The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
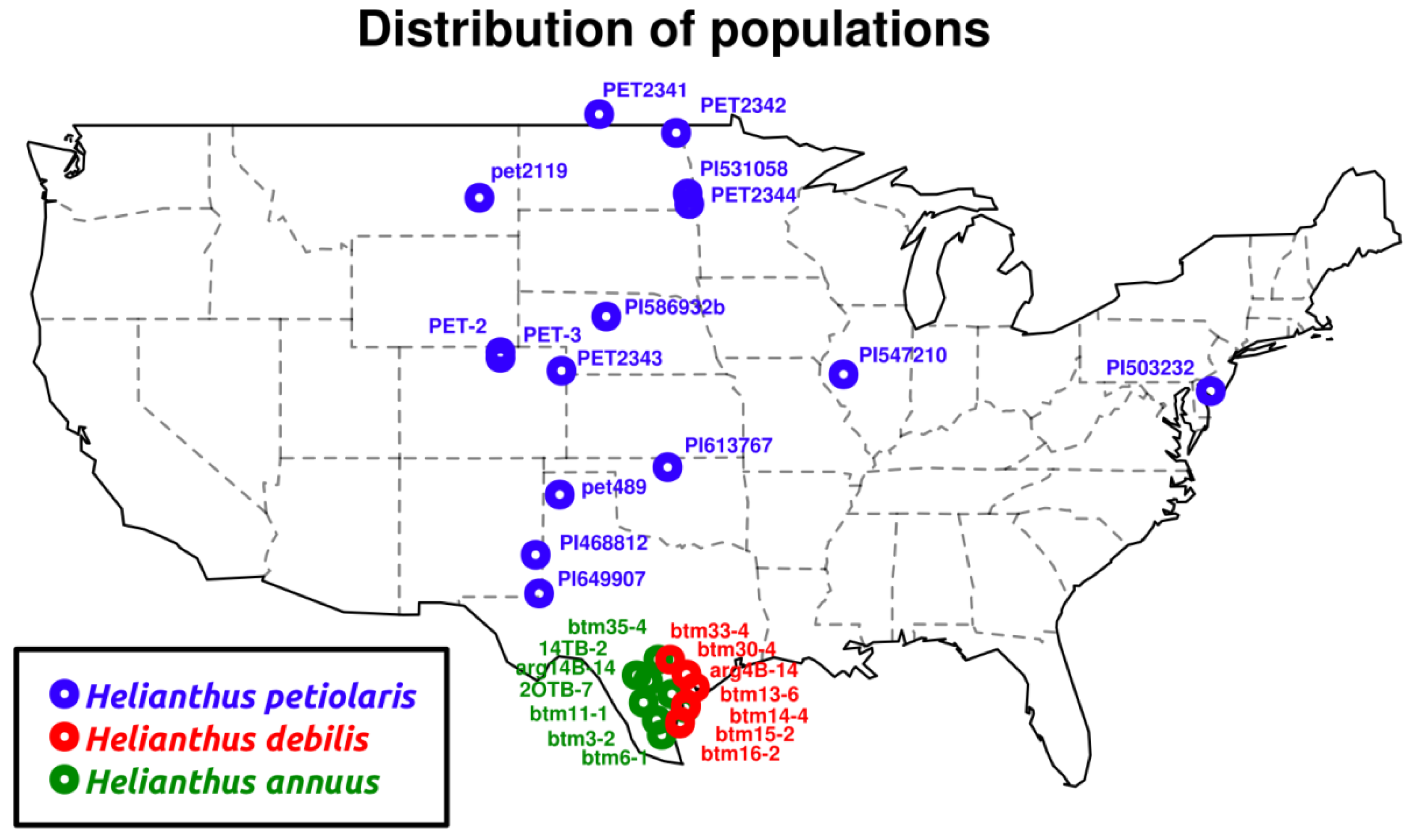

{kind=link}
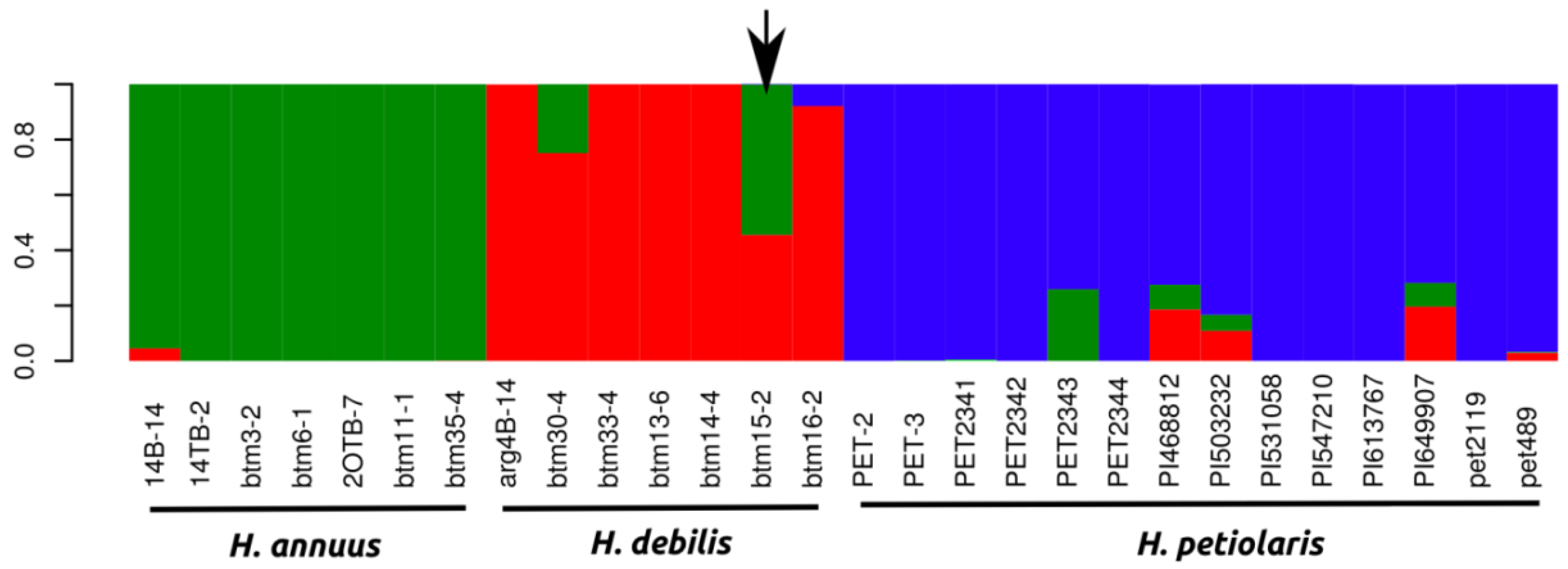{kind=link}
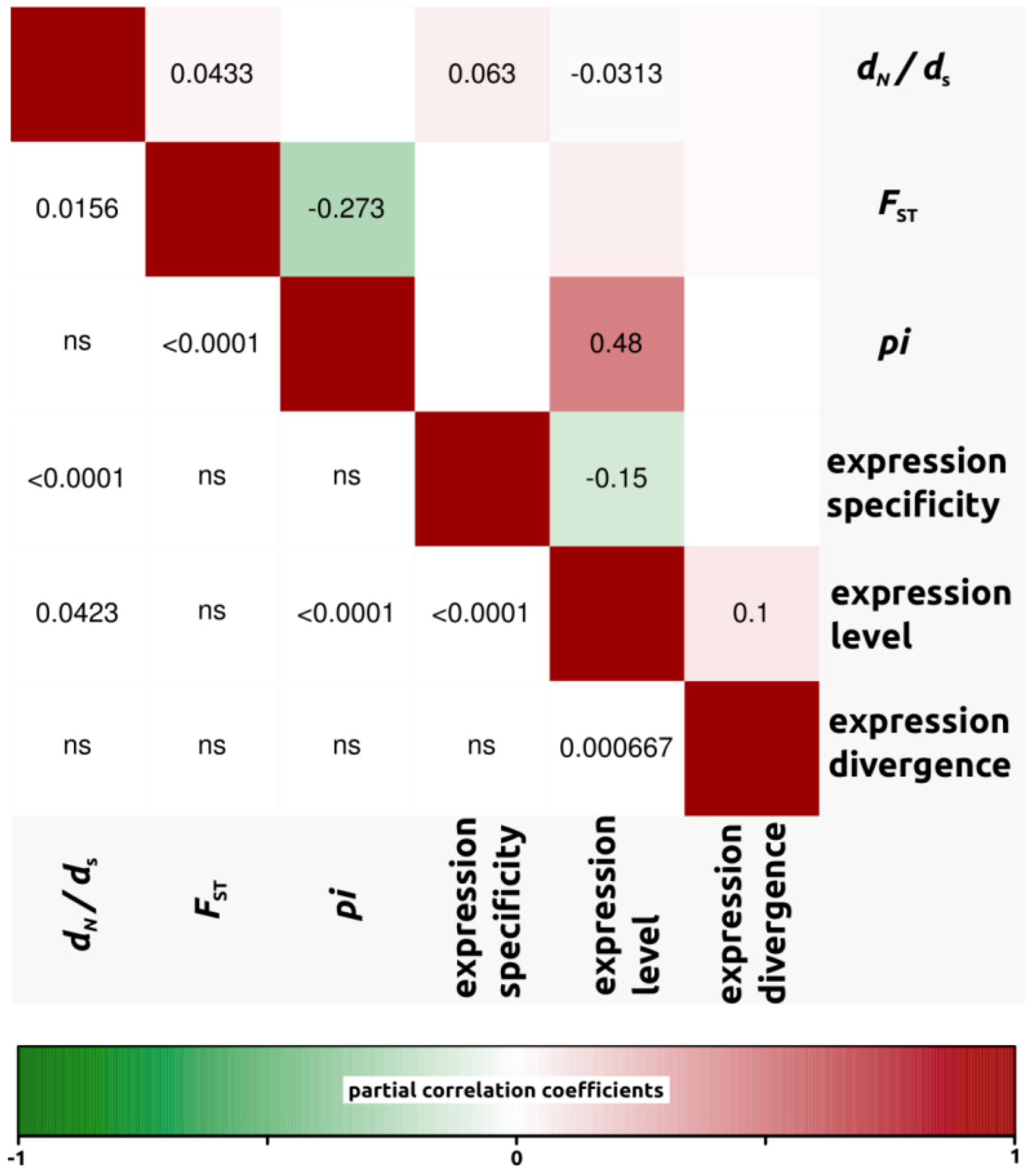{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total number of reference contigs = 16,312 | |||
| Total number of polymorphic contigs = 13,412 | |||
| Total number of polymorphic sites = 433,257 | |||
| Samples | H. annuus | H. petiolaris | H. debilis |
| Mean number of reads aligned per contig a | 382 | 798 | 894 |
| Median number of reads aligned per contig a | 109 | 155 | 198 |
| Mean depth per polymorphic site a | 75 | 102 | 107 |
| Median depth per polymorphic site a | 23 | 32 | 32 |
| Species | Sample name a | Raw (GB b) | Clean c (GB) | Aligned (GB) | % aligned/total | % aligned/clean | Median coverage per contig |
|---|---|---|---|---|---|---|---|
| H. annuus | 14B-14 | 3.58 | 1.38 | 0.72 | 20 | 52 | 100 |
| H. annuus | 14TB-2 | 3.38 | 1.24 | 0.53 | 16 | 43 | 70 |
| H. annuus | btm3-2 | 3.48 | 1.38 | 0.69 | 20 | 50 | 123 |
| H. annuus | btm6-1 | 3.88 | 1.52 | 0.8 | 21 | 53 | 127 |
| H. annuus | 2OTB-7 | 3.64 | 1.42 | 0.73 | 20 | 51 | 123 |
| H. annuus | btm11-1 | 2.46 | 1.0 | 0.54 | 22 | 54 | 88 |
| H. annuus | btm35-4 | 3.64 | 1.44 | 0.76 | 21 | 53 | 131 |
| H. debilis | Arg4B-14 | 5.54 | 3.16 | 1.69 | 31 | 53 | 204 |
| H. debilis | btm30-4 | 5.24 | 3.38 | 1.9 | 36 | 56 | 244 |
| H. debilis | btm33-4 | 3.64 | 1.84 | 0.97 | 27 | 53 | 113 |
| H. debilis (praecox) | btm13-6 | 4.86 | 3.12 | 1.62 | 33 | 52 | 225 |
| H. debilis (praecox) | btm14-4 | 3.94 | 2.46 | 1.35 | 34 | 55 | 152 |
| H. debilis (praecox) | btm15-2 | 6.74 | 3.44 | 1.75 | 26 | 51 | 281 |
| H. debilis (praecox) | btm16-2 | 4.78 | 2.7 | 1.44 | 30 | 53 | 171 |
| H. petiolaris | pet489 | 4.84 | 2.98 | 1.57 | 32 | 53 | 201 |
| H. petiolaris | pet2119 | 5.52 | 3.46 | 1.71 | 31 | 49 | 275 |
| H. petiolaris | PET2341 | 4.66 | 3.32 | 1.78 | 38 | 54 | 204 |
| H. petiolaris | PET2342 | 3.88 | 3.06 | 1.67 | 43 | 55 | 175 |
| H. petiolaris | PET2343 | 4.62 | 2.42 | 1.26 | 27 | 52 | 185 |
| H. petiolaris | PET2344 | 4.96 | 2.66 | 1.4 | 28 | 53 | 166 |
| H. petiolaris | PI468812 | 3.7 | 2.66 | 1.51 | 41 | 57 | 142 |
| H. petiolaris | PI503232 | 5.24 | 2.52 | 1.29 | 25 | 51 | 165 |
| H. petiolaris | PI531058 | 3.04 | 2.44 | 1.29 | 42 | 53 | 132 |
| H. petiolaris | PI547210 | 3.82 | 1.92 | 1.02 | 27 | 53 | 120 |
| H. petiolaris | PI586932 d | 1.38 | --- | --- | --- | --- | --- |
| H. petiolaris | PI613767 | 5.58 | 3.02 | 1.54 | 28 | 51 | 180 |
| H. petiolaris | PI649907 | 5.32 | 2.54 | 1.36 | 26 | 54 | 165 |
| H. petiolaris | PET-2 | 3.36 | 2.38 | 1.3 | 39 | 55 | 78 |
| H. petiolaris | PET-3 | 3.1 | 2.2 | 1.08 | 35 | 49 | 86 |
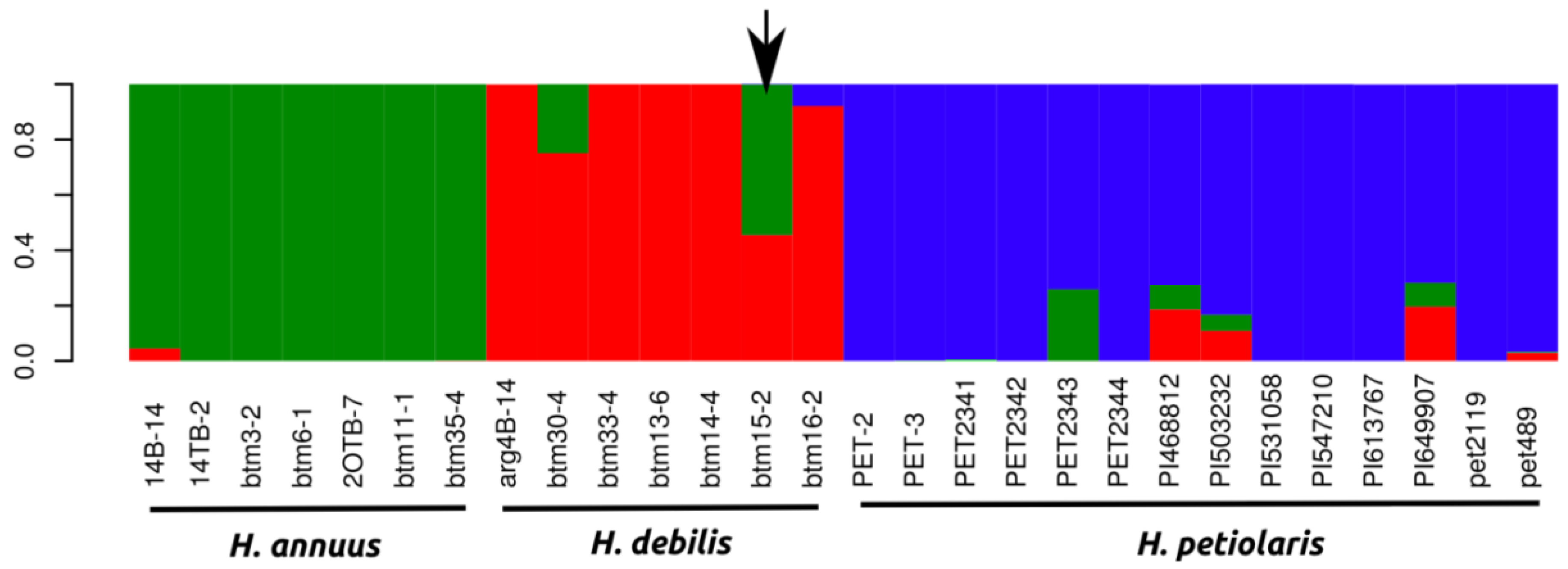
2.1.1. Population Genetics
2.1.2. Protein Coding Evolution and Effect of Selection
| Comparison | H. annuus-H. debilis | H. annuus-H. petiolaris | H. debilis-H. petiolaris |
|---|---|---|---|
| Mean FST per gene (13,412 genes) | 0.28 | 0.23 | 0.25 |
| Median FST per gene (13,412 genes) | 0.26 | 0.22 | 0.21 |
| Coding SNPs (12,310 Open Reading Frames) | |||
| Synonymous polymorphic SNPs (Ps) | 147,193 | 162,867 | 174,480 |
| Non-synonymous polymorphic SNPs (Pn) | 104,969 | 116,701 | 126,668 |
| Synonymous fixed (FST > 0.9) SNPs (Ds) | 3,339 | 2,612 | 865 |
| Non-synonymous fixed (FST > 0.9) SNPs (Dn) | 3,581 | 2,935 | 873 |
| alpha (proportion of base substitutions fixed by natural selection) and confidence intervals | 0.26 | 0.29 | 0.19 |
| [0.22–0.31] | [0.24–0.34] | [0.11–0.29] | |
| G-statistic (p-value) * | 239 | 231 | 23 |
| (2.2e–16) | (2.2e–16) | (1.4e–6) | |
| dN/dS | 0.22 | 0.24 | 0.22 |
| alpha | |||||
|---|---|---|---|---|---|
| Description | Number of genes | Gene ontology category (GO) | H. annuus/H. debilis | H. annuus/H. petiolaris | H. debilis/H. petiolaris |
| All annotated genes | 11,081 (total) | 0.26 ± 0.04 | 0.29 ± 0.05 | 0.19 ± 0.09 | |
| apoptosis | 84 | GO:0006915 | 0.28 ± 0.03 | 0.3 ± 0.04 | 0.79 ± 0.03 |
| defense response to fungus | 63 | GO:0050832 | 0.33 ± 0.05 | 0.56 ± 0.03 | 1 ± 0 |
| flower development | 94 | GO:0009908 | 0.54 ± 0.04 | 0.63 ± 0.03 | 0.4 ± 0.12 |
| glycolysis | 76 | GO:0006096 | 0.83 ± 0.02 | 0.71 ± 0.02 | 0.72 ± 0.04 |
| GTP catabolic process | 103 | GO:0006184 | 0.55 ± 0.02 | 0.29 ± 0.04 | −0.14 ± 0.08 |
| ion transmembrane transport | 85 | GO:0034220 | 0.45 ± 0.03 | 0.65 ± 0.03 | −0.41 ± 0.14 |
| lipid catabolic process | 130 | GO:0016042 | 0.45 ± 0.03 | 0.52 ± 0.03 | 0.76 ± 0.02 |
| lipid metabolic process | 243 | GO:0006629 | 0.45 ± 0.03 | 0.25 ± 0.04 | 0.88 ± 0.02 |
| lipid transport | 94 | GO:0006869 | 0.58 ± 0.02 | 0.48 ± 0.02 | −0.04 ± 0.23 |
| mitosis | 79 | GO:0007067 | 0.59 ± 0.02 | 0.59 ± 0.04 | 0.64 ± 0.04 |
| response to biotic stimulus | 60 | GO:0009607 | 0.63 ± 0.02 | 0.8 ± 0.01 | 1 ± 0 |
| translational initiation | 89 | GO:0006413 | 0.67 ± 0.03 | 0.03 ± 0.06 | −0.93 ± 0.14 |
| ubiquitin-dependent protein catabolic process | 105 | GO:0006511 | 0.43 ± 0.06 | 0.65 ± 0.03 | 0.14 ± 0.2 |
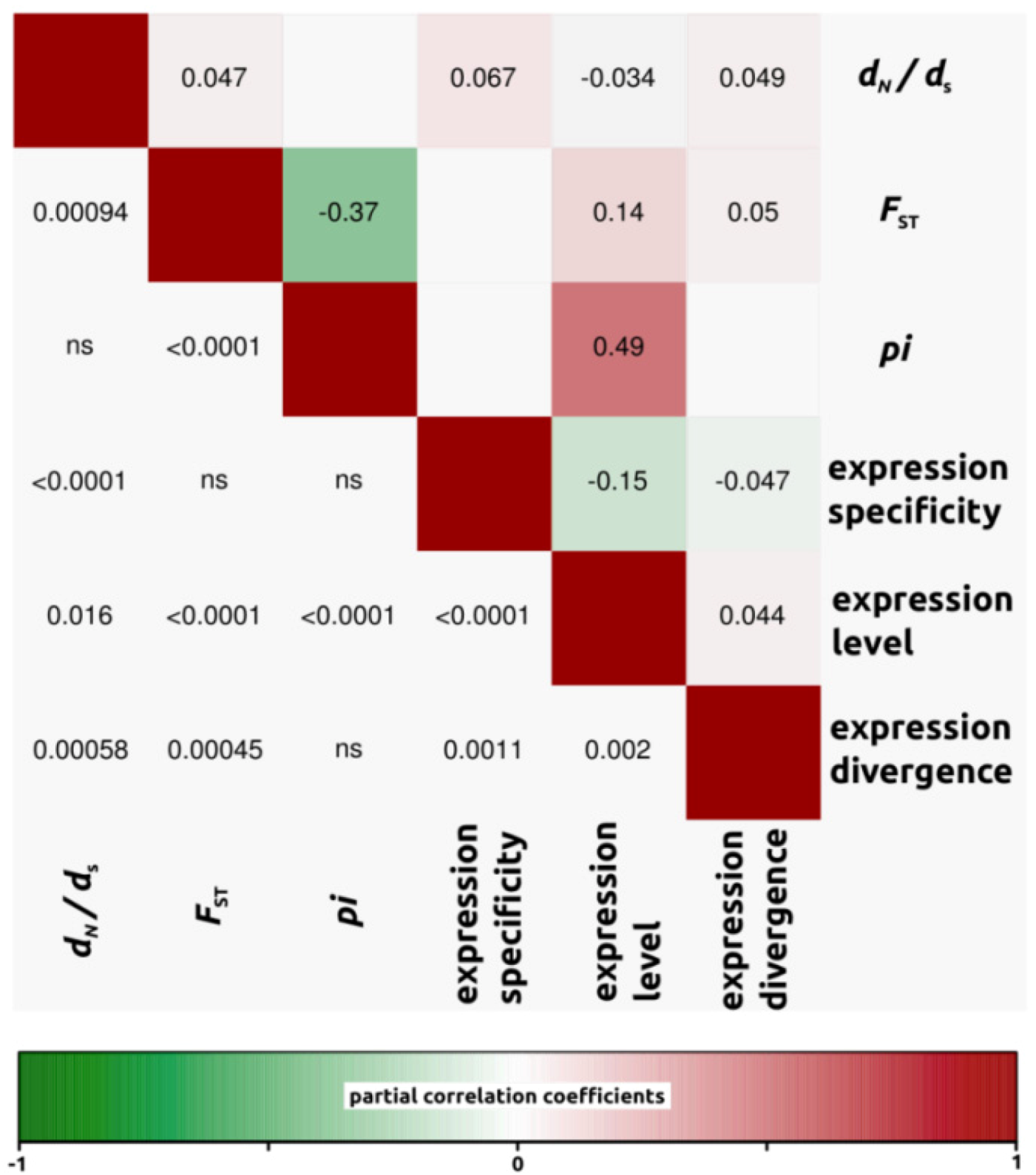
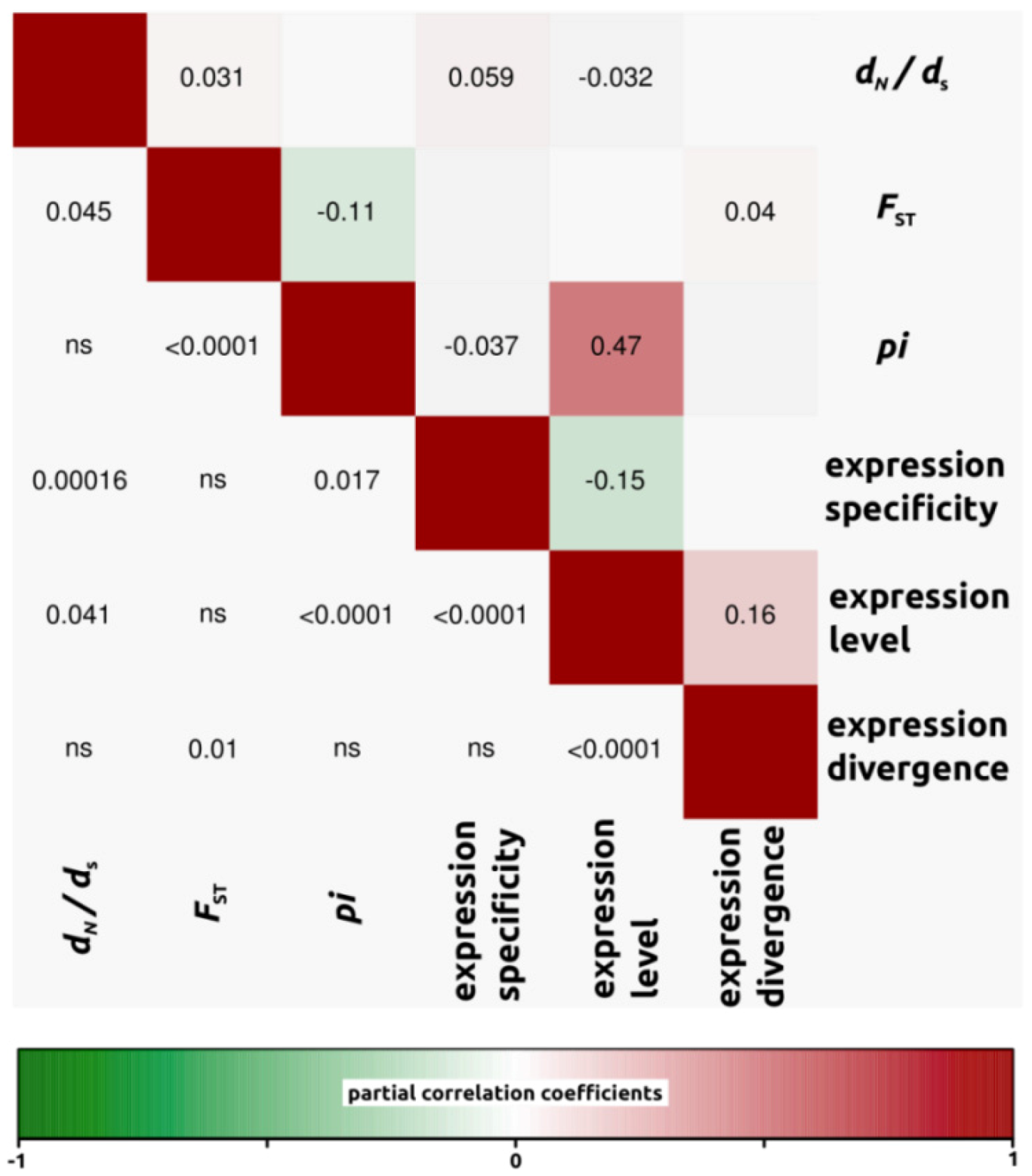
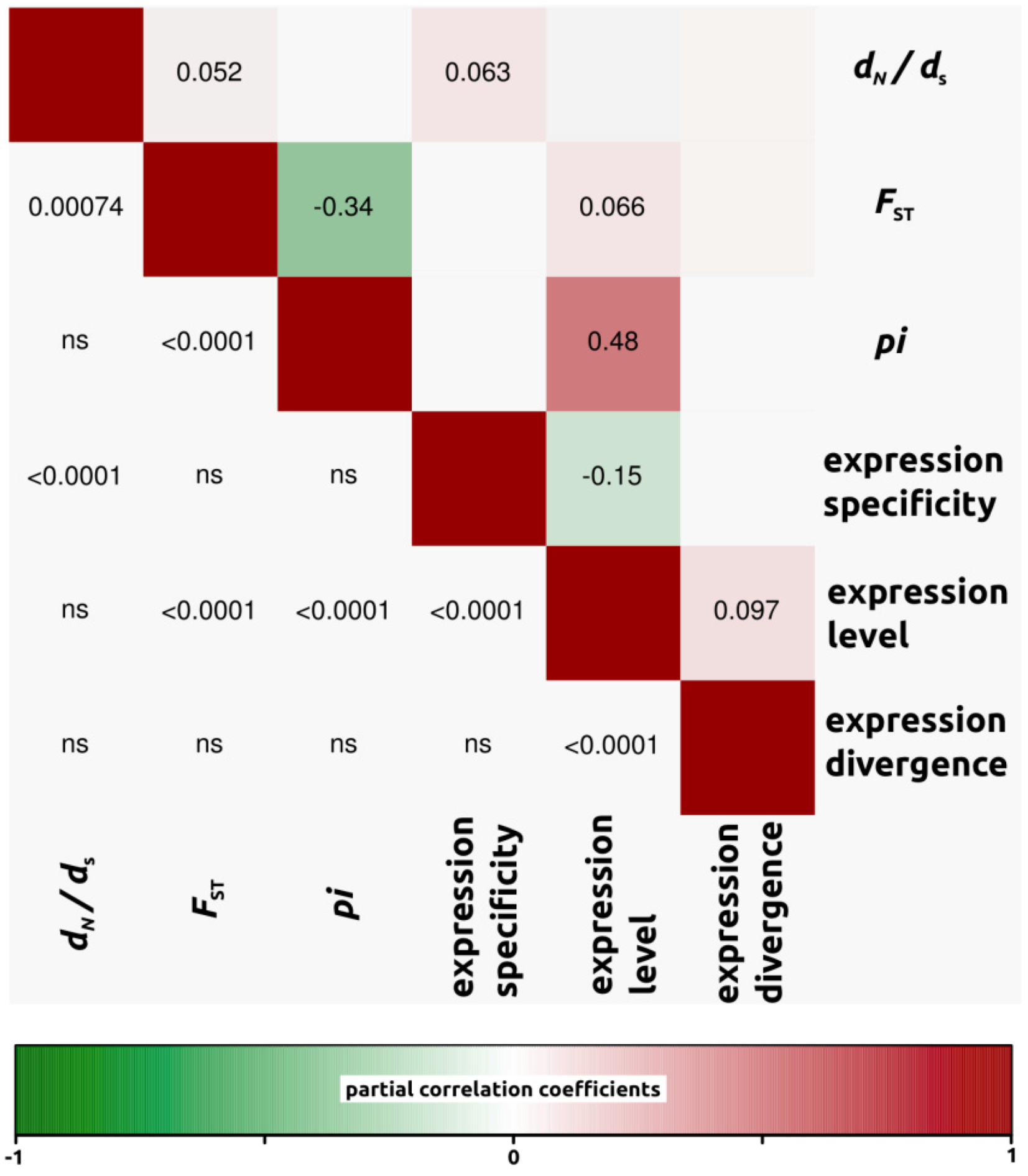
2.1.3. Genomic Determinants of Rates of Protein Evolution
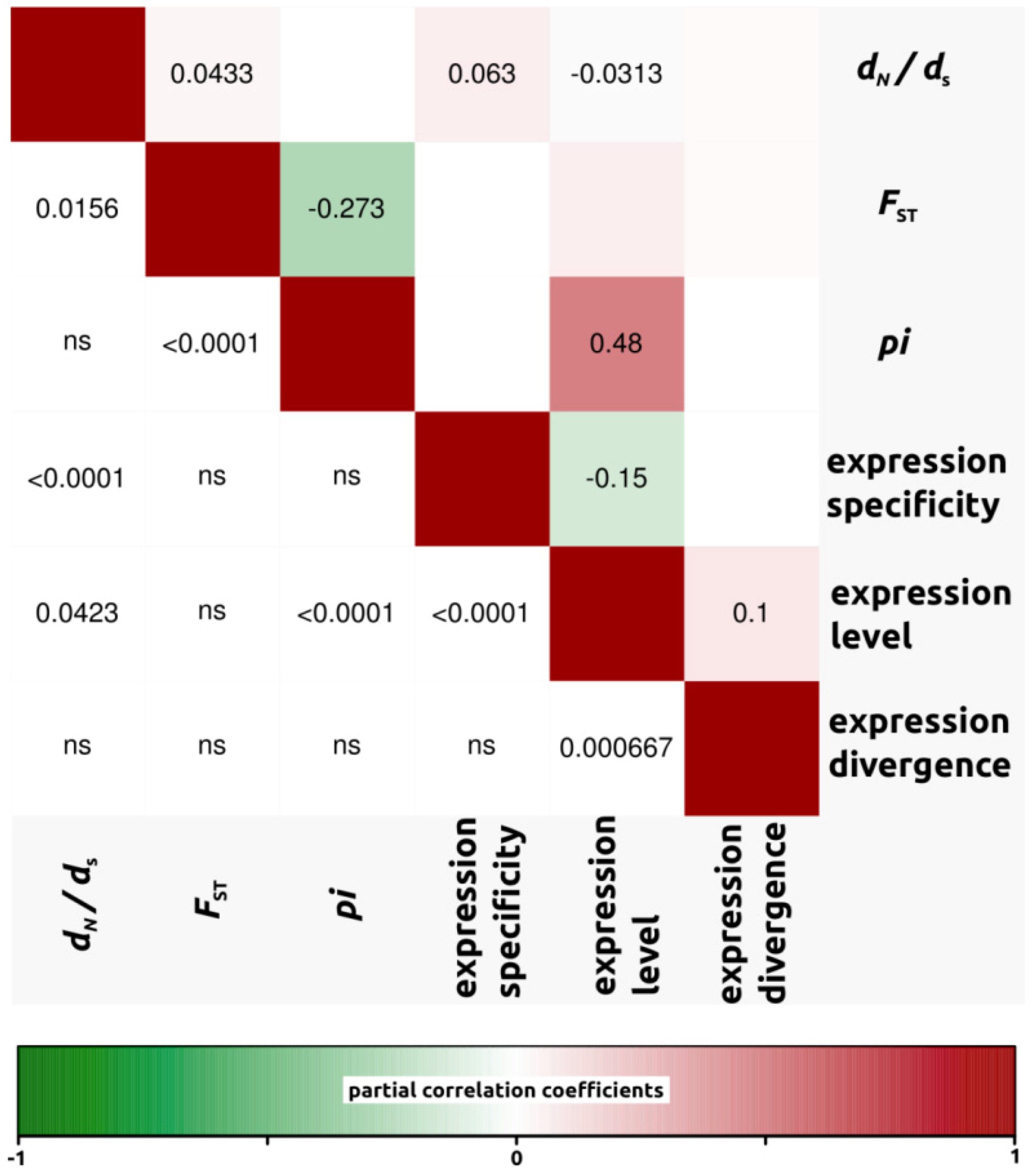
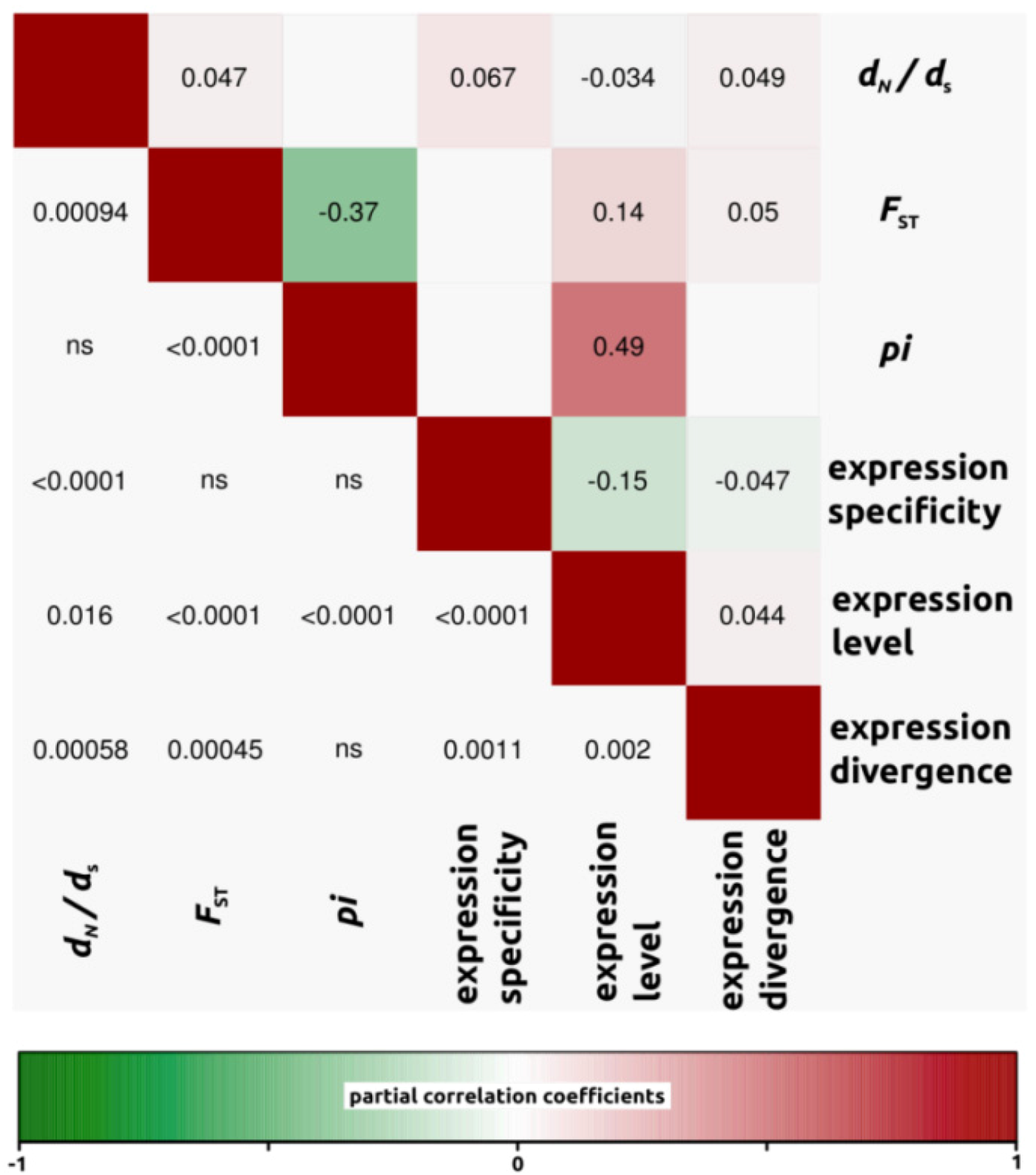
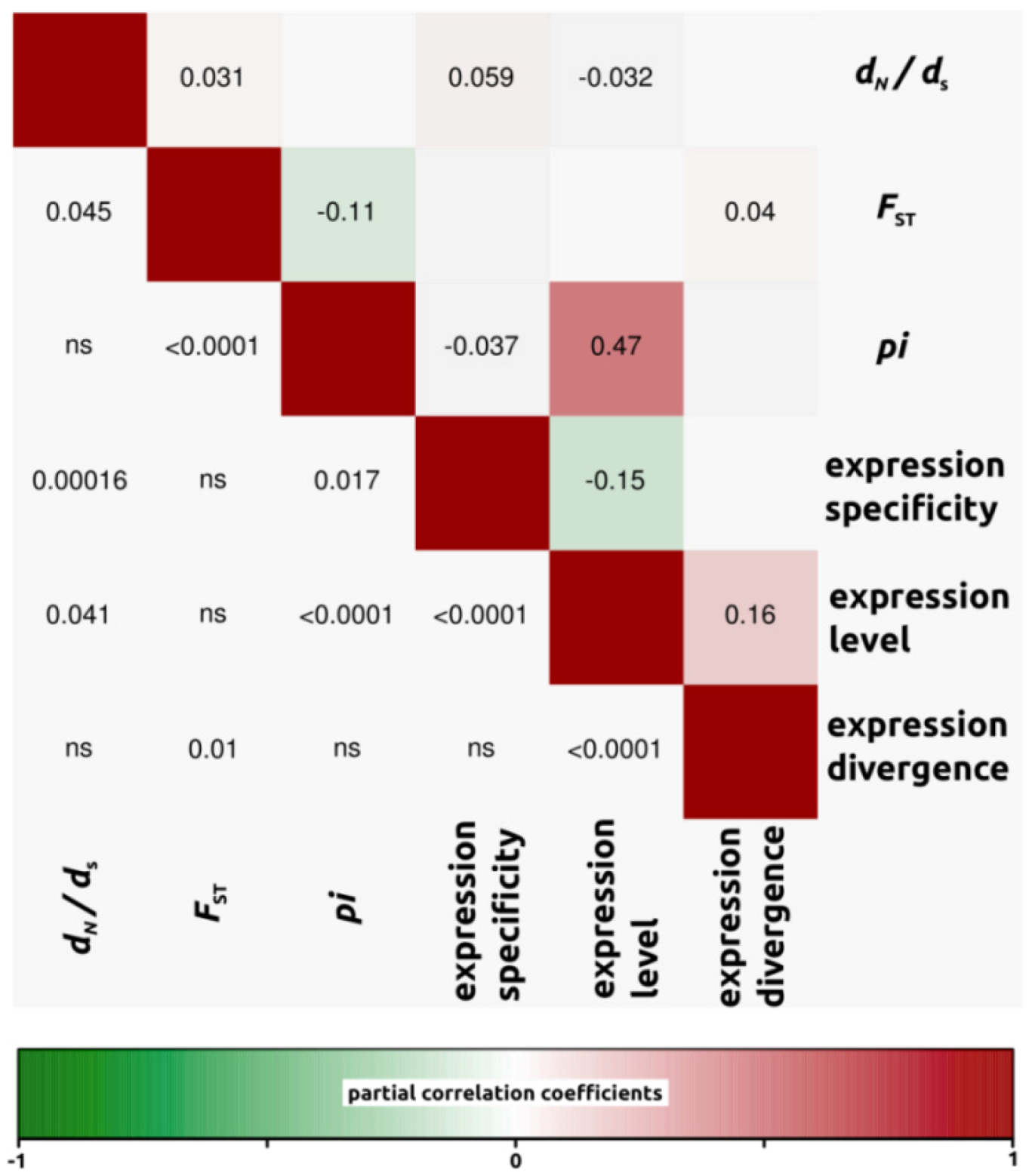
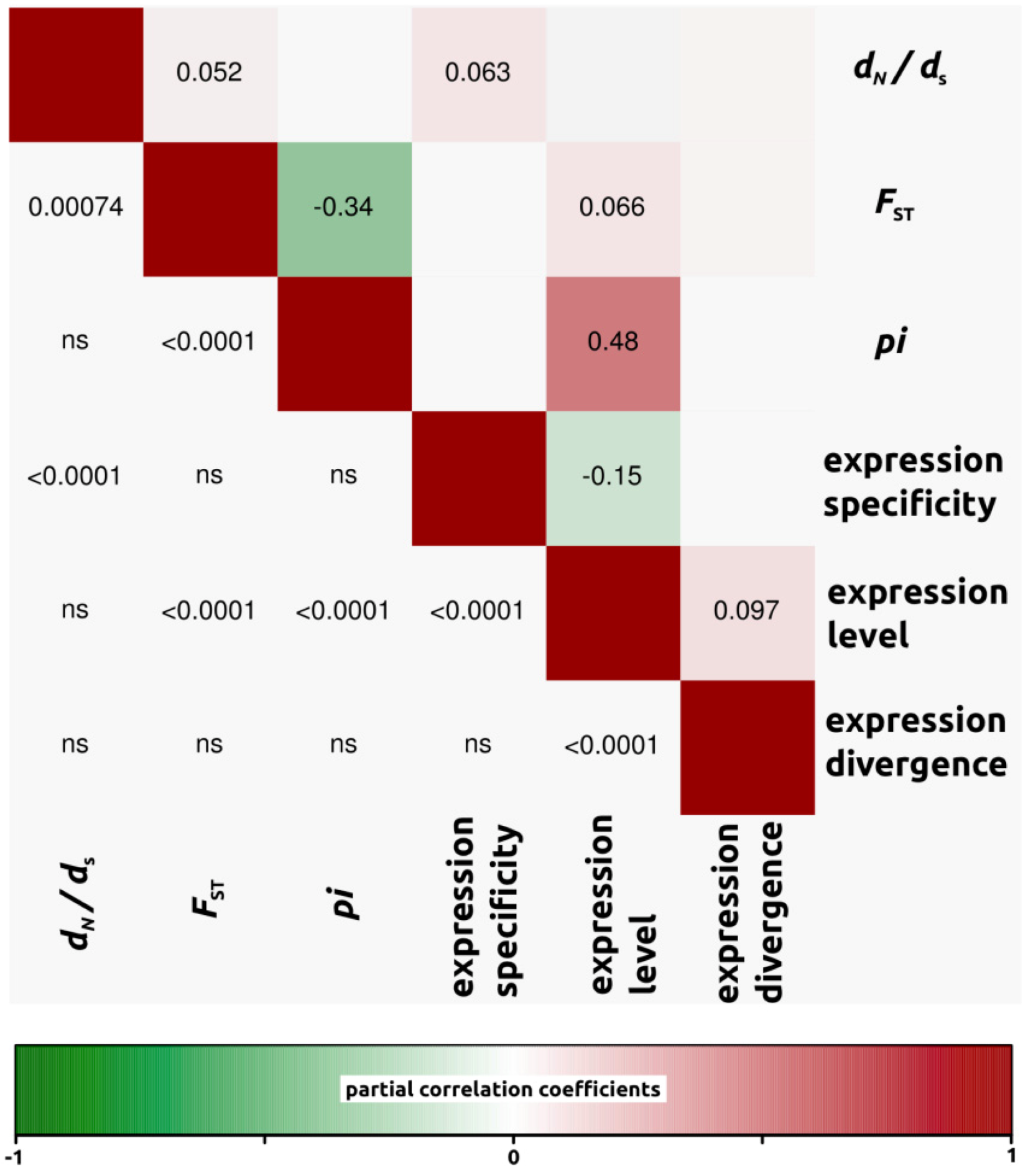
2.2. Discussion
2.2.1. Next-Generation Sequencing Era
2.2.2. Population Genetics
2.2.3. Protein Coding Evolution
2.2.4. Genomic Determinants of Rates of Protein Evolution
3. Experimental Section
3.1. Sample Preparation, Sequencing and Reference
3.2. Alignments
3.3. Population Genetics
3.4. Protein Coding Evolution and Effect of Selection
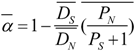
3.5. Genomic Determinants of Protein Evolution
4. Conclusions
Acknowledgments
References and Notes
- Larracuente, A.M.; Sackton, T.B.; Greenberg, A.J.; Wong, A.; Singh, N.D.; Sturgill, D.S.; Zhang, Y.; Oliver, B.; Clark, A.G. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008, 24, 114–123. [Google Scholar] [CrossRef]
- Gilad, Y.; Pritchard, J.; Thornton, K.; Thornton, K. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 2009, 25, 463–471. [Google Scholar] [CrossRef]
- Stapley, J.; Reger, J.; Feulner, P.G.D.; Smadja, C.; Galindo, J.; Ekblom, R.; Bennison, C.; Ball, A.D.; Beclerman, A.P.; Slate, J. Adaptation genomics: The next generation. Trends Ecol. Evol. 2010, 25, 705–712. [Google Scholar] [CrossRef]
- Kryazhimskiy, S.; Plotkin, J.B. The Population Genetics of dN/dS. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef]
- Duret, L.; Mouchiroud, D. Determinants of Substitution Rates in Mammalian Genes: Expression Pattern Affects Selection Intensity but Not Mutation Rate. Mol. Biol. Evol. 2000, 17, 68–74. [Google Scholar] [CrossRef]
- Pal, C.; Papp, B.; Lercher, M.J. An integrated view of protein evolution. Nat. Rev. Genet. 2006, 7, 337–348. [Google Scholar] [CrossRef]
- Ingvarsson, P.K. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol. Biol. Evol. 2007, 24, 836–844. [Google Scholar] [CrossRef]
- Slotte, T.; Bataillon, T.; Hansen, T.T.; Onge, K.S.; Wright, S.I.; Schierup, M.H. Genomic determinants of protein evolution and polymorphism in Arabidopsis. Genome Biol. Evol. 2011, 3, 1210–1219. [Google Scholar] [CrossRef]
- Nuzhdin, S.; Wayne, M.; Harmon, K.; McIntyre, L. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol. Biol. Evol. 2004, 21, 1308–1317. [Google Scholar] [CrossRef]
- Khaitovich, P. Parallel Patterns of Evolution in the Genomes and Transcriptomes of Humans and Chimpanzees. Science 2005, 309, 1850–1854. [Google Scholar] [CrossRef]
- Tirosh, I.; Barkai, N. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 2008, 24, 109–113. [Google Scholar] [CrossRef]
- Jeukens, J.; Renaut, S.; St-Cyr, J.; Nolte, A.W.; Bernatchez, L. The transcriptomics of sympatric dwarf and normal lake whitefish (Coregonus clupeaformis spp., Salmonidae) divergence as revealed by next-generation sequencing. Mol. Ecol. 2010, 19, 5389–5403. [Google Scholar] [CrossRef]
- Kane, N.C.; Barker, M.S.; Zhan, S.H.; Rieseberg, L.H. Molecular Evolution across the Asteraceae: Micro- and Macroevolutionary Processes. Mol. Biol. Evol. 2011, 28, 3225–3235. [Google Scholar] [CrossRef]
- Gaut, B.; Yang, L.; Takuno, S.; Eguiarte, L.E. The Patterns and Causes of Variation in Plant Nucleotide Substitution Rates. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 245–266. [Google Scholar] [CrossRef]
- Rieseberg, L.; Willis, J. Plant Speciation. Science 2007, 5840, 910–914. [Google Scholar] [CrossRef]
- Kane, N.C.; King, M.G.; Barker, M.S.; Raduski, A.; Karrenberg, S.; Yatabe, Y.; Knapp, S.; Rieseberg, L.H. Comparative genomic and population genetic analyses indicate highly porous genomes and high levels of gene flow between divergent Helianthus species. Evolution 2009, 63, 2061–2075. [Google Scholar] [CrossRef]
- Sambatti, J.; Strasburg, J.L.; Ortiz-Barrientos, D.; Baack, E.J.; Rieseberg, L.H. Reconciling extremely strong barriers with high levels of gene exchange in annual sunflowers. Evolution 2012, 66, 1459–1473. [Google Scholar] [CrossRef]
- Heiser, C.B. Hybridization in the Annual Sunflowers-Helianthus-annuus X H-argophyllus. Am. Nat. 1951, 85, 65–72. [Google Scholar]
- Strasburg, J.L.; Rieseberg, L.H. Molecular demographic history of the annual sunflowers Helianthus annuus and H. petiolaris-Large effective population sizes and rates of long-term gene flow. Evolution 2008, 62, 1936–1950. [Google Scholar] [CrossRef]
- Scascitelli, M.; Whitney, K.D.; Randell, R.A.; King, M.; Buerkle, C.A.; Rieseberg, L.H. Genome scan of hybridizing sunflowers from Texas (Helianthus annuus and H debilis) reveals asymmetric patterns of introgression and small islands of genomic differentiation. Mol. Ecol. 2010, 19, 521–541. [Google Scholar] [CrossRef]
- Heiser, C.B., Jr. Hybridization between the sunflower species Helianthus annuus and H. petiolaris. petiolaris. Evolution 1947, 1, 249–262. [Google Scholar]
- Heiser, C.B., Jr. Hybridization in the annual sunflowers: Helianthus annuus × H. debilis var. cucumerifolius. Evolution 1951, 5, 42–51. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Lai, Z.; Kane, N.C.; Kozik, A.; Hodgins, K.A.; Dlugosch, K.M.; Barker, M.S.; Matvienko, M.; Yu, Q.; Turner, K.G.; Pearl, S.A.; et al. Genomics of Compositae weeds: EST libraries, microarrays, and evidence of introgression. Am. J. Bot. 2012, 99, 209–218. [Google Scholar] [CrossRef]
- Dryad. Available online: http://dx.doi.org/10.5061/dryad.rs4k0/ (accessed on 8 October 2012).
- United States Department of Agriculture. Available online: http://www.arsgrin.gov/npgs/acc/acc_queries.html/ (accessed on 8 October 2012).
- McDonald, J.H.; Kreitman, M. Adaptive protein evolution at the adh locus in Drosophila. Nature 1991, 351, 652–654. [Google Scholar] [CrossRef]
- Yang, L.; Gaut, B.S. Factors that Contribute to Variation in Evolutionary Rate among Arabidopsis Genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar] [CrossRef]
- Ptitsyn, A. Stochastic Resonance Reveals “Pilot Light” Expression in Mammalian Genes. PLoS One 2008. [Google Scholar] [CrossRef]
- Kane, N.C.; Gill, N.; King, M.G.; Bowers, J.E.; Berges, H.; Gouzy, J.; Bachlava, E.; Langlade, N.B.; Lai, Z.; Stewar, M.; et al. Progress towards a reference genome for sunflower. Botany 2011, 89, 429–437. [Google Scholar] [CrossRef]
- Khalturin, K.; Hemmrich, G.; Fraune, S.; Augustin, R. More than just orphans: Are taxonomically-restricted genes important in evolution? Trends Genet. 2009, 25, 404–413. [Google Scholar] [CrossRef]
- Rieseberg, L.H. Homoploid reticulate evolution in Helianthus (Asteraceae): Evidence from ribosomal genes. Am. J. Bot. 1991, 78, 1218–1237. [Google Scholar] [CrossRef]
- Timme, R.E.; Simpson, B.B.; Linder, C.R. High-resolution phylogeny for Helianthus (Asteraceae) using the 18S-26S ribosomal DNA external transcribed spacer. Am. J. Bot. 2007, 94, 1837–1852. [Google Scholar] [CrossRef]
- Rieseberg, L.; Whitton, J.; Gardner, K. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 1999, 152, 713–727. [Google Scholar]
- Heiser, C.B. Variation and Subspeciation in the Common Sunflower, Helianthus Annuus. Am. Midl. Nat. 1954, 51, 287–305. [Google Scholar] [CrossRef]
- Linder, C.R. Adaptive Evolution of Seed Oils in Plants: Accounting for the Biogeographic Distribution of Saturated and Unsaturated Fatty Acids in Seed Oils. Am. Nat. 2000, 156, 442–458. [Google Scholar] [CrossRef]
- Whitney, K.D.; Randell, R.A.; Rieseberg, L.H. Adaptive Introgression of Herbivore Resistance Traits in the Weedy Sunflower Helianthus annuus. Am. Nat. 2006, 167, 794–807. [Google Scholar] [CrossRef]
- Chapman, M.A.; Leebens-Mack, J.H.; Burke, J.M. Positive Selection and expression divergence following gene duplication in the sunflower cycloidea gene family. Mol. Biol. Evol. 2008, 25, 1260–1273. [Google Scholar] [CrossRef]
- Blackman, B.K.; Rasmussen, D.A.; Strasburg, J.L.; Raduski, A.R.; Burke, J.M.; Knapp, S.J.; Michaels, S.D.; Rieseberg, L.H. Contributions of Flowering Time Genes to Sunflower Domestication and Improvement. Genetics 2011, 187, 271–287. [Google Scholar] [CrossRef]
- Chapman, M.A.; Tang, S.; Draeger, D.; Nambeesan, S.; Shaffer, H.; Barb, J.G.; Knapp, S.J.; Burke, J.M. Genetic Analysis of Floral Symmetry in Van Gogh’s Sunflowers Reveals Independent Recruitment of CYCLOIDEA Genes in the Asteraceae. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Schluter, D.; Nagel, L.M. Parallel Speciation by Natural Selection. Am. Nat. 1995, 146, 292–301. [Google Scholar]
- Pavlidis, P.; Jensen, J.D.; Stephan, W.; Stamatakis, A. A Critical Assessment of Storytelling: Gene Ontology Categories and the Importance of Validating Genomic Scans. Mol. Biol. Evol. 2012, 29, 3237–3248. [Google Scholar]
- Slotte, T.; Foxe, J.P.; Hazzouri, K.M.; Wright, S.I. Genome-Wide Evidence for Efficient Positive and Purifying Selection in Capsella grandiflora, a Plant Species with a Large Effective Population Size. Mol. Biol. Evol. 2010, 27, 1813–1821. [Google Scholar] [CrossRef]
- Russo, C.A.; Takezaki, N.; Nei, M. Molecular phylogeny and divergence times of drosophilid species. Mol. Biol. Evol. 1995, 12, 391–404. [Google Scholar]
- Erwin, D.H. Macroevolution is more than repeated rounds of microevolution. Evol. Dev. 2000, 2, 78–84. [Google Scholar] [CrossRef]
- Gilad, Y.; Rifkin, S.; Pritchard, J. Revealing the architecture of gene regulation: The promise of eQTL studies. Trends Genet. 2008, 24, 408–415. [Google Scholar] [CrossRef]
- Heiser, C.B. Biosystematics of Helianthus debilis. Madrono 1956, 37, 145–176. [Google Scholar]
- Heesacker, A.; Kishore, V.K.; Gao, W.; Tang, S.; Kolkman, J.M.; Gingle, A.; Matvienko, M.; Kozik, A.; Michelmore, R.M.; LaI, Z.; et al. SSRs and INDELs mined from the sunflower EST database: Abundance, polymorphisms, and cross-taxa utility. Theor. Appl. Genet. 2008, 117, 1021–1029. [Google Scholar] [CrossRef]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.; Müller, W.E.G.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Bowers, J.E.; Bachlava, E.; Brunick, R.L.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Development of a 10,000 Locus Genetic Map of the Sunflower Genome Based on Multiple Crosses. G3 Genes Genomes Genet. 2012, 2, 721–729. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hannal, M.; Banksl, E.; Sivchenko, A.; Cibulskisl, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriell, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, N.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup7. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Pritchard, J.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 20 0724, 1586–1591. [Google Scholar]
- Smith, N.G.C.; Eyre-Walker, A. Adaptive protein evolution in Drosophila. Nature 2002, 415, 1022–1024. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstei, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- UniProt. Available online: http://www.ebi.ac.uk/uniprot/ (accessed on 08 October 2012).
- R Development Core Team, R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012.
- Kim, S.H.; Yi, S.V. Understanding relationship between sequence and functional evolution in yeast proteins. Genetica 2007, 131, 151–156. [Google Scholar] [CrossRef]
- SITES (Hey Lab Distributed Software). Available online: http://genfaculty.rutgers.edu/hey/%E2%80%A8software#SITES/ (accessed on 08 October 2012).
- Watterson, G.A. Theoretical Population Biology-On the number of segregating sites in genetical models without recombination. Theoret. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Weir, B. Genetic Data Analysis II; Sinauer Associates, Inc.: Sunderland, MA, USA, 1996. [Google Scholar]
- Goudet, J. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 2010, 11, 94–94. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Renaut, S.; Grassa, C.J.; Moyers, B.T.; Kane, N.C.; Rieseberg, L.H. The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq. Biology 2012, 1, 575-596. https://doi.org/10.3390/biology1030575
Renaut S, Grassa CJ, Moyers BT, Kane NC, Rieseberg LH. The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq. Biology. 2012; 1(3):575-596. https://doi.org/10.3390/biology1030575
Chicago/Turabian StyleRenaut, Sébastien, Christopher J. Grassa, Brook T. Moyers, Nolan C. Kane, and Loren H. Rieseberg. 2012. "The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq" Biology 1, no. 3: 575-596. https://doi.org/10.3390/biology1030575
APA StyleRenaut, S., Grassa, C. J., Moyers, B. T., Kane, N. C., & Rieseberg, L. H. (2012). The Population Genomics of Sunflowers and Genomic Determinants of Protein Evolution Revealed by RNAseq. Biology, 1(3), 575-596. https://doi.org/10.3390/biology1030575