Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
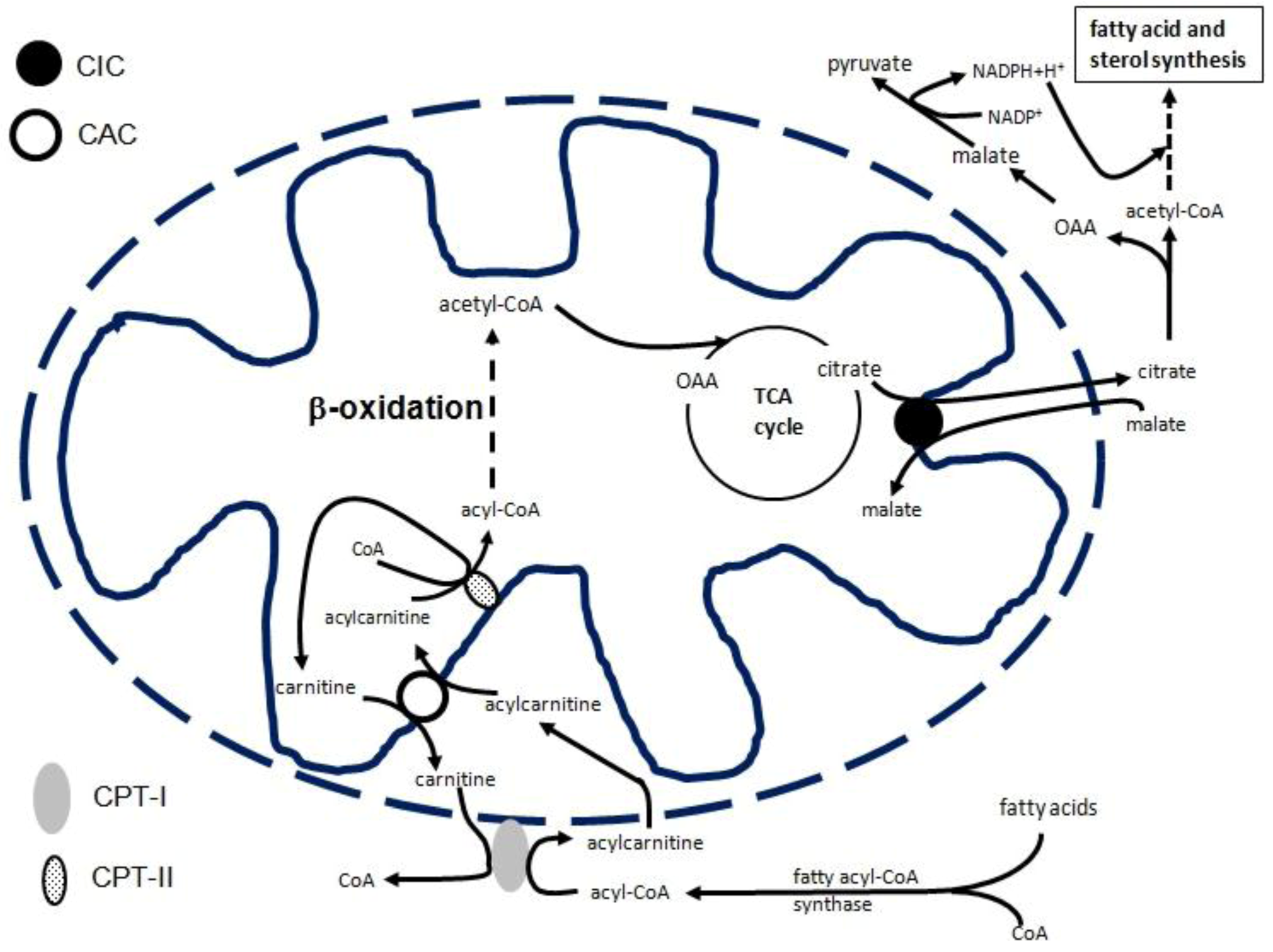
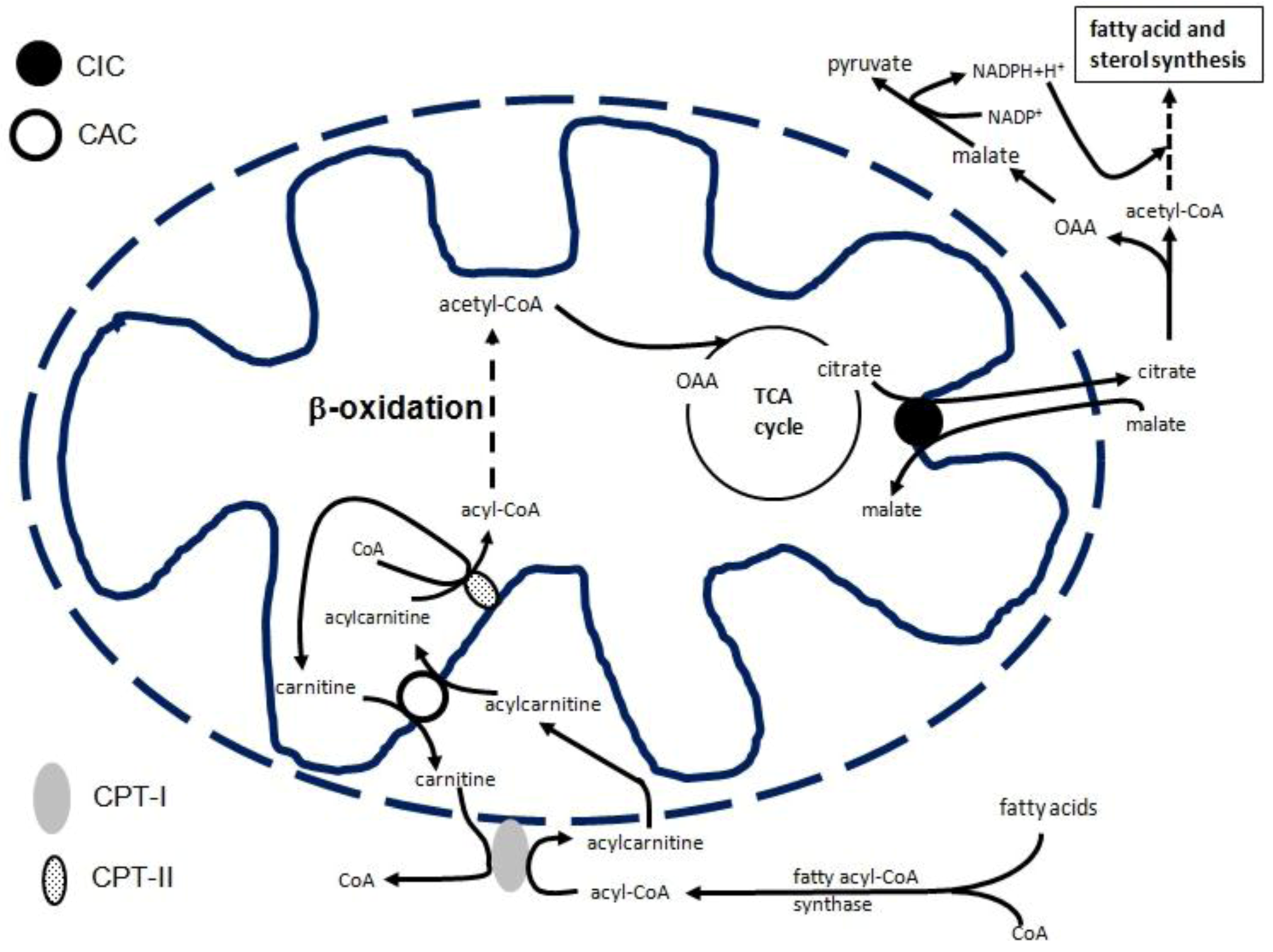
2. CIC Gene Transcriptional Regulation
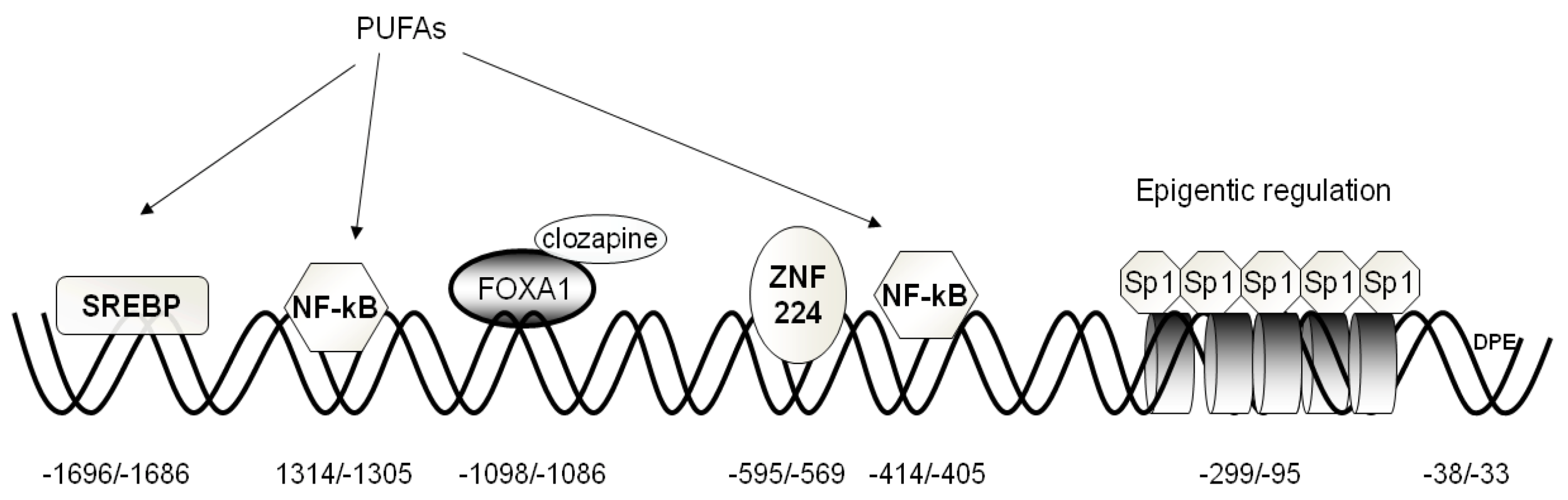
2.1. Activators
2.2. Repressors
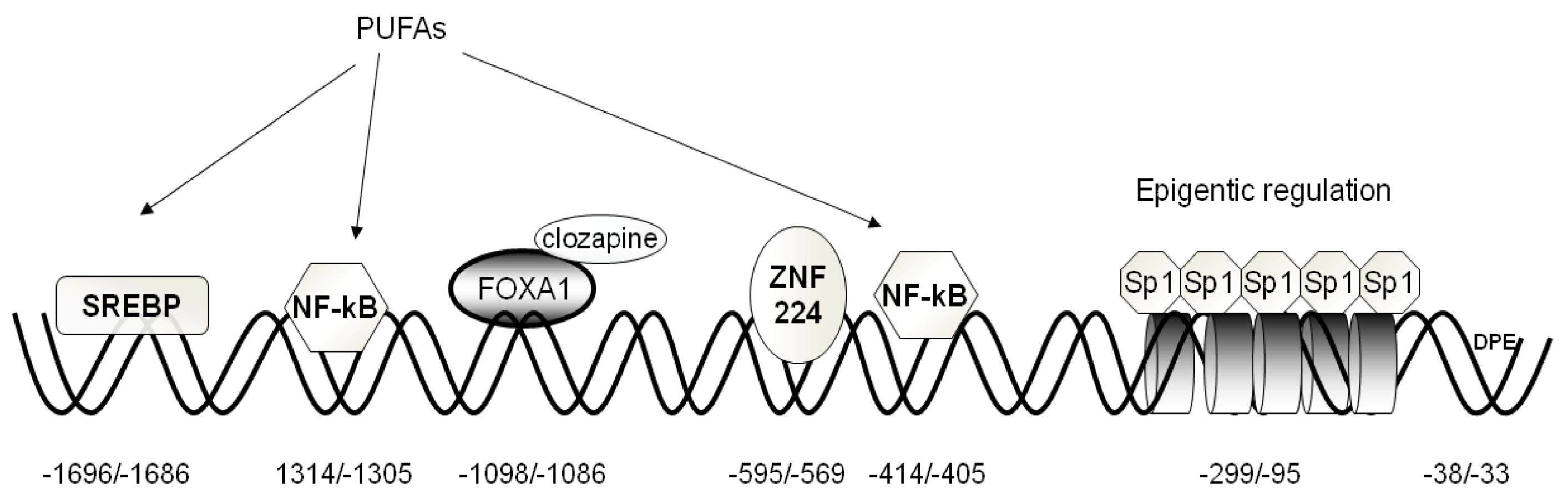
2.3. Epigenetic Mechanisms Regulating CIC Expression
2.4. Hormones Regulating CIC Expression
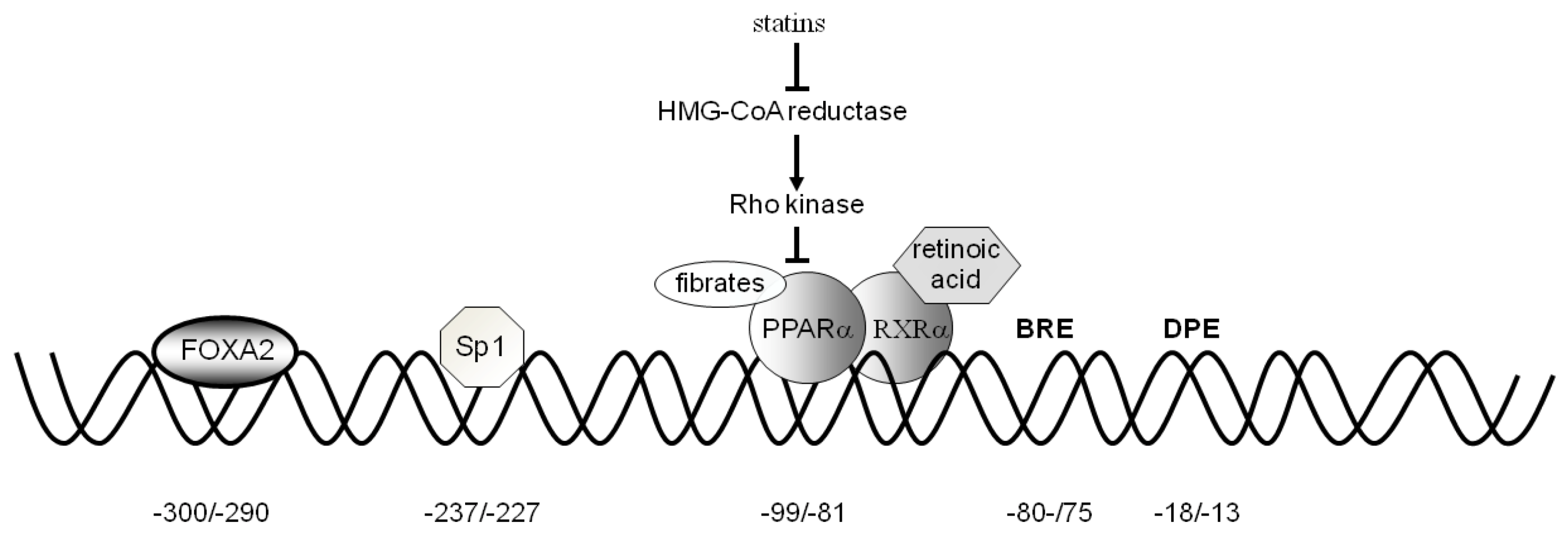
3. CAC Gene Transcriptional Regulation
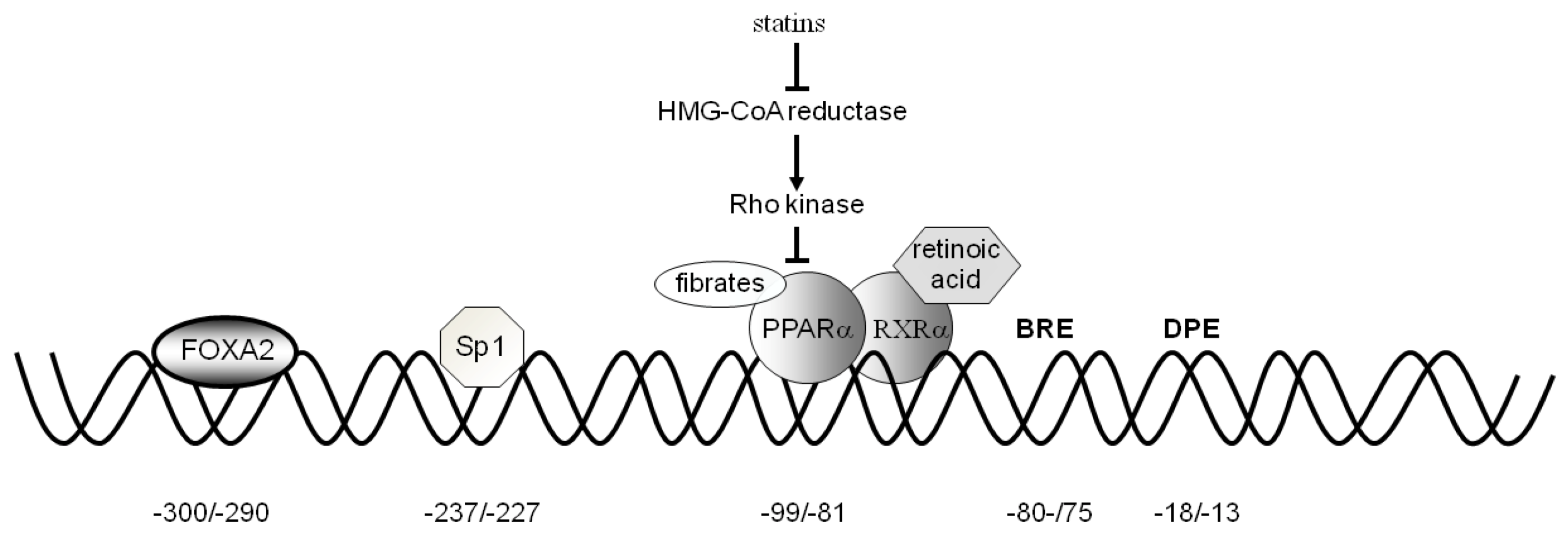
3.1. Activators
4. Regulation of CIC and CAC Expression by Fatty Acids
5. Conclusions
Acknowledgments
References and Notes
- Desvergne, B.; Liliane, M.; Wahali, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Shimano, H.; Horton, J.D.; Hammer, R.E.; Shimomura, I.; Brown, M.S.; Goldstein, J.L. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest. 1996, 98, 1575–1584. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I. Sterol regulatory element-binding proteins: Activators of cholesterol and fatty acid biosynthesis. Curr. Opin. Lipidol. 1999, 10, 143–150. [Google Scholar] [CrossRef]
- Siu, F.; Chen, C.; Zhong, C.; Kilberg, M.S. CCAAT/enhancer-binding protein-beta is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J. Biol. Chem. 2001, 276, 48100–48107. [Google Scholar]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Identification, properties and physiopathology. Mol. Aspects Med. 2012. [Google Scholar] [CrossRef]
- Li, K.; Hodge, J.A.; Wallace, D.C. OXBOX, a positive transcriptional element of the heart-skeletal muscle ADP/ATP translocator gene. J. Biol. Chem. 1990, 265, 20585–20588. [Google Scholar]
- Li, R.; Hodny, Z.; Luciakova, K.; Barath, P.; Nelson, B.D. Sp1 activates and inhibits transcription from separate elements in the proximal promoter of the human adenine nucleotide translocase 2 (ANT2) gene. J. Biol. Chem. 1996, 27, 18925–18930. [Google Scholar]
- Barath, P.; Poliakova, D.; Luciakova, K.; Nelson, B.D. Identification of NF1 as silencer protein of the human adenine nucleotide translocase-2 gene. Eur. J. Biochem. 2004, 271, 1781–1788. [Google Scholar] [CrossRef]
- Luciakova, K.; Barath, P.; Poliakova, D.; Persson, A.; Nelson, B.D. Repression of the human adenine nucleotide translocase-2 gene in growth-arrested human diploid cells: the role of nuclear factor-1. J. Biol. Chem. 2003, 278, 30624–30633. [Google Scholar]
- Kehoe, S.M.; Oka, M.; Hankowski, K.E.; Reichert, N.; Garcia, S.; McCarrey, J.R.; Gaubatz, S.; Terada, N. A conserved E2F6-binding element in murine meiosis-specific gene promoters. Biol. Reprod. 2008, 79, 921–930. [Google Scholar] [CrossRef]
- Medvedev, A.V.; Snedden, S.K.; Raimbault, S.; Ricquier, D.; Collins, S. Transcriptional regulation of the mouse uncoupling protein-2 gene. Double E-box motif is required for peroxisome proliferator-activated receptor-gamma-dependent activation. J. Biol. Chem. 2001, 276, 10817–10823. [Google Scholar]
- Yubero, P.; Manchado, C.; Cassard-Doulcier, M.; Mampel, T.; Viñas, O.; Iglesias, R.; Giralt, M.; Villarroya, F. CCAAT/enhancer binding proteins a and b are transcriptiona activators of the brown fat uncoupling protein gene promoter. Biochem. Biophys. Res. Commun. 1994, 198, 653–659. [Google Scholar] [CrossRef]
- Kozak, U.C.; Kopecky, J.; Teisinger, J.; Enerback, S.; Boyer, B.; Kozak, L.P. An upstream enhancer regulating brow-fat- specific expression of the mitochondrial uncoupling protein gene. Mol. Cell. Biol. 1994, 14, 59–67. [Google Scholar]
- Sluse, F.E.; Jarmuszkiewicz, W.; Navet, R.; Douette, P.; Mathy, G.; Sluse-Goffart, C.M. MitochondrialUCPs: New insights into regulation and impact. Biochim. Biophys. Acta 2006, 1757, 480–485. [Google Scholar] [CrossRef]
- Yonezawa, T.; Kurata, R.; Hosomichi, K.; Kono, A.; Kimura, M.; Inoko, H. Nutritional and hormonal regulation of uncoupling protein 2. IUBMB Life 2009, 61, 1123–1131. [Google Scholar] [CrossRef]
- Bugge, A.; Siersbaek, M.; Madsen, M.S.; Göndör, A.; Rougier, C.; Mandrup, S. A novel intronic peroxisome proliferator-activated receptor gamma enhancer in the uncoupling protein (UCP) 3 gene as a regulator of both UCP2 and -3 expression in adipocytes. J. Biol. Chem. 2010, 285, 1731–1737. [Google Scholar]
- Iacobazzi, V.; Infantino, V.; Costanzo, P.; Izzo, P.; Palmieri, F. Functional analysis of the promoter of the mitochondrial phosphate carrier human gene: Identification of activator and repressor elements and their transcription factors. Biochem. J. 2005, 391, 613–621. [Google Scholar] [CrossRef]
- Pegorier, J.P.; Le May, C.; Girard, J. Control of Gene Expression by Fatty Acids. J. Nutr. 2004, 134, 2444S–2449S. [Google Scholar]
- Heisterkamp, N.; Mulder, M.P.; Langeveld, A.; ten Hoeve, J.; Wang, Z.; Roe, B.A.; Groffen, J. Localization of the human mitochondrial citrate transporter protein gene tochromosome22Q11 in the DiGeorge syndrome critical region. Genomics 1995, 29, 451–456. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Lauria, G.; Palmieri, F. Organization and sequence of the human gene for the mitochondrial citrate transport protein. DNA Seq. 1997, 7, 127–139. [Google Scholar]
- Huizing, M.; Ruitenbeek, W.; van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.; Palmieri, F.; Trijbels, J.M. Human mitochondrial transmembrane metabolite carriers: tissue distribution and its implication for mitochondrial disorders. J. Bioenerg Biomembr. 1998, 30, 277–284. [Google Scholar] [CrossRef]
- Quina, A.S.; Buschbeck, M.; Di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Palmieri, F. Epigenetic mechanisms and Sp1 regulate mitochondrial citrate carrier gene expression. Biochem. Biophys. Res. Commun. 2008, 376, 15–20. [Google Scholar] [CrossRef]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Bisaccia, F.; Castegna, A.; Palmieri, F. Role of FOXA in mitochondrial citrate carrier gene expression and insulin secretion. Biochem. Biophys. Res. Commun. 2009, 385, 220–224. [Google Scholar] [CrossRef]
- Friedman, J.R.; Kaestner, K.H. The Foxa family of transcription factors in development and metabolism. Cell Mol. Life Sci. 2006, 63, 2317–2328. [Google Scholar] [CrossRef]
- Shih, D.Q.; Navas, M.A.; Kuwajima, S.; Duncan, S.A.; Stoffel, M. Impaired glucose homeostasis and neonatal mortality in hepatocyte nuclear factor 3alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1999, 96, 10152–10157. [Google Scholar] [CrossRef]
- Vatamaniuk, M.Z.; Gupta, R.K.; Lantz, K.A.; Doliba, N.M.; Matschinsky, F.M.; Kaestner, K.H. Foxa1-deficient mice exhibit impaired insulin secretion due to uncoupled oxidative phosphorylation. Diabetes 2006, 55, 2730–2736. [Google Scholar] [CrossRef]
- Joseph, J.; Jensen, M.; Ilkayeva, O.; Palmieri, F.; Alárcon, C.; Rhodes, C.; Newgard, C. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. [Google Scholar]
- Menga, A.; Infantino, V.; Iacobazzi, F.; Convertini, P.; Palmieri, F.; Iacobazzi, V. Insight into mechanism of in vitro insulin secretion increase induced by antipsychotic clozapine: Role of FOXA1 and mitochondrial citrate carrier. Eur. Neuropsychopharmacol. 2012, in press. [Google Scholar]
- Sasaki, N.; Iwase, M.; Uchizono, Y.; Nakamura, U.; Imoto, H.; Abe, S.; Iida, M. The atypical antipsychotic clozapine impairs insulin secretion by inhibiting glucose metabolism and distal steps in rat pancreatic islets. Diabetologia 2006, 49, 2930–2938. [Google Scholar] [CrossRef]
- Infantino, V.; Iacobazzi, V.; De Santis, F.; Mastrapasqua, M.; Palmieri, F. Transcription of the mitochondrial citrate carrier gene: Role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem. Biophys. Res. Commun. 2007, 356, 249–254. [Google Scholar] [CrossRef]
- Osborne, T. F Sterol regulatory element-binding proteins (SREBPs): Key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000, 275, 32379–32382. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Invest. 1998, 101, 2331–2339. [Google Scholar] [CrossRef]
- Goodridge, A.G. Dietary regulation of gene expression: enzymes involved in carbohydrate and lipid metabolism. Annu. Rev. Nutr. 1987, 7, 157–185. [Google Scholar] [CrossRef]
- Hillgartner, F.B.; Salati, L.M.; Goodridge, A.G. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev. 1995, 75, 47–76. [Google Scholar]
- Zhang, Y.; Hillgartner, F.B. Starvation and feeding a high-carbohydrate, low-fat diet regulate the expression sterol regulatory element-binding protein-1 in chickens. J. Nutr. 2004, 134, 2205–2210. [Google Scholar]
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661. [Google Scholar] [CrossRef]
- Foretz, M.; Guichard, C.; Ferré, P.; Foufelle, F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc. Natl. Acad. Sci. 1999, 96, 12737–12740. [Google Scholar]
- Carrillo, J.J.; Ibares, B.; Esteban-Gamboa, A.; Feliu, J.E. Involvement of both phosphatidylinositol 3-kinase and p44/p42 mitogenactivated protein kinase pathways in the short-term regulation of pyruvate kinase L by insulin. Endocrinology 2001, 142, 1057–1064. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar]
- O'Neill, L.A. A critical role forcitratemetabolism inLPSsignalling. Biochem. J. 2011, 438, 5–6. [Google Scholar]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Citrate carrier promoter is target of peroxisome proliferator-activated receptor alpha and gamma in hepatocytes and adipocytes. Int J. Biochem. Cell. Biol. 2012, 44, 659–668. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V.; Convertini, P.; Vozza, A.; Agrimi, G.; Palmieri, F. Transcription of the mitochondrial citrate carrier gene: identification of a silencer and its binding protein ZNF224. Biochem. Biophys. Res. Commun. 2009, 386, 186–191. [Google Scholar] [CrossRef]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef]
- Herrera, E.; Amusquivar, E. Lipid metabolism in the fetus and the newborn. Diabetes Metab. Res. Rev. 2000, 16, 202–210. [Google Scholar] [CrossRef]
- Van Aerde, J.E.; Feldman, M.; Clandinin, M.T. Fetal and Neonatal Physiology, 2nd; Polin, R.A., Fox, W.W., Eds.; Elsevier Health Sciences: Philadelphia, PA, USA, 1998; pp. 458–477. [Google Scholar]
- Kaplan, R.S.; Oliveira, D.L.; Wilson, G.L. Streptozotocin-induced alterations in the levels of functional mitochondrial anion transport proteins. Arch. Biochem. Biophys. 1990, 280, 1811–1891. [Google Scholar]
- Damiano, F.; Mercuri, E.; Stanca, E.; Gnoni, G.V.; Siculella, L. Streptozotocin-induced diabetes affects in rat liver citrate carrier gene expression by transcriptional and posttranscriptional mechanisms. Int. J. Biochem. Cell. Biol. 2011, 43, 1621–1629. [Google Scholar] [CrossRef]
- Kaplan, R.S.; Mayor, J.A.; Blackwell, R.; Maughon, R.H.; Wilson, G.L. The effect of insulin supplementation on diabetes-induced alterations in the extractable levels of functional mitochondrial anion transport proteins. Arch. Biochem Biophys. 1991, 287, 305–311. [Google Scholar] [CrossRef]
- Giudetti, A.M.; Leo, M.; Siculella, L.; Gnoni, G.V. Hypothyroidism down-regulates mitochondrial citrate carrier activity and expression in rat liver. Biochim. Biophys Acta. 2006, 1761, 484–491. [Google Scholar] [CrossRef]
- Siculella, L.; Sabetta, S.; Giudetti, A.M.; Gnoni, G.V. Hypothyroidism reduces tricarboxylate carrier activity and expression in rat liver mitochondria by reducing nuclear transcription rate and splicing efficiency. J. Biol. Chem. 2006, 281, 19072–19080. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Gullberg, H.; Fernandez, L.; Ståhlberg, N.; Lee, N.H.; Vennström, B.; Norstedt, G. Patterns of liver gene expression governed by TRbeta. Mol. Endocrinol. 2002, 16, 1257–1260. [Google Scholar] [CrossRef]
- Mynatt, R.L.; Park, E.A.; Thorngate, F.E.; Das, H.K.; Cook, G.A. Changes in carnitine palmitoyltransferase-I mRNA abundance produced by hyperthyroidism and hypothyroidism parallel changes in activity. Biochem. Biophys. Res. Commun. 1994, 201, 932–937. [Google Scholar] [CrossRef]
- Viggiano, L.; Iacobazzi, V.; Marzella, R.; Cassano, C.; Rocchi, M.; Palmieri, F. Assignment of the carnitine/acylcarnitine translocase gene (CACT) to human chromosome band 3p21.31 by in situ hybridization. Cytogenet. Cell. Genet. 1997, 79, 62–63. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Naglieri, M.A.; Stanley, C.A.; Wanders, R.J.; Palmieri, F. The structure and organization of the human carnitine/acylcarnitine translocase (CACT) gene. Biochem. Biophys. Res. Commun. 1998, 252, 770–774. [Google Scholar] [CrossRef]
- Huizing, M.; Iacobazzi, V.; Ijlst, L.; Savelkoul, P.; Ruitenbeek, W.; van den Heuvel, L.; Indiveri, C.; Smeitink, J.; Trijbels, F.; Wanders, R.; Palmieri, F. Cloning of the human carnitine–acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am. J. Hum. Genet. 1997, 61, 1239–1245. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Convertini, P.; Infantino, V.; Scarcia, P.; Todisco, S.; Palmieri, F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem. Biophys. Res. Commun. 2009, 388, 643–647. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; Moore, J.T.; Willson, T.M. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. 2001, 98, 13919–13924. [Google Scholar]
- Peters, J.M.; Hennuyer, N.; Staels, B.; Fruchart, J.C.; Fievet, C.; Gonzalez, F.J.; Auwerx, J. Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J. Biol. Chem. 1997, 272, 27307–27312. [Google Scholar]
- Martin, G.; Duez, H.; Blanquart, C.; Berezowski, V.; Poulain, P.; Fruchart, J.C.; Najib-Fruchart, J.; Glineur, C.; Staels, B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J. Clin. Invest. 2001, 107, 1423–1432. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Orphan nuclear receptors: shifting endocrinology into reverse. Science 1999, 284, 757–760. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Garavaglia, B.; Bartuli, A.; Invernizzi, F.; DiDonato, S.; Sabetta, G.; Kahler, S.G.; Millington, D.S. Carnitine–acylcarnitine translocase deficiency: Benign course without cardiac involvement. Pediatr. Res. 1995, 37, 147. [Google Scholar]
- Olpin, S.E.; Bonham, J.R.; Downing, M.; Manning, N.J.; Pollitt, R.J.; Sharrard, M.J.; Tanner, M.S. Carnitine–acylcarnitine translocase deficiency- a mild phenotype. J. Inherit. Metab. Dis. 1997, 20, 714–715. [Google Scholar] [CrossRef]
- Morris, A.A.; Olpin, S.E.; Brivet, M.; Turnbull, D.M.; Jones, R.A.; Leonard, J.V. A patient with carnitine–acylcarnitine translocase deficiency with a mild Phenotype. J. Pediatr. 1998, 132, 514–516. [Google Scholar] [CrossRef]
- Lopriore, E.; Gemke, R.J.; Verhoeven, N.M.; Jakobs, C.; Wanders, R.J.; Roeleveld-Versteeg, A.B. Carnitine–acylcarnitine translocase deficiency: Phenotype, residual enzyme activity and outcome. Eur. J. Pediatr. 2001, 160, 101–104. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Invernizzi, F.; Baratta, S.; Pons, R.; Chung, W.; Garavaglia, B.; Dionisi-Vici, C.; Ribes, A.; Parini, R.; Huertas, M.D.; Roldan, S.; Lauria, G.; Palmieri, F.; Taroni, F. Molecular and functional analysis of SLC25A20 mutations causing carnitine–acylcarnitine translocase deficiency. Hum. Mutat. 2004, 24, 312–320. [Google Scholar] [CrossRef]
- Convertini, P.; Infantino, V.; Bisaccia, F.; Palmieri, F.; Iacobazzi, V. Role of FOXA and Sp1 in mitochondrial acylcarnitine carrier gene expression in different cell lines. Biochem. Biophys. Res. Commun. 2011, 404, 376–381. [Google Scholar] [CrossRef]
- Wolfrum, C.; Asilmaz, E.; Luca, E.; Friedman, J.M.; Stoffel, M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004, 432, 1027–1032. [Google Scholar] [CrossRef]
- Silva, J.P.; von Meyenn, F.; Howell, J.; Thorens, B.; Wolfrum, C.; Stoffel, M. Regulation of adaptive behaviour during fasting by hypothalamic Foxa2. Nature 2009, 462, 646–650. [Google Scholar] [CrossRef]
- York, B.; O'Malley, B.W. Steroid receptor coactivator (SRC) family: Masters of systems biology. J. Biol. Chem. 2010, 285, 38743–38750. [Google Scholar] [CrossRef]
- York, B.; Reineke, E.L.; Sagen, J.V.; Nikolai, B.C.; Zhou, S.; Louet, J.F.; Chopra, A.R.; Chen, X.; Reed, G.; Noebels, J.; Adesina, A.M.; Yu, H.; Wong, L.J.; Tsimelzon, A.; Hilsenbeck, S.; Stevens, R.D.; Wenner, B.R.; Ilkayeva, O.; Xu, J.; Newgard, C.B.; O'Malley, B.W. Ablation of steroid receptor coactivator-3 resembles the human CACT metabolic myopathy. Cell Metab. 2012, 15, 752–763. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Tonazzi, A.; Giangregorio, N.; Infantino, V.; Convertini, P.; Console, L.; Palmieri, F. The mitochondrial carnitine/acylcarnitine carrier: function, structure and physiopathology. Mol. Aspects Med. 2011, 32, 223–233. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; Lopaschuk, G.D.; Muoio, D.M. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Gacias, M.; Pérez-Martí, A.; Pujol-Vidal, M.; Marrero, P.F.; Haro, D.; Relat, J. PGC-1β regulates mouse carnitine–acylcarnitine translocase through estrogen-related receptor α. Biochem. Biophys. Res. Commun. 2012, 423, 838–843. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar]
- Dufour, C.R.; Wilson, B.J.; Huss, J.M.; Kelly, D.P.; Alaynick, W.A.; Downes, M.; Evans, R.M.; Blanchette, M.; Giguère, V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007, 5, 345–356. [Google Scholar] [CrossRef]
- Jump, D.B.; Botolin, D.; Wang, Y.; Xu, J.; Christian, B.; Demeure, O. Fatty acid regulation of hepatic gene transcription. J. Nutr. 2005, 135, 2503–2506. [Google Scholar]
- Clarke, S.D.; Jump, D.B. Dietary polyunsaturated fatty acid regulation of gene transcription. Annu. Rev. Nutr. 1994, 14, 83–98. [Google Scholar] [CrossRef]
- Ferramosca, A.; Conte, A.; Burri, L.; Berge, K.; De Nuccio, F.; Giudetti, A.M. A krill oil supplemented diet suppresses hepatic steatosis in high-fat fed rats. PLoS One 2012, 7, e38797. [Google Scholar]
- Damiano, F.; Gnoni, G.V.; Siculella, L. Functional analysis of rat livercitratecarrier promoter: differential responsiveness to polyunsaturated fatty acids. Biochem. J. 2009, 417, 561–571. [Google Scholar] [CrossRef]
- Siculella, L.; Sabetta, S.; Damiano, F.; Giudetti, A.M.; Gnoni, G.V. Different dietary fatty acids have dissimilar effects on activity and gene expression of mitochondrial tricarboxylate carrier in rat liver. FEBS Lett. 2004, 578, 280–284. [Google Scholar] [CrossRef]
- Jump, D.B.; Depner, C.M.; Tripathy, S. Omega-3 Fatty Acid Supplementation and Cardiovascular Disease. J. Lipid Res. 2012, 309, 27. [Google Scholar]
- Priore, P.; Stanca, E.; Gnoni, G.V.; Siculella, L. Dietary fat types differently modulate the activity and expression of mitochondrial carnitine/acylcarnitine translocase in rat liver. Biochim. Biophys. Acta 2012, 1821, 1341–1349. [Google Scholar] [CrossRef]
- Chen, W.; Esselman, W.J.; Jump, D.B.; Busik, J.V. Anti-inflammatory effect of docosahexaenoic acid on cytokine induced adhesion molecule expression in human retinal vascular endothelial cells. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4342–4347. [Google Scholar] [CrossRef]
- Schumann, J.; Fuhrmann, H. Impairment of NFkappaB activity by unsaturated fatty acids. Int. Immunopharmacol. 2010, 10, 978–984. [Google Scholar] [CrossRef]
- Bisaccia, F.; De Palma, A.; Palmieri, F. Identification and purification of the tricarboxylate carrier from rat liver mitochondria. Biochim. Biophys. Acta. 1989, 977, 171–176. [Google Scholar] [CrossRef]
- Kaplan, R.S.; Mayor, J.A.; Wood, D.O. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J. Biol. Chem. 1993, 268, 13682–13690. [Google Scholar]
- Kaplan, R.S.; Mayor, J.A.; Gremse, D.A.; Wood, D.O. High level expression and characterization of the mitochondrialcitratetransport protein from theyeastSaccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 4108–4110. [Google Scholar]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification andpurificationof thecarnitinecarrier from rat liver mitochondria. Biochim. Biophys. Acta. 1990, 1020, 81–86. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. The mitochondrialcarnitinecarrier protein: cDNA cloning, primary structure and comparison with other mitochondrial transport proteins. Biochem. J. 1997, 321, 713–719. [Google Scholar]
- Palmieri, L.; Lasorsa, F.M.; Iacobazzi, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae. FEBS Lett. 1999, 462, 472–476. [Google Scholar] [CrossRef]
- Indiveri, C.; Iacobazzi, V.; Giangregorio, N.; Palmieri, F. Bacterial overexpression, purification and reconstitution of the carnitine/acylcarnitine carrier from rat liver mitochondria. Biochemical. Biophys. Res. Commun. 1998, 249, 589–594. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Prezioso, G.; Palmieri, F. Kinetic characterization of the reconstituted carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta. 1991, 1065, 231–238. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Characterization of the unidirectional transport of carnitine catalyzed by the reconstituted carnitine carrier from rat liver mitochondria. Biochim. Biophys. Acta. 1991, 1069, 110–116. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. The reconstituted carnitine carrier from rat liver mitochondria. Evidence for a transport mechanism different from that of the other mitochondrial translocators. Biochim. Biophys. Acta 1994, 1189, 65–73. [Google Scholar] [CrossRef]
- Indiveri, C.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Site-directed mutagenesis and chemical modification of the six native cysteine residues of the rat mitochondrial carnitine carrier: implications for the role of cysteine-136. Biochemistry 2002, 41, 8649–8656. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis and chemical modification of three vicinal cysteine residues in rat mitochondrial carnitine/acylcarnitine transporter. J. Biol. Chem. 2005, 280, 19607–19612. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Indiveri, C.; Palmieri, F. Conformation-dependent accessibility of Cys-136 and Cys-155 of the mitochondrial rat carnitine/acylcarnitine carrier to membrane-impermeable SH reagents. Biochim. Biophys. Acta. 2007, 1767, 1331–1339. [Google Scholar] [CrossRef]
- de Lucas, J.R.; Indiveri, C.; Tonazzi, A.; Perez, P.; Giangregorio, N.; Iacobazzi, V.; Palmieri, F. Functional characterisation of residues within the carnitine/acylcarnitine translocase RX2PANAAXF distinct motif. Mol. Membr Biol. 2008, 25, 152–163. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Site-directed mutagenesis of the His residues of the rat mitochondrial carnitine/acylcarnitine carrier: implications for the role of His-29 in the transport pathway. Biochim. Biophys. Acta 2009, 1787, 1009–1015. [Google Scholar] [CrossRef]
- Giangregorio, N.; Tonazzi, A.; Console, L.; Indiveri, C.; Palmieri, F. Site-directed mutagenesis of charged amino acids of the human mitochondrial carnitine/acylcarnitine carrier: Insight into the molecular mechanism of transport. Biochim. Biophys. Acta 2010, 1797, 839–845. [Google Scholar] [CrossRef]
- Tonazzi, A.; Console, L.; Giangregorio, N.; Indiveri, C.; Palmieri, F. Identification by site-directed mutagenesis of a hydrophobic binding site of the mitochondrial carnitine/acylcarnitine carrier involved in the interaction with acyl groups. Biochim. Biophys. Acta 2012, 1817, 697–704. [Google Scholar] [CrossRef]
- Xu, Y.; Kakhniashvili, D.A.; Gremse, D.A.; Wood, D.O.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. The yeast mitochondrial citrate transport protein. Probing the roles of cysteines. J. Biol. Chem. 2000, 275, 7117–7124. [Google Scholar]
- Kaplan, R.S.; Mayor, J.A.; Brauer, D.; Kotaria, R.; Walters, D.E.; Dean, A.M. The yeast mitochondrial citrate protein. Probing the secondary structure of transmembrane domain IV and identification of residues that likely coprise a portion of the citrate translocation pathway. J. Biol. Chem. 2000, 275, 12009–12016. [Google Scholar]
- Kaplan, R.S.; Mayor, J.A.; Kotaria, R.; Walters, D.E.; McHaourab, H.S. The yeast mitochondrial citrate transport protein: Determination of secondary structure and solvent accessibility of transmembrane domain IV using site-directed spin labeling. Biochemistry 2000, 39, 9157–9163. [Google Scholar] [CrossRef]
- Ma., C.; Kotaria, R.; Mayor, J.A.; Eriks, L.R.; Dean, A.M.; Walters, D.E.; Kaplan, R.S. The mitochondrial citrate transport protein: probing the secondary structure of transmembrane domain III, identification of residues that likely comprise a portion of the citrate transport pathway, and development of a model for the putative TMDIII-TMDIII' interface. J. Biol. Chem. 2004, 279, 1533–1540. [Google Scholar]
- Ma, C.; Kotaria, R.; Mayor, J.A.; Remani, S.; Walters, D.E.; Kaplan, R.S. The yeast mitochondrial citrate transport protein: characterization of transmembrane domain III residue involvement in substrate translocation. J. Biol. Chem. 2005, 280, 2331–2340. [Google Scholar]
- Ma, C.; Remani, S.; Sun, J.; Kotaria, R.; Mayor, J.A.; Walters, D.E.; Kaplan, R.S. Identification of the substrate binding sites within the yeast mitochondrial citrate transport protein. J. Biol. Chem. 2007, 282, 17210–17220. [Google Scholar]
- Aluvila, S.; Sun, J.; Harrison, D.H.; Walters, D.E.; Kaplan, R.S. Inhibitors of the mitochondrial citrate transport protein: Validation of the role of substrate binding residues and discovery of the first purely competitive inhibitor. Mol. Pharmacol. 2010, 77, 26–34. [Google Scholar] [CrossRef]
- Aluvila, S.; Kotaria, R.; Sun, J.; Mayor, J.A.; Walters, D.E.; Harrison, D.H.; Kaplan, R.S. The yeast mitochondrial citrate transport protein: Molecular determinants of its substrate specificity. J. Biol. Chem. 2010, 285, 27314–27326. [Google Scholar]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; Avantaggiati, M.L. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iacobazzi, V.; Infantino, V.; Palmieri, F. Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation. Biology 2013, 2, 284-303. https://doi.org/10.3390/biology2010284
Iacobazzi V, Infantino V, Palmieri F. Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation. Biology. 2013; 2(1):284-303. https://doi.org/10.3390/biology2010284
Chicago/Turabian StyleIacobazzi, Vito, Vittoria Infantino, and Ferdinando Palmieri. 2013. "Transcriptional Regulation of the Mitochondrial Citrate and Carnitine/Acylcarnitine Transporters: Two Genes Involved in Fatty Acid Biosynthesis and β-oxidation" Biology 2, no. 1: 284-303. https://doi.org/10.3390/biology2010284