Next-Generation Sequencing: From Understanding Biology to Personalized Medicine


{kind=link}
Abstract
:1. From Genes, Biology and Disease
Why Four Letters of Genetic Code Are So Complicated
2. Next-Generation Sequencing—Towards Understanding Biology
3. Genomics
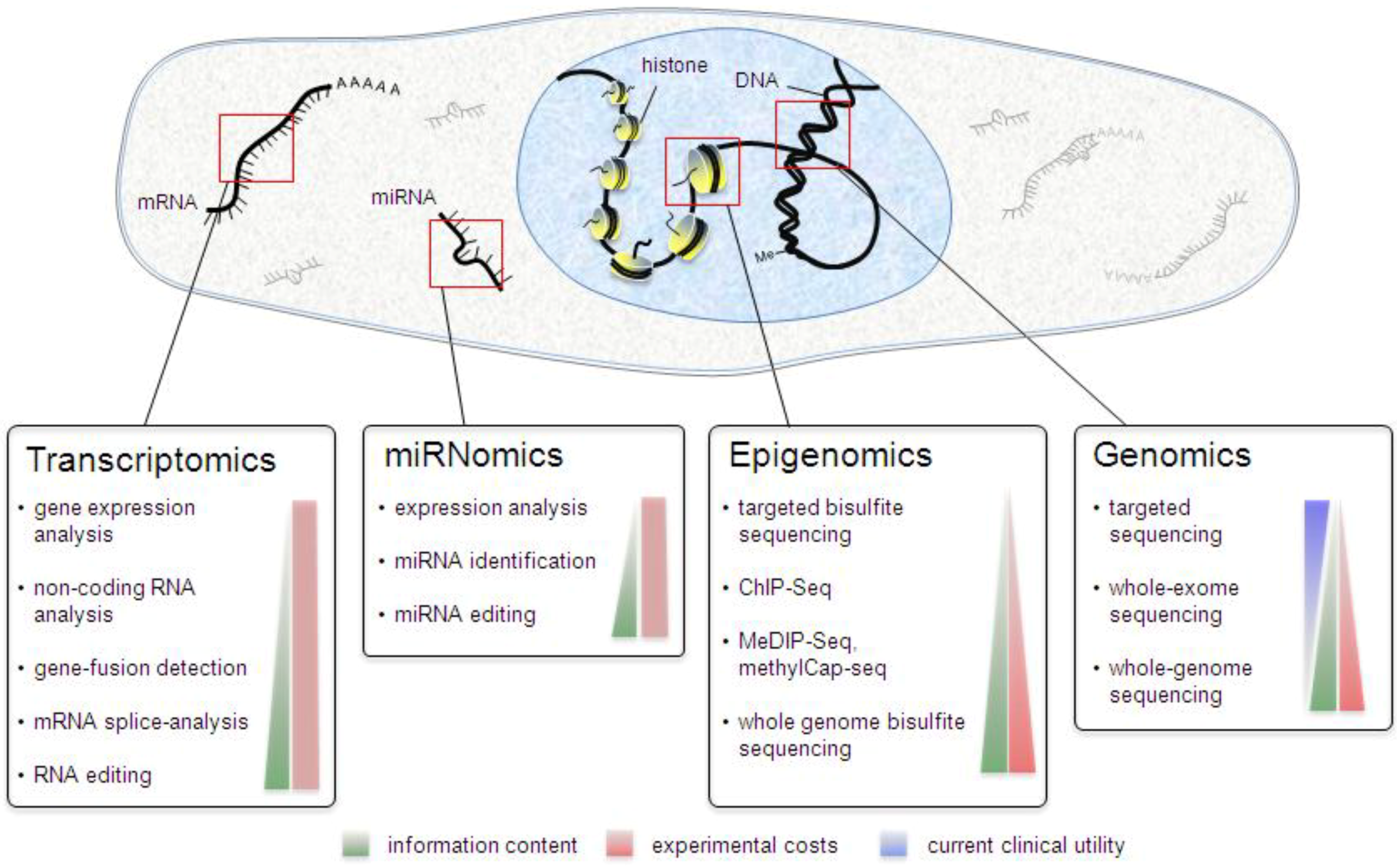
3.1. Exome Sequencing
3.2. Whole-Genome Sequencing
4. Transcriptomics
4.1. Defective RNA Processing and Disease
4.2. NGS Methods to Study the Transcriptome
4.3. Gene-Expression Analyses and mRNA Splicing
4.4. RNA Editing and Non-Coding RNAs
5. Epigenomics
5.1. Role of Epigenetic Alterations
5.2. NGS Methods to Assess the Epigenome
6. NGS—Towards Personalized Medicine
Acknowledgements
References and Notes
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Sastre, L. Clinical implications of the encode project. Clin. Transl. Oncol. 2012, 14, 801–802. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A. Decoding the human genome. Genome Res. 2012, 22, 1599–1601. [Google Scholar] [CrossRef]
- Database of Genomic Variants. Available online: http://projects.tcag.ca/ (accessed on 5 July 2012).
- Ecker, J.R.; Bickmore, W.A.; Barroso, I.; Pritchard, J.K.; Gilad, Y.; Segal, E. Genomics: Encode explained. Nature 2012, 489, 52–55. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar]
- Genomeweb. Available online: http://genomeweb.com/ (accessed on 12 December 2012).
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef]
- Braslavsky, I.; Hebert, B.; Kartalov, E.; Quake, S.R. Sequence information can be obtained from single DNA molecules. Proc. Natl. Acad. Sci. USA 2003, 100, 3960–3964. [Google Scholar]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef]
- Astier, Y.; Braha, O.; Bayley, H. Toward single molecule DNA sequencing: Direct identification of ribonucleoside and deoxyribonucleoside 5'-monophosphates by using an engineered protein nanopore equipped with a molecular adapter. J. Am. Chem. Soc. 2006, 128, 1705–1710. [Google Scholar] [CrossRef]
- Rusk, N. Focus on next-generation sequencing data analysis. Nat. Methods 2009, 6, S1. [Google Scholar] [CrossRef]
- Lee, H.C.; Lai, K.; Lorenc, M.T.; Imelfort, M.; Duran, C.; Edwards, D. Bioinformatics tools and databases for analysis of next-generation sequence data. Brief Funct. Genomics 2012, 11, 12–24. [Google Scholar] [CrossRef]
- Torri, F.; Dinov, I.D.; Zamanyan, A.; Hobel, S.; Genco, A.; Petrosyan, P.; Clark, A.P.; Liu, Z.; Eggert, P.; Pierce, J.; et al. Next generation sequence analysis and computational genomics using graphical pipeline workflows. Genes 2012, 3, 545–575. [Google Scholar] [CrossRef]
- Afgan, E.; Chapman, B.; Taylor, J. Cloudman as a platform for tool, data, and analysis distribution. BMC Bioinformatics 2012, 13, 315. [Google Scholar] [CrossRef]
- Schadt, E.E.; Linderman, M.D.; Sorenson, J.; Lee, L.; Nolan, G.P. Computational solutions to large-scale data management and analysis. Nat. Rev. Genet. 2010, 11, 647–657. [Google Scholar]
- Abeel, T.; van Parys, T.; Saeys, Y.; Galagan, J.; van de Peer, Y. Genomeview: A next-generation genome browser. Nucleic Acids Res. 2012, 40, e12. [Google Scholar] [CrossRef]
- Bao, S.; Jiang, R.; Kwan, W.; Wang, B.; Ma, X.; Song, Y.Q. Evaluation of next-generation sequencing software in mapping and assembly. J. Hum. Genet. 2011, 56, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Cordero, F.; Beccuti, M.; Donatelli, S.; Calogero, R.A. Large disclosing the nature of computational tools for the analysis of next generation sequencing data. Curr. Top. Med. Chem. 2012, 12, 1320–1330. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Siegfried, J.D. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll Cardiol. 2011, 57, 1641–1649. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar]
- Ng, S.B.; Turner, E.H.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar]
- Hood, R.L.; Lines, M.A.; Nikkel, S.M.; Schwartzentruber, J.; Beaulieu, C.; Nowaczyk, M.J.; Allanson, J.; Kim, C.A.; Wieczorek, D.; Moilanen, J.S.; et al. Mutations in srcap, encoding snf2-related crebbp activator protein, cause floating-harbor syndrome. Am. J. Hum. Genet. 2012, 90, 308–313. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies mll2 mutations as a cause of kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar]
- Wang, J.L.; Yang, X.; Xia, K.; Hu, Z.M.; Weng, L.; Jin, X.; Jiang, H.; Zhang, P.; Shen, L.; Guo, J.F.; et al. Tgm6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010, 133, 3510–3518. [Google Scholar] [CrossRef]
- Tariq, M.; Belmont, J.W.; Lalani, S.; Smolarek, T.; Ware, S.M. Shroom3 is a novel candidate for heterotaxy identified by whole exome sequencing. Genome Biol. 2011, 12, R91. [Google Scholar] [CrossRef]
- Lara-Pezzi, E.; Dopazo, A.; Manzanares, M. Understanding cardiovascular disease: A journey through the genome (and what we found there). Dis. Model Mech. 2012, 5, 434–443. [Google Scholar] [CrossRef]
- Musunuru, K.; Pirruccello, J.P.; Do, R.; Peloso, G.M.; Guiducci, C.; Sougnez, C.; Garimella, K.V.; Fisher, S.; Abreu, J.; Barry, A.J.; et al. Exome sequencing, angptl3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 2010, 363, 2220–2227. [Google Scholar]
- Meder, B.; Haas, J.; Keller, A.; Heid, C.; Just, S.; Borries, A.; Boisguerin, V.; Scharfenberger-Schmeer, M.; Stahler, P.; Beier, M.; et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ. Cardiovasc. Genet. 2011, 4, 110–122. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of ttn, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar]
- Galmiche, L.; Serre, V.; Beinat, M.; Assouline, Z.; Lebre, A.S.; Chretien, D.; Nietschke, P.; Benes, V.; Boddaert, N.; Sidi, D.; et al. Exome sequencing identifies mrpl3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011, 32, 1225–1231. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in bag3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]
- Clark, M.J.; Chen, R.; Lam, H.Y.; Karczewski, K.J.; Euskirchen, G.; Butte, A.J.; Snyder, M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011, 29, 908–914. [Google Scholar]
- Norton, N.; Li, D.; Hershberger, R.E. Next-generation sequencing to identify genetic causes of cardiomyopathies. Curr. Opin. Cardiol. 2012, 27, 214–220. [Google Scholar] [CrossRef]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Lupski, J.R.; Reid, J.G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D.C.; Nazareth, L.; Bainbridge, M.; Dinh, H.; Jing, C.; Wheeler, D.A.; et al. Whole-genome sequencing in a patient with charcot-marie-tooth neuropathy. N. Engl. J. Med. 2010, 362, 1181–1191. [Google Scholar] [CrossRef]
- Smith, K.A.; Joziasse, I.C.; Chocron, S.; van Dinther, M.; Guryev, V.; Verhoeven, M.C.; Rehmann, H.; van der Smagt, J.J.; Doevendans, P.A.; Cuppen, E.; et al. Dominant-negative alk2 allele associates with congenital heart defects. Circulation 2009, 119, 3062–3069. [Google Scholar]
- Meder, B.; Laufer, C.; Hassel, D.; Just, S.; Marquart, S.; Vogel, B.; Hess, A.; Fishman, M.C.; Katus, H.A.; Rottbauer, W. A single serine in the carboxyl terminus of cardiac essential myosin light chain-1 controls cardiomyocyte contractility in vivo. Circ. Res. 2009, 104, 650–659. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar]
- Chacko, E.; Ranganathan, S. Comprehensive splicing graph analysis of alternative splicing patterns in chicken, compared to human and mouse. BMC Genomics 2009, 10, S5. [Google Scholar]
- Modrek, B.; Lee, C. A genomic view of alternative splicing. Nat. Genet. 2002, 30, 13–19. [Google Scholar] [CrossRef]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar]
- Bentley, D. Coupling rna polymerase ii transcription with pre-mRNA processing. Curr. Opin. Cell Biol. 1999, 11, 347–351. [Google Scholar]
- Minvielle-Sebastia, L.; Keller, W. mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol. 1999, 11, 352–357. [Google Scholar] [CrossRef]
- Maniatis, T.; Reed, R. An extensive network of coupling among gene expression machines. Nature 2002, 416, 499–506. [Google Scholar]
- Philips, A.V.; Cooper, T.A. RNA processing and human disease. Cell. Mol. Life Sci. 2000, 57, 235–249. [Google Scholar] [CrossRef]
- Stoss, O.; Olbrich, M.; Hartmann, A.M.; Konig, H.; Memmott, J.; Andreadis, A.; Stamm, S. The star/gsg family protein rslm-2 regulates the selection of alternative splice sites. J. Biol. Chem. 2001, 276, 8665–8673. [Google Scholar]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Mendell, J.T.; Dietz, H.C. When the message goes awry: Disease-producing mutations that influence mrna content and performance. Cell 2001, 107, 411–414. [Google Scholar] [CrossRef]
- Caceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Nissim-Rafinia, M.; Kerem, B. Splicing regulation as a potential genetic modifier. Trends Genet. 2002, 18, 123–127. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef]
- Hammond, S.M.; Wood, M.J. Genetic therapies for rna mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef]
- Tang, F.; Lao, K.; Surani, M.A. Development and applications of single-cell transcriptome analysis. Nat. Methods 2011, 8, S6–S11. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Raghavachari, N.; Barb, J.; Yang, Y.; Liu, P.; Woodhouse, K.; Levy, D.; O'Donnell, C.J.; Munson, P.J.; Kato, G.J. A systematic comparison and evaluation of high density exon arrays and rna-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC Med. Genomics 2012, 5, 28. [Google Scholar]
- Feng, J.; Li, W.; Jiang, T. Inference of isoforms from short sequence reads. J. Comput. Biol. 2011, 18, 305–321. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Herbert, A.; Rich, A. RNA processing in evolution. The logic of soft-wired genomes. Ann. N. Y. Acad. Sci. 1999, 870, 119–132. [Google Scholar] [CrossRef]
- Kwan, T.; Benovoy, D.; Dias, C.; Gurd, S.; Serre, D.; Zuzan, H.; Clark, T.A.; Schweitzer, A.; Staples, M.K.; Wang, H.; et al. Heritability of alternative splicing in the human genome. Genome Res. 2007, 17, 1210–1218. [Google Scholar] [CrossRef]
- Cheung, V.G.; Spielman, R.S.; Ewens, K.G.; Weber, T.M.; Morley, M.; Burdick, J.T. Mapping determinants of human gene expression by regional and genome-wide association. Nature 2005, 437, 1365–1369. [Google Scholar]
- Veyrieras, J.B.; Kudaravalli, S.; Kim, S.Y.; Dermitzakis, E.T.; Gilad, Y.; Stephens, M.; Pritchard, J.K. High-resolution mapping of expression-qtls yields insight into human gene regulation. PLoS Genet. 2008, 4, e1000214. [Google Scholar]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with rna sequencing. Nature 2010, 464, 768–772. [Google Scholar]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar]
- Backes, C.; Meese, E.; Lenhof, H.P.; Keller, A. A dictionary on micrornas and their putative target pathways. Nucleic Acids Res. 2010, 38, 4476–4486. [Google Scholar]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef]
- Biesiadecki, B.J.; Elder, B.D.; Yu, Z.B.; Jin, J.P. Cardiac troponin t variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J. Biol. Chem. 2002, 277, 50275–50285. [Google Scholar] [CrossRef]
- Neagoe, C.; Kulke, M.; del Monte, F.; Gwathmey, J.K.; de Tombe, P.P.; Hajjar, R.J.; Linke, W.A. Titin isoform switch in ischemic human heart disease. Circulation 2002, 106, 1333–1341. [Google Scholar]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a cug-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Poon, K.L.; Tan, K.T.; Wei, Y.Y.; Ng, C.P.; Colman, A.; Korzh, V.; Xu, X.Q. RNA-binding protein rbm24 is required for sarcomere assembly and heart contractility. Cardiovasc. Res. 2012, 94, 418–427. [Google Scholar] [CrossRef]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator rbm20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012, 9, 390–396. [Google Scholar]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll Cardiol. 2009, 54, 930–941. [Google Scholar]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. Rbm20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar]
- Bass, B.L. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 1997, 22, 157–162. [Google Scholar] [CrossRef]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor glur-b mrna in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell Biol. 1995, 15, 5376–5388. [Google Scholar]
- Kawahara, Y.; Ito, K.; Sun, H.; Aizawa, H.; Kanazawa, I.; Kwak, S. Glutamate receptors: RNA editing and death of motor neurons. Nature 2004, 427, 801. [Google Scholar]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G. Adenosine-to-inosine rna editing meets cancer. Carcinogenesis 2011, 32, 1569–1577. [Google Scholar] [CrossRef]
- Seeburg, P.H.; Higuchi, M.; Sprengel, R. RNA editing of brain glutamate receptor channels: Mechanism and physiology. Brain Res. Brain Res. Rev. 1998, 26, 217–229. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread a-to-i RNA editing of alu-containing mrnas in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [Green Version]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant a-to-i editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Costa, F.F. Non-coding rnas: Meet thy masters. Bioessays 2010, 32, 599–608. [Google Scholar] [CrossRef]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef]
- Costa, V.; Angelini, C.; de Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-seq. J. Biomed. Biotechnol. 2010, 2010, 853916. [Google Scholar]
- Fratkin, E.; Bercovici, S.; Stephan, D.A. The implications of encode for diagnostics. Nat. Biotechnol. 2012, 30, 1064–1065. [Google Scholar]
- Tilgner, H.; Knowles, D.G.; Johnson, R.; Davis, C.A.; Chakrabortty, S.; Djebali, S.; Curado, J.; Snyder, M.; Gingeras, T.R.; Guigo, R. Deep sequencing of subcellular rna fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncrnas. Genome Res. 2012, 22, 1616–1625. [Google Scholar] [CrossRef]
- Amaral, P.P.; Mattick, J.S. Noncoding rna in development. Mamm. Genome 2008, 19, 454–492. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mehler, M.F. RNA regulation of epigenetic processes. Bioessays 2009, 31, 51–59. [Google Scholar] [CrossRef]
- Mattick, J.S. The genetic signatures of noncoding rnas. PLoS Genet. 2009, 5, e1000459. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding rnas: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Struhl, K. Transcriptional noise and the fidelity of initiation by rna polymerase II. Nat. Struct. Mol. Biol. 2007, 14, 103–105. [Google Scholar] [CrossRef]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef]
- Orom, U.A.; Derrien, T.; Guigo, R.; Shiekhattar, R. Long noncoding rnas as enhancers of gene expression. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 325–331. [Google Scholar] [CrossRef]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding rnas with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar] [CrossRef]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding rnas within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding rna induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef]
- Christov, C.P.; Trivier, E.; Krude, T. Noncoding human y RNAs are overexpressed in tumours and required for cell proliferation. Br. J. Cancer 2008, 98, 981–988. [Google Scholar]
- Angeloni, D.; ter Elst, A.; Wei, M.H.; van der Veen, A.Y.; Braga, E.A.; Klimov, E.A.; Timmer, T.; Korobeinikova, L.; Lerman, M.I.; Buys, C.H. Analysis of a new homozygous deletion in the tumor suppressor region at 3p12. 3 reveals two novel intronic noncoding rna genes. Genes Chromosomes Cancer 2006, 45, 676–691. [Google Scholar] [CrossRef]
- Koob, M.D.; Moseley, M.L.; Schut, L.J.; Benzow, K.A.; Bird, T.D.; Day, J.W.; Ranum, L.P. An untranslated ctg expansion causes a novel form of spinocerebellar ataxia (sca8). Nat. Genet. 1999, 21, 379–384. [Google Scholar]
- Lee, J.H.; Gao, C.; Peng, G.; Greer, C.; Ren, S.; Wang, Y.; Xiao, X. Analysis of transcriptome complexity through rna sequencing in normal and failing murine hearts. Circ. Res. 2011, 109, 1332–1341. [Google Scholar] [CrossRef]
- Harismendy, O.; Notani, D.; Song, X.; Rahim, N.G.; Tanasa, B.; Heintzman, N.; Ren, B.; Fu, X.D.; Topol, E.J.; Rosenfeld, M.G.; et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature 2011, 470, 264–268. [Google Scholar]
- Ishii, N.; Ozaki, K.; Sato, H.; Mizuno, H.; Saito, S.; Takahashi, A.; Miyamoto, Y.; Ikegawa, S.; Kamatani, N.; Hori, M.; et al. Identification of a novel non-coding RNA, miat, that confers risk of myocardial infarction. J. Hum. Genet. 2006, 51, 1087–1099. [Google Scholar] [CrossRef]
- Jarinova, O.; Stewart, A.F.; Roberts, R.; Wells, G.; Lau, P.; Naing, T.; Buerki, C.; McLean, B.W.; Cook, R.C.; Parker, J.S.; et al. Functional analysis of the chromosome 9p21.3. coronary artery disease risk locus. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1671–1677. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microrna biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar]
- Ramsingh, G.; Koboldt, D.C.; Trissal, M.; Chiappinelli, K.B.; Wylie, T.; Koul, S.; Chang, L.W.; Nagarajan, R.; Fehniger, T.A.; Goodfellow, P.; et al. Complete characterization of the micrornaome in a patient with acute myeloid leukemia. Blood 2010, 116, 5316–5326. [Google Scholar]
- Latronico, M.V.; Condorelli, G. Micrornas and cardiac pathology. Nat. Rev. Cardiol. 2009, 6, 419–429. [Google Scholar]
- Calin, G.A.; Croce, C.M. Investigation of microrna alterations in leukemias and lymphomas. Methods Enzymol. 2007, 427, 193–213. [Google Scholar]
- Keller, A.; Backes, C.; Leidinger, P.; Kefer, N.; Boisguerin, V.; Barbacioru, C.; Vogel, B.; Matzas, M.; Huwer, H.; Katus, H.A.; et al. Next-generation sequencing identifies novel micrornas in peripheral blood of lung cancer patients. Mol. Biosyst. 2011, 7, 3187–3199. [Google Scholar] [CrossRef]
- Keller, A.; Leidinger, P.; Bauer, A.; Elsharawy, A.; Haas, J.; Backes, C.; Wendschlag, A.; Giese, N.; Tjaden, C.; Ott, K.; et al. Toward the blood-borne mirnome of human diseases. Nat. Methods 2011, 8, 841–843. [Google Scholar]
- Meder, B.; Keller, A.; Vogel, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Just, S.; Borries, A.; Rudloff, J.; Leidinger, P.; et al. Microrna signatures in total peripheral blood as novel biomarkers for acute myocardial infarction. Basic Res. Cardiol. 2011, 106, 13–23. [Google Scholar]
- Calin, G.A.; Pekarsky, Y.; Croce, C.M. The role of microrna and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract. Res. Clin. Haematol. 2007, 20, 425–437. [Google Scholar] [CrossRef]
- Calin, G.A.; Liu, C.G.; Ferracin, M.; Hyslop, T.; Spizzo, R.; Sevignani, C.; Fabbri, M.; Cimmino, A.; Lee, E.J.; Wojcik, S.E.; et al. Ultraconserved regions encoding ncrnas are altered in human leukemias and carcinomas. Cancer Cell 2007, 12, 215–229. [Google Scholar] [CrossRef]
- Rossi, S.; Sevignani, C.; Nnadi, S.C.; Siracusa, L.D.; Calin, G.A. Cancer-associated genomic regions (cagrs) and noncoding RNAs: Bioinformatics and therapeutic implications. Mamm. Genome 2008, 19, 526–540. [Google Scholar] [CrossRef]
- Braconi, C.; Kogure, T.; Valeri, N.; Huang, N.; Nuovo, G.; Costinean, S.; Negrini, M.; Miotto, E.; Croce, C.M.; Patel, T. Microrna-29 can regulate expression of the long non-coding RNA gene meg3 in hepatocellular cancer. Oncogene 2011, 30, 4750–4756. [Google Scholar] [CrossRef]
- Jeggari, A.; Marks, D.S.; Larsson, E. Mircode: A map of putative microrna target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar]
- Chen, X.; Liang, H.; Zhang, C.Y.; Zen, K. Mirna regulates noncoding RNA: A noncanonical function model. Trends Biochem. Sci. 2012, 37, 457–459. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Ordovas, J.M.; Smith, C.E. Epigenetics and cardiovascular disease. Nat. Rev. Cardiol. 2010, 7, 510–519. [Google Scholar] [CrossRef]
- Szyf, M. The role of DNA hypermethylation and demethylation in cancer and cancer therapy. Curr. Oncol. 2008, 15, 72–75. [Google Scholar] [CrossRef]
- Szyf, M.; Pakneshan, P.; Rabbani, S.A. DNA methylation and breast cancer. Biochem. Pharmacol. 2004, 68, 1187–1197. [Google Scholar]
- Banerjee, H.N.; Verma, M. Epigenetic mechanisms in cancer. Biomark Med. 2009, 3, 397–410. [Google Scholar] [CrossRef]
- Vinci, M.C.; Polvani, G.; Pesce, M. Epigenetic programming and risk: The birthplace of cardiovascular disease? Stem Cell Rev. 2012. [Google Scholar] [CrossRef]
- Barker, D.J. Fetal programming of coronary heart disease. Trends Endocrinol. Metab. 2002, 13, 364–368. [Google Scholar] [CrossRef]
- Movassagh, M.; Choy, M.K.; Knowles, D.A.; Cordeddu, L.; Haider, S.; Down, T.; Siggens, L.; Vujic, A.; Simeoni, I.; Penkett, C.; et al. Distinct epigenomic features in end-stage failing human hearts. Circulation 2011, 124, 2411–2422. [Google Scholar]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 1–17. [Google Scholar]
- Bjornsson, H.T.; Fallin, M.D.; Feinberg, A.P. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004, 20, 350–358. [Google Scholar] [CrossRef]
- Leung, A.; Schones, D.E.; Natarajan, R. Using epigenetic mechanisms to understand the impact of common disease causing alleles. Curr. Opin. Immunol. 2012, 24, 558–563. [Google Scholar]
- Hackenberg, M.; Barturen, G.; Oliver, J.L. Ngsmethdb: A database for next-generation sequencing single-cytosine-resolution DNA methylation data. Nucleic Acids Res. 2011, 39, D75–D79. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim do, Y.; Park, J.Y.; Chang, H.Y.; Lee, M.H.; Han, K.H.; Chon, C.Y.; Ahn, S.H. Clinical features and prognosis of hepatocellular carcinoma with respect to pre-s deletion and basal core promoter mutations of hepatitis b virus genotype c2. J. Med. Virol. 2011, 83, 2088–2095. [Google Scholar]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Beck, S.; Rakyan, V.K. The methylome: Approaches for global DNA methylation profiling. Trends Genet. 2008, 24, 231–237. [Google Scholar] [CrossRef]
- Fouse, S.D.; Nagarajan, R.O.; Costello, J.F. Genome-scale DNA methylation analysis. Epigenomics 2010, 2, 105–117. [Google Scholar] [CrossRef]
- Jacinto, F.V.; Ballestar, E.; Esteller, M. Methyl-DNA immunoprecipitation (medip): Hunting down the DNA methylome. Biotechniques 2008, 44, 35, 37, 39 passim. [Google Scholar]
- Yu, W.; Jin, C.; Lou, X.; Han, X.; Li, L.; He, Y.; Zhang, H.; Ma, K.; Zhu, J.; Cheng, L.; et al. Global analysis of DNA methylation by methyl-capture sequencing reveals epigenetic control of cisplatin resistance in ovarian cancer cell. PLoS One 2011, 6, e29450. [Google Scholar]
- Farthing, C.R.; Ficz, G.; Ng, R.K.; Chan, C.F.; Andrews, S.; Dean, W.; Hemberger, M.; Reik, W. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008, 4, e1000116. [Google Scholar]
- Dindot, S.V.; Person, R.; Strivens, M.; Garcia, R.; Beaudet, A.L. Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions. Genome Res. 2009, 19, 1374–1383. [Google Scholar] [CrossRef]
- Down, T.A.; Rakyan, V.K.; Turner, D.J.; Flicek, P.; Li, H.; Kulesha, E.; Graf, S.; Johnson, N.; Herrero, J.; Tomazou, E.M.; et al. A bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat. Biotechnol. 2008, 26, 779–785. [Google Scholar]
- Koga, Y.; Pelizzola, M.; Cheng, E.; Krauthammer, M.; Sznol, M.; Ariyan, S.; Narayan, D.; Molinaro, A.M.; Halaban, R.; Weissman, S.M. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 2009, 19, 1462–1470. [Google Scholar] [CrossRef]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of cpg island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using medip-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar]
- Weinmann, A.S.; Yan, P.S.; Oberley, M.J.; Huang, T.H.; Farnham, P.J. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and cpg island microarray analysis. Genes Dev. 2002, 16, 235–244. [Google Scholar] [CrossRef]
- Ballestar, E.; Paz, M.F.; Valle, L.; Wei, S.; Fraga, M.F.; Espada, J.; Cigudosa, J.C.; Huang, T.H.; Esteller, M. Methyl-cpg binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003, 22, 6335–6345. [Google Scholar] [CrossRef]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of stat1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar]
- Euskirchen, G.M.; Rozowsky, J.S.; Wei, C.L.; Lee, W.H.; Zhang, Z.D.; Hartman, S.; Emanuelsson, O.; Stolc, V.; Weissman, S.; Gerstein, M.B.; et al. Mapping of transcription factor binding regions in mammalian cells by chip: Comparison of array- and sequencing-based technologies. Genome Res. 2007, 17, 898–909. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar]
- Fokstuen, S.; Munoz, A.; Melacini, P.; Iliceto, S.; Perrot, A.; Ozcelik, C.; Jeanrenaud, X.; Rieubland, C.; Farr, M.; Faber, L.; et al. Rapid detection of genetic variants in hypertrophic cardiomyopathy by custom DNA resequencing array in clinical practice. J. Med. Genet. 2011, 48, 572–576. [Google Scholar]
- Worthey, E.A.; Mayer, A.N.; Syverson, G.D.; Helbling, D.; Bonacci, B.B.; Decker, B.; Serpe, J.M.; Dasu, T.; Tschannen, M.R.; Veith, R.L.; et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011, 13, 255–262. [Google Scholar]
- Chou, L.S.; Liu, C.S.; Boese, B.; Zhang, X.; Mao, R. DNA sequence capture and enrichment by microarray followed by next-generation sequencing for targeted resequencing: Neurofibromatosis type 1 gene as a model. Clin. Chem. 2010, 56, 62–72. [Google Scholar] [CrossRef]
- Vogel, B.; Keller, A.; Frese, K.; Kloos, W.; Kayvanpour, E.; Sedaghat-Hamedani, F.; Hassel, S.; Marquart, S.; Beier, M.; Giannitis, E.; et al. Refining diagnostic microrna signatures by whole-mirnome kinetic analysis in acute myocardial infarction. Clin. Chem. 2012, 59, 410–418. [Google Scholar]
- Davies Thirty Groups Enter Clarity Clinical Genome Interpretation Challenge. Available online: http://genes.childrenshospital.org/ (accessed on 12 July 2012).
- Soden, S.E.; Farrow, E.G.; Saunders, C.J.; Lantos, J.D. Genomic medicine: Evolving science, evolving ethics. Per. Med. 2012, 9, 523–528. [Google Scholar]
- De Lecea, M.G.; Rossbach, M. Translational genomics in personalized medicine—Scientific challenges en route to clinical practice. HUGO J. 2012, 6, 2. [Google Scholar] [CrossRef]
- Tester, D.J.; Ackerman, M.J. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2011, 123, 1021–1037. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frese, K.S.; Katus, H.A.; Meder, B. Next-Generation Sequencing: From Understanding Biology to Personalized Medicine. Biology 2013, 2, 378-398. https://doi.org/10.3390/biology2010378
Frese KS, Katus HA, Meder B. Next-Generation Sequencing: From Understanding Biology to Personalized Medicine. Biology. 2013; 2(1):378-398. https://doi.org/10.3390/biology2010378
Chicago/Turabian StyleFrese, Karen S., Hugo A. Katus, and Benjamin Meder. 2013. "Next-Generation Sequencing: From Understanding Biology to Personalized Medicine" Biology 2, no. 1: 378-398. https://doi.org/10.3390/biology2010378