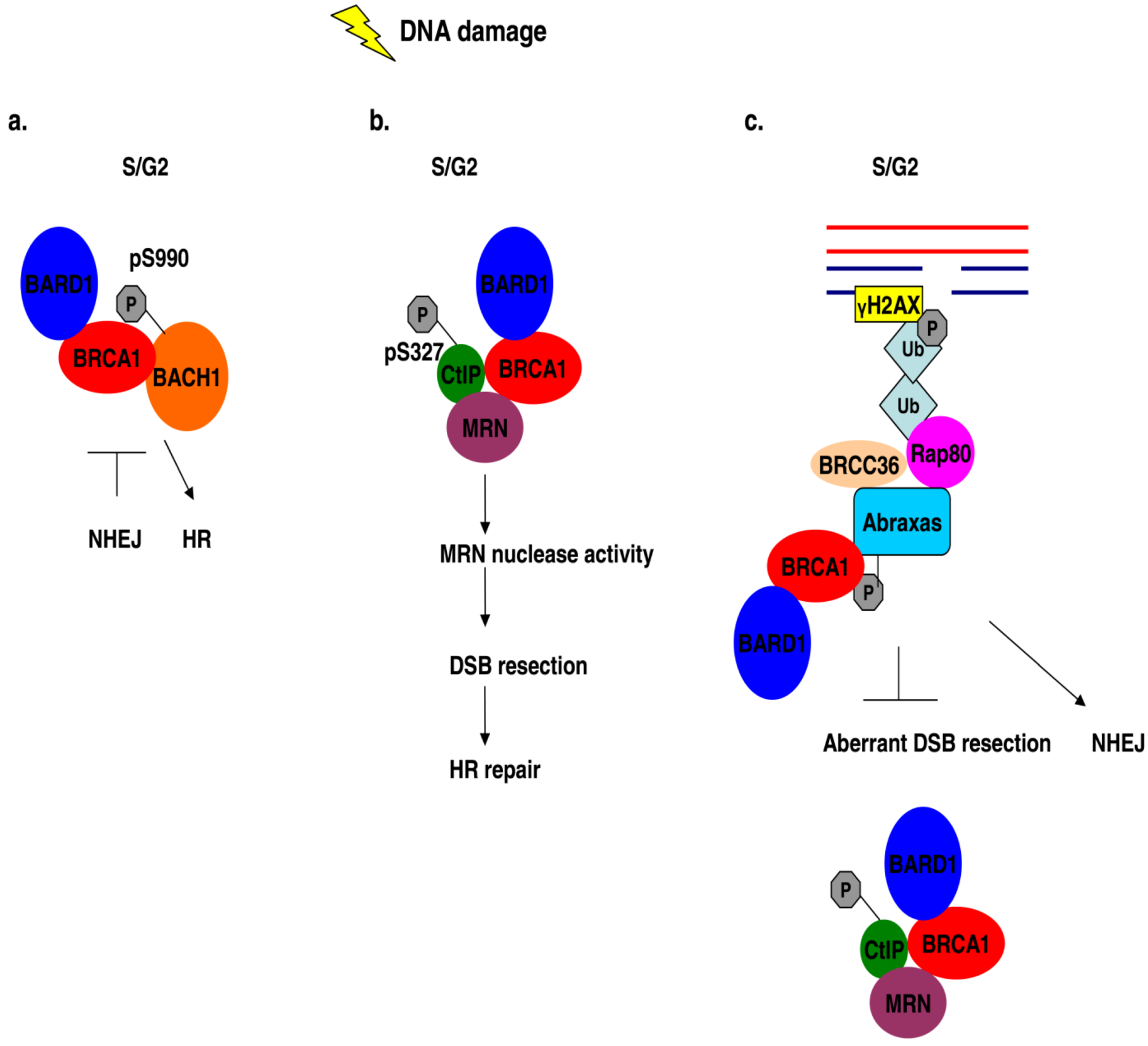
9. BRCA1 Interacts with the Phosphoproteins Abraxas (CCDC98) and Rap80
10. BRCA1 Interaction with the Mismatch Repair Proteins MSH2, MSH3, MSH6 and MLH1
11. Conclusions
Acknowledgments
References
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y.; et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar]
- Chapman, M.S.; Verma, I.M. Transcriptional activation by BRCA1. Nature 1996, 382, 678–679. [Google Scholar] [CrossRef]
- Chen, Y.; Farmer, A.A.; Chen, C.F.; Jones, D.C.; Chen, P.L.; Lee, W.H. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res. 1996, 56, 3168–3172. [Google Scholar]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar] [CrossRef]
- Shao, N.; Chai, Y.L.; Shyam, E.; Reddy, P.; Rao, V.N. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 1996, 13, 1–7. [Google Scholar]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar]
- Freemont, P.S. The RING finger. A novel protein sequence motif related to the zinc finger. Ann. NY Acad. Sci. 1993, 684, 174–192. [Google Scholar] [CrossRef]
- Joazeiro, C.A.; Weissman, A.M. RING finger proteins: Mediators of ubiquitin ligase activity. Cell 2000, 102, 549–552. [Google Scholar] [CrossRef]
- Saurin, A.J.; Borden, K.L.; Boddy, M.N.; Freemont, P.S. Does this have a familiar RING? Trends Biochem. Sci. 1996, 21, 208–214. [Google Scholar]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef]
- Scully, R.; Ganesan, S.; Brown, M.; De Caprio, J.A.; Cannistra, S.A.; Feunteun, J.; Schnitt, S.; Livingston, D.M. Location of BRCA1 in human breast and ovarian cancer cells. Science 1996, 272, 123–126. [Google Scholar] [CrossRef]
- Leung, C.C.; Glover, J.N. BRCT domains: easy as one, two, three. Cell Cycle 2011, 10, 2461–2470. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; Songyang, Z. BRCT domains: Phosphopeptide binding and signaling modules. Front. Biosci. 2008, 13, 5905–5915. [Google Scholar] [CrossRef]
- BIC, B.C.I.C.D.B. Available online: http://research.nhgri.nih.gov/projects/bic/ (accessed on 25 November 2012).
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev.Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef]
- Tashiro, S.; Kotomura, N.; Shinohara, A.; Tanaka, K.; Ueda, K.; Kamada, N. S phase specific formation of the human Rad51 protein nuclear foci in lymphocytes. Oncogene 1996, 12, 2165–2170. [Google Scholar]
- Baumann, P.; Benson, F.E.; West, S.C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef]
- Baumann, P.; West, S.C. The human Rad51 protein: polarity of strand transfer and stimulation by hRP-A. EMBO J. 1997, 16, 5198–5206. [Google Scholar] [CrossRef]
- Snouwaert, J.N.; Gowen, L.C.; Latour, A.M.; Mohn, A.R.; Xiao, A.; DiBiase, L.; Koller, B.H. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene 1999, 18, 7900–7907. [Google Scholar]
- Dong, Y.; Hakimi, M.A.; Chen, X.; Kumaraswamy, E.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 2003, 12, 1087–1099. [Google Scholar] [CrossRef]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; Scully, R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar]
- Chen, P.L.; Chen, C.F.; Chen, Y.; Xiao, J.; Sharp, Z.D.; Lee, W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. USA 1998, 95, 5287–5292. [Google Scholar] [CrossRef]
- Mizuta, R.; LaSalle, J.M.; Cheng, H.L.; Shinohara, A.; Ogawa, H.; Copeland, N.; Jenkins, N.A.; Lalande, M.; Alt, F.W. RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6927–6932. [Google Scholar]
- Wong, A.K.; Pero, R.; Ormonde, P.A.; Tavtigian, S.V.; Bartel, P.L. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem. 1997, 272, 31941–31944. [Google Scholar]
- Carreira, A.; Hilario, J.; Amitani, I.; Baskin, R.J.; Shivji, M.K.; Venkitaraman, A.R.; Kowalczykowski, S.C. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 2009, 136, 1032–1043. [Google Scholar] [CrossRef]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar]
- Shivji, M.K.; Davies, O.R.; Savill, J.M.; Bates, D.L.; Pellegrini, L.; Venkitaraman, A.R. A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res. 2006, 34, 4000–4011. [Google Scholar] [CrossRef]
- Galkin, V.E.; Esashi, F.; Yu, X.; Yang, S.; West, S.C.; Egelman, E.H. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc. Natl. Acad. Sci. USA 2005, 102, 8537–8542. [Google Scholar]
- Chen, C.F.; Chen, P.L.; Zhong, Q.; Sharp, Z.D.; Lee, W.H. Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J. Biol. Chem. 1999, 274, 32931–32935. [Google Scholar] [CrossRef]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar]
- Cousineau, I.; Abaji, C.; Belmaaza, A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: Implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 2005, 65, 11384–11391. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Ray Chaudhuri, A.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.L.; Riley, D.J.; Lee, W.H.; Allred, D.C.; Osborne, C.K. Response: Location of BRCA1 in human breast and ovarian cancer cells. Science 1996, 272, 125–126. [Google Scholar]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef]
- Gatei, M.; Scott, S.P.; Filippovitch, I.; Soronika, N.; Lavin, M.F.; Weber, B.; Khanna, K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000, 60, 3299–3304. [Google Scholar]
- Tibbetts, R.S.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef]
- Lee, J.S.; Collins, K.M.; Brown, A.L.; Lee, C.H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar]
- Ouchi, M.; Fujiuchi, N.; Sasai, K.; Katayama, H.; Minamishima, Y.A.; Ongusaha, P.P.; Deng, C.; Sen, S.; Lee, S.W.; Ouchi, T. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J. Biol. Chem. 2004, 279, 19643–19648. [Google Scholar]
- Okada, S.; Ouchi, T. Cell cycle differences in DNA damage-induced BRCA1 phosphorylation affect its subcellular localization. J. Biol. Chem. 2003, 278, 2015–2020. [Google Scholar] [CrossRef]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef]
- Lindsay, H.D.; Griffiths, D.J.; Edwards, R.J.; Christensen, P.U.; Murray, J.M.; Osman, F.; Walworth, N.; Carr, A.M. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998, 12, 382–395. [Google Scholar] [CrossRef]
- Zeng, Y.; Forbes, K.C.; Wu, Z.; Moreno, S.; Piwnica-Worms, H.; Enoch, T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature 1998, 395, 507–510. [Google Scholar]
- Tomlinson, G.E.; Chen, T.T.; Stastny, V.A.; Virmani, A.K.; Spillman, M.A.; Tonk, V.; Blum, J.L.; Schneider, N.R.; Wistuba, I.I.; Shay, J.W.; et al. Characterization of a breast cancer cell line derived from a germ-line BRCA1 mutation carrier. Cancer Res. 1998, 58, 3237–3242. [Google Scholar]
- Marumoto, T.; Hirota, T.; Morisaki, T.; Kunitoku, N.; Zhang, D.; Ichikawa, Y.; Sasayama, T.; Kuninaka, S.; Mimori, T.; Tamaki, N.; et al. Roles of aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells 2002, 7, 1173–1182. [Google Scholar] [CrossRef]
- Hirota, T.; Kunitoku, N.; Sasayama, T.; Marumoto, T.; Zhang, D.; Nitta, M.; Hatakeyama, K.; Saya, H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 2003, 114, 585–598. [Google Scholar] [CrossRef]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef]
- Meza, J.E.; Brzovic, P.S.; King, M.C.; Klevit, R.E. Mapping the functional domains of BRCA1. Interaction of the ring finger domains of BRCA1 and BARD1. J. Biol. Chem. 1999, 274, 5659–5665. [Google Scholar]
- Ohta, T.; Michel, J.J.; Schottelius, A.J.; Xiong, Y. ROC1, a homolog of APC11, represents a family of cullin partners with an associated ubiquitin ligase activity. Mol. Cell 1999, 3, 535–541. [Google Scholar] [CrossRef]
- Tan, P.; Fuchs, S.Y.; Chen, A.; Wu, K.; Gomez, C.; Ronai, Z.; Pan, Z.Q. Recruitment of a ROC1-CUL1 ubiquitin ligase by Skp1 and HOS to catalyze the ubiquitination of I kappa B alpha. Mol. Cell 1999, 3, 527–533. [Google Scholar] [CrossRef]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G., Jr.; Elledge, S.J.; Conaway, R.C.; Harper, J.W.; Conaway, J.W. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar]
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003, 278, 5255–5263. [Google Scholar]
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef]
- Herrmann, J.; Lerman, L.O.; Lerman, A. Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res. 2007, 100, 1276–1291. [Google Scholar] [CrossRef]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar]
- Nishikawa, H.; Ooka, S.; Sato, K.; Arima, K.; Okamoto, J.; Klevit, R.E.; Fukuda, M.; Ohta, T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 3916–3924. [Google Scholar]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef]
- Cooper, E.M.; Cutcliffe, C.; Kristiansen, T.Z.; Pandey, A.; Pickart, C.M.; Cohen, R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009, 28, 621–631. [Google Scholar] [CrossRef]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.S.; deRooij, D.G.; Hirsch, S.; Ravi, K.; Hicks, J.B.; et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 2011, 334, 525–528. [Google Scholar]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Haber, J.E. The many interfaces of Mre11. Cell 1998, 95, 583–586. [Google Scholar] [CrossRef]
- Wu, X.; Petrini, J.H.; Heine, W.F.; Weaver, D.T.; Livingston, D.M.; Chen, J. Independence of R/M/N focus formation and the presence of intact BRCA1. Science 2000, 289, 11. [Google Scholar] [CrossRef]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar]
- Wong, A.K.; Ormonde, P.A.; Pero, R.; Chen, Y.; Lian, L.; Salada, G.; Berry, S.; Lawrence, Q.; Dayananth, P.; Ha, P.; et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene 1998, 17, 2279–2285. [Google Scholar]
- Li, S.; Ting, N.S.; Zheng, L.; Chen, P.L.; Ziv, Y.; Shiloh, Y.; Lee, E.Y.; Lee, W.H. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000, 406, 210–215. [Google Scholar]
- Harkin, D.P.; Bean, J.M.; Miklos, D.; Song, Y.H.; Truong, V.B.; Englert, C.; Christians, F.C.; Ellisen, L.W.; Maheswaran, S.; Oliner, J.D.; et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell 1999, 97, 575–586. [Google Scholar] [CrossRef]
- Takekawa, M.; Maeda, T.; Saito, H. Protein phosphatase 2Calpha inhibits the human stress-responsive p38 and JNK MAPK pathways. EMBO J. 1998, 17, 4744–4752. [Google Scholar] [CrossRef]
- Thangaraju, M.; Kaufmann, S.H.; Couch, F.J. BRCA1 facilitates stress-induced apoptosis in breast and ovarian cancer cell lines. J. Biol. Chem. 2000, 275, 33487–33496. [Google Scholar] [CrossRef]
- Gilmore, P.M.; McCabe, N.; Quinn, J.E.; Kennedy, R.D.; Gorski, J.J.; Andrews, H.N.; McWilliams, S.; Carty, M.; Mullan, P.B.; Duprex, W.P.; et al. BRCA1 interacts with and is required for paclitaxel-induced activation of mitogen-activated protein kinase kinase kinase 3. Cancer Res. 2004, 64, 4148–4154. [Google Scholar]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef]
- Yu, X.; Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; Livingston, D.M. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Gu, L.; Yan, N.; Shi, Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol. Cell 2004, 14, 405–412. [Google Scholar] [CrossRef]
- Dohrn, L.; Salles, D.; Siehler, S.Y.; Kaufmann, J.; Wiesmuller, L. BRCA1-mediated repression of mutagenic end-joining of DNA double-strand breaks requires complex formation with BACH1. Biochem. J. 2012, 441, 919–926. [Google Scholar]
- Kim, H.; Huang, J.; Chen, J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol. 2007, 14, 710–715. [Google Scholar]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef]
- Feng, L.; Huang, J.; Chen, J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar]
- Bian, C.; Wu, R.; Cho, K.; Yu, X. Loss of BRCA1-A complex function in RAP80 null tumor cells. PLoS One 2012. [Google Scholar] [CrossRef]
- Solyom, S.; Aressy, B.; Pylkas, K.; Patterson-Fortin, J.; Hartikainen, J.M.; Kallioniemi, A.; Kauppila, S.; Nikkila, J.; Kosma, V.M.; Mannermaa, A.; et al. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci. Transl. Med. 2012, 4, 122–123. [Google Scholar]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar]
- Hu, X.; Paul, A.; Wang, B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J. Biol. Chem. 2012, 287, 25510–25519. [Google Scholar]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar]
- Fishel, R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998, 12, 2096–2101. [Google Scholar]
- Fishel, R. Signaling mismatch repair in cancer. Nat. Med. 1999, 5, 1239–1241. [Google Scholar]
- Hakem, R.; de la Pompa, J.L.; Mak, T.W. Developmental studies of Brca1 and Brca2 knock-out mice. J. Mammary Gland Biol. Neoplasia 1998, 3, 431–445. [Google Scholar]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40-63. https://doi.org/10.3390/biology2010040
Christou CM, Kyriacou K. BRCA1 and Its Network of Interacting Partners. Biology. 2013; 2(1):40-63. https://doi.org/10.3390/biology2010040
Chicago/Turabian StyleChristou, Charita M., and Kyriacos Kyriacou. 2013. "BRCA1 and Its Network of Interacting Partners" Biology 2, no. 1: 40-63. https://doi.org/10.3390/biology2010040
APA StyleChristou, C. M., & Kyriacou, K. (2013). BRCA1 and Its Network of Interacting Partners. Biology, 2(1), 40-63. https://doi.org/10.3390/biology2010040