Genotyping-by-Sequencing in Plants

Abstract
:1. Introduction
2. Genotyping Applications
3. Ultra-high Throughput Genotyping Applications
4. Ultra High Throughput DNA Sequencing

{kind=link}
| Sequencing Platform | Sequencing Chemistry | Detection Chemistry | Run Timea | Read Length (bp) | Reads per Run (million) | Throughput per Run (Gbp) |
|---|---|---|---|---|---|---|
| Roche 454 FLX Titanium | Sequencing by Synthesis | Light | 23 hours | ~800 | ~1 | ~0.7 |
| Illumina MiSeq | Sequencing by Synthesis | Fluorescence | 39 hours | 2 × 250 b | ~1 | ~8 |
| Illumina HiSeq2500 | Sequencing by Synthesis | Fluorescence | 11 days (high output)/27 hours (rapid run) | 2 × 100 b | ~3,000 | ~600 (high output)/~120 (rapid run) |
| Life Technologies 5500xl | Sequencing byLigation | Fluorescence | 8 days | 75 + 35 b | ~5,000 | ~310 |
| Ion Torrent PGM | Sequencing bySynthesis | pH | 4 hours | 100 | 1 | ~0.1 |
5. Applications of NGS Technologies to Genotyping-by-Sequencing
6. Polymorphism Detection from NGS Data
| Software | Link | Input Format | Primary Function |
|---|---|---|---|
| MAQ | http://maq.sourceforge.net/ | FASTA, FASTQ | Mapping and Assembly |
| SAMtools | http://samtools.sourceforge.net/ | SAM, BAM | Alignments |
| GATK | http://www.broadinstitute.org/gatk/ | SAM | Alignments |
| SOAPsnp | http://soap.genomics.org.cn/soapsnp.html | SOAP | Mapping and Assembly |
| SNIP-Seq | http://polymorphism.scripps.edu/~vbansal/software/SNIP-Seq/ | Pileup | Alignments |
| MapNext | http://evolution.sysu.edu.cn/english/software/mapnext.htm | FASTA, FASTQ | Alignments |
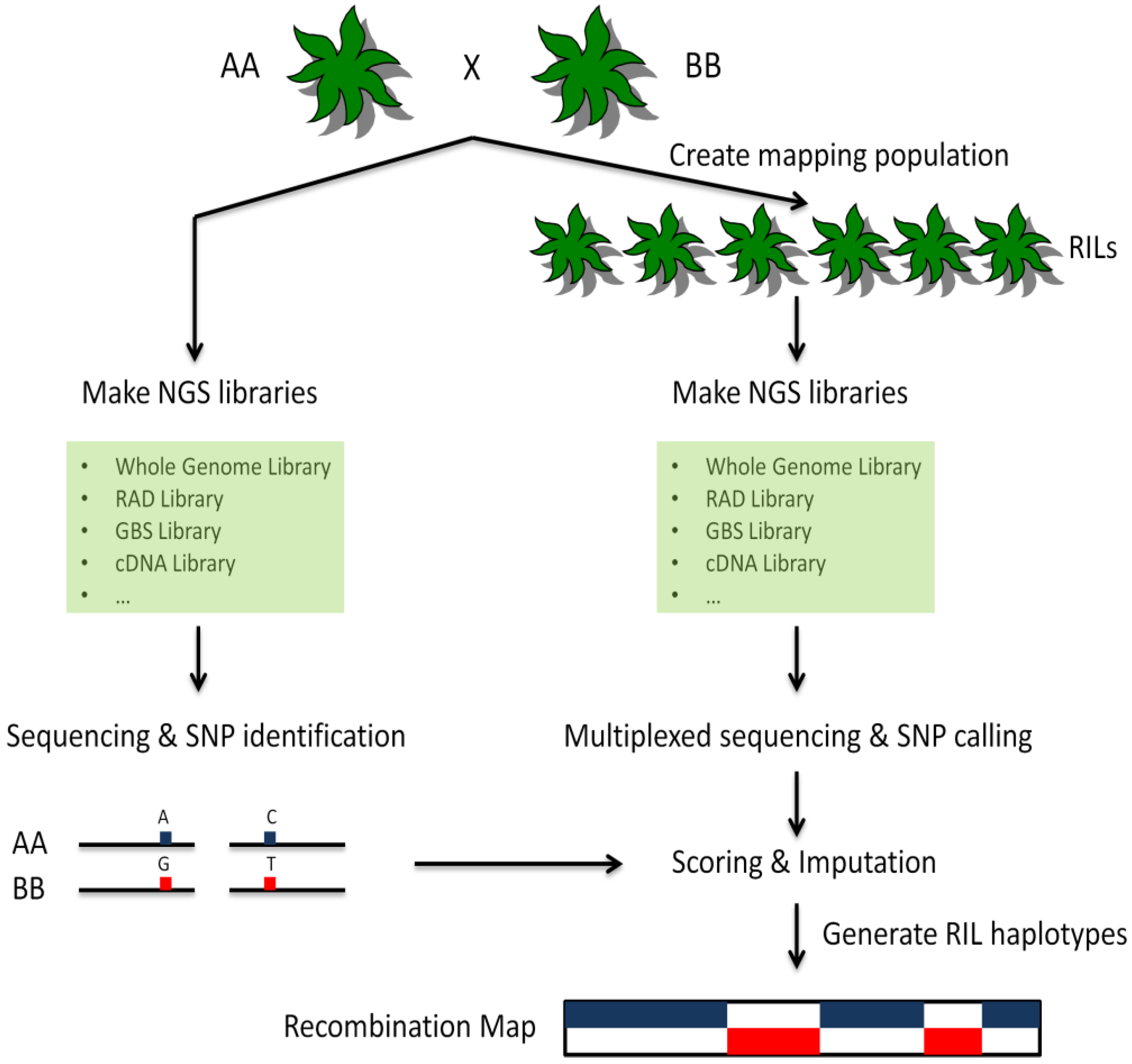
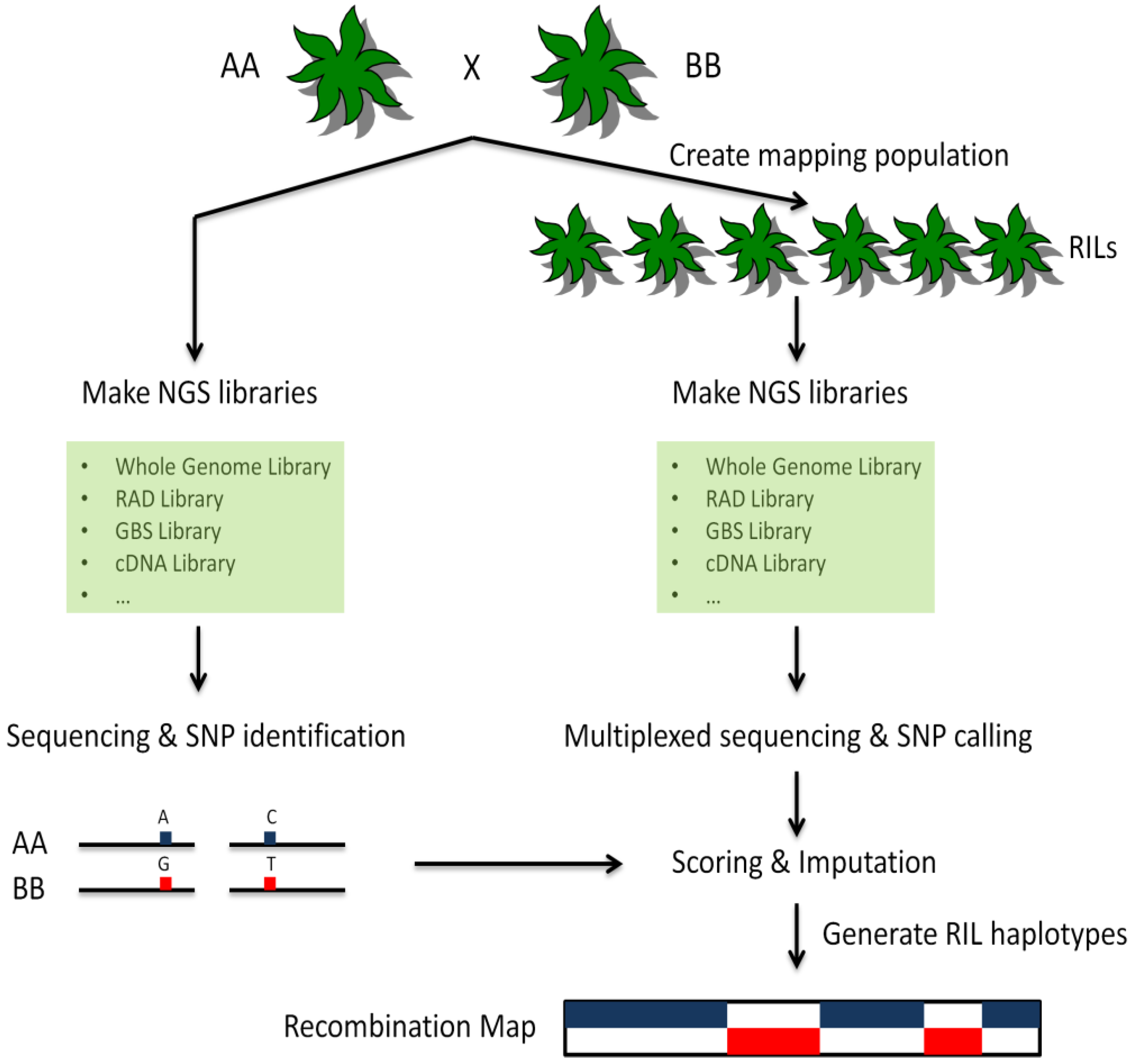
7. Genotyping-by-Sequencing in Plants
8. Conclusions
References and Notes
- Rafalski, A. Applications of single nucleotide polymorphisms in crop genetics. Curr. Opin. Plant Biol. 2002, 5, 94–100. [Google Scholar] [CrossRef]
- Bernatsky, R.; Tanksley, S. Toward a saturated linkage map in tomato based on isozymes and random cDNA sequences. Genetics 1986, 112, 887–898. [Google Scholar]
- Litt, M.; Luty, J.A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am. J. Hum. Genet. 1986, 44, 397–401. [Google Scholar]
- Williams, J.G.; Kubelik, A.R.; Livak, K.J.; Rafalski, J.A.; Tingey, S.V. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990, 18, 6531–6535. [Google Scholar]
- Paran, I.; Michelmore, R.W. Development of reliable PCR-based markers linked to downy mildew resistance genes in lettuce. Theor. Appl. Genet. 1993, 85, 985–993. [Google Scholar]
- Konieczny, A.; Ausubel, F.M. A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 1993, 4, 403–410. [Google Scholar]
- Salimath, S.S.; De Oliveira, A.C.; Bennetzen, J.; Godwin, I.D. Assessment of genomic origin and genetic diversity in the genus Eleusine with DNA markers. Genome 1995, 38, 757–763. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Desmarais, E.; Lanneluc, I.; Lagnel, J. Direct amplification of length polymorphisms (DALP), or how to get and characterize new genetic markers in many species. Nucleic Acids Res. 1998, 26, 1458–1465. [Google Scholar] [CrossRef]
- Wang, D.G.; Fan, J-B.; Siao, C.J.; Berno, A.; Young, P.; Sapolsky, R.; Ghandour, G.; Perkins, N.; Winchester, E.; Spencer, J. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998, 280, 1077–1082. [Google Scholar] [CrossRef]
- Täpp, I.; Malmberg, L.; Rennel, E.; Wik, M.; Syvanen, A.C. Homogeneous scoring of single-nucleotide polymorphisms: comparison of the 5'-nuclease TaqMan assay and molecular beacon probes. Biotechniques 2000, 28, 732–738. [Google Scholar]
- Prince, J.A.; Feuk, L.; Howell, W.M.; Jobs, M.; Emahazion, T.; Blennow, K.; Brookes, A.J. Robust and accurate single nucleotide polymorphism genotyping by dynamic allele-specific hybridization (DASH): design criteria and assay validation. Genome Res. 2001, 11, 152–162. [Google Scholar] [CrossRef]
- Storm, N.; Darnhofer-Patel, B.; van den Boom, D.; Rodi, C.P. MALDI-TOF mass spectrometry-based SNP genotyping. Methods Mol. Biol. 2003, 212, 241–262. [Google Scholar]
- Livak, K.J. SNP genotyping by the 50-nuclease reaction. Methods Mol. Biol. 2003, 212, 129–147. [Google Scholar]
- Olivier, M. The Invader assay for SNP genotyping. Mutat. Res. 2005, 573, 103–110. [Google Scholar] [CrossRef]
- Ragoussis, J. Genotyping technologies for all. Drug Discov. Today Technol. 2006, 3, 115–122. [Google Scholar] [CrossRef]
- Procunier, J.D.; Prashar, S.; Chen, G.; Wolfe, D.; Fox, S.; Ali, M.L.; Gray, M.; Zhou, Y.; Shillinglaw, M.; Roeven, R.; Ron DePauw, R. Rapid ID technology (RIDT) in plants: High-speed DNA fingerprinting in grain seeds for the identification, segregation, purity, and traceability of varieties using lab automation robotics. J. Lab. Automat. 2009, 14, 221–231. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S.; Mir, R.R. Array-based high-throughput DNA markers for crop improvement. Heredity 2008, 101, 5–18. [Google Scholar] [CrossRef]
- Nybon, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Arif, I.A.; Bakir, M.A.; Khan, H.A.; Al Farhan, A.H.; Al Homaidan, A.A.; Bahkali, A.H.; Al Sadoon, M.; Shobrak, M. A brief review of molecular techniques to assess plant diversity. Int. J. Mol. Sci. 2010, 11, 2079–2096. [Google Scholar] [CrossRef]
- Zheng, P.; Allen, W.B.; Roesler, K.; Williams, M.E.; Zhang, S.; Li, J.; Glassman, K.; Ranch, J.; Nubel, D.; Solawetz, W.; Bhattramakki, D.; et al. A phenylalanine in DGAT is a key determinant of oil content and composition in maize. Nat. Genet. 2008, 40, 367–372. [Google Scholar]
- Ruta, N.; Liedgens, M.; Fracheboud, Y.; Stamp, P.; Hund, A. QTLs for the elongation of axile and lateral roots of maize in response to low water potential. Theor. Appl. Genet. 2010, 120, 621–631. [Google Scholar] [CrossRef]
- Yano, M.; Harushima, Y.; Nagamura, Y.; Kurata, N.; Minobe, Y.; Sasaki, T. Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor. Appl. Genet. 1997, 95, 1025–1032. [Google Scholar] [CrossRef]
- El-Din El-Assal, S.; Alonso-Blanco, C.; Peeters, A.J.; Raz, V.; Koornneef, M. The cloning of a flowering time QTL reveals a novel allele of CRY2. Nat. Genet. 2001, 29, 435–440. [Google Scholar]
- Liu, J.; van Eck, J.; Cong, B.; Tanksley, S.D. A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc. Natl. Acad. Sci. USA 2002, 99, 13302–13306. [Google Scholar]
- Salvi, S.; Tuberosa, R. To clone or not to clone plant QTLs: Present and future challenges. Trends Plant Sci. 2005, 10, 297–304. [Google Scholar] [CrossRef]
- Frary, A.; Nesbitt, T.C.; Grandillo, S.; Knaap, E.; Cong, B.; Liu, J.; Meller, J.; Elber, R.; Alpert, K.B.; Tanksley, S.D. fw2.2: A quantitative trait locus key to the evolution of tomato fruit size. Science 2000, 289, 85–88. [Google Scholar]
- Rafalski, A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Schneeberger, K.; Weigel, D. Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 2011, 16, 282–288. [Google Scholar] [CrossRef]
- Risch, N.; Merikangas, K. The future of genetic studies of complex human diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotech. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Beló, A.; Zheng, P.; Luck, S.; Shen, B.; Meyer, D.J.; Li, B.; Tingey, S.; Rafalski, A. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol. Genet. Genomics 2008, 279, 1–10. [Google Scholar] [CrossRef]
- Nordborg, M.; Weigel, D. Next-generation genetics in plants. Nature 2008, 456, 720–723. [Google Scholar]
- Thornsberry, J.M.; Goodman, M.M.; Doebley, J.; Kresovich, S.; Nielsen, D.; Buckler, E.S. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 2001, 28, 286–289. [Google Scholar]
- Hansen, M.; Kraft, T.; Ganestam, S.; Säll, T.; Nilsson, N.O. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet. Res. 2001, 77, 61–66. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Buckler, E.S. Structure of linkage disequilibrium in plants. Ann. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thuillet, A.C.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high-resolution platform for quantitative tgrait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef]
- Sharbel, T.F.; Haubold, B.; Mitchell-Olds, T. Genetic isolation by distance in Arabidopsis thaliana: biogeography and postglacial colonization of Europe. Mol. Ecol. 2000, 9, 2109–2118. [Google Scholar] [CrossRef]
- Zhu, C.; Gore, M.; Buckler, E.S.; Yu, J. Status and prospects of association mapping in plants. Plant Gen. 2008, 1, 5–20. [Google Scholar] [CrossRef]
- Damerval, C.; Maurice, A.; Josse, J.M.; de Vienne, D. Quantitative trait loci underlying gene product variation: A novel perspective for analyzing genome expression. Genetics 1994, 137, 289–301. [Google Scholar]
- Holloway, B.; Li, B. Expression QTLs: Applications for crop improvement. Mol. Breeding 2010, 26, 381–391. [Google Scholar] [CrossRef]
- Holloway, B.; Luck, S.; Beatty, M.; Rafalski, J.A.; Li, B. Genome-wide expression quantitative trait loci (eQTL) analysis in maize. BMC Genomics 2011, 12, 1–14. [Google Scholar]
- West, M.A.; Kim, K.; Kliebenstein, D.J.; van Leeuwen, H.; Michelmore, R.W.; Doerge, R.W.; St Clair, D.A. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 2007, 175, 1441–1450. [Google Scholar]
- Swanson-Wagner, R.A.; DeCook, R.; Jia, Y.; Bancroft, T.; Ji, T.; Zhao, X.; Nettleton, D.; Schnable, P.S. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 2009, 326, 1118–1120. [Google Scholar]
- Becker, A.; Chao, D.Y.; Zhang, X.; Salt, D.E.; Baxter, I. Bulk Segregant Analysis Using Single Nucleotide Polymorphism Microarrays. PLoS One 2011, 6, e15993. [Google Scholar]
- Wolyn, D.J.; Borevitz, J.O.; Loudet, O.; Schwartz, C.; Maloof, J.; Ecker, J.R.; Berry, C.C.; Chory, J. Light-response quantitative trait loci identified with composite interval and eXtreme array mapping in Arabidopsis thaliana. Genetics 2004, 167, 907–917. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Morgante, M.; de Paoli, E.; Radovic, S. Transposable elements and the plant pangenomes. Curr. Opin. Plant Biol. 2007, 10, 149–155. [Google Scholar] [CrossRef]
- Llaca, V.; Campbell, M.; Deschamps, S. Genome diversity in maize. J. Botany 2011, 104172, 1–10. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Hunkapiller, T.; Kaiser, R.J.; Koop, B.F.; Hood, L. Large-scale and automated DNA sequence determination. Science 1991, 254, 59–67. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2011, 409, 860–921. [Google Scholar]
- Goff, S.A.; Ricke, D.; Lan, T.H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar]
- Rostoks, N.; Mudie, S.; Cardle, L.; Russell, J.; Ramsay, L.; Booth, A.; Svensson, J.T.; Wanamaker, S.I.; Walia, H.; Rodriguez, E.M.; et al. Genome-wide SNP discovery and linkage analysis in barley based on genes responsive to abiotic stress. Mol. Genet. Genomics 2005, 274, 515–527. [Google Scholar] [CrossRef]
- Choi, I.Y.; Hyten, D.L.; Matukumalli, L.K.; Song, Q.; Chaky, J.M.; Quigley, C.V.; Chase, K.; Lark, K.G.; Reiter, R.S.; Yoon, M.S.; et al. A soybean transcript map: Gene distribution, haplotype and single nucleotide polymorphism analysis. Genetics 2007, 176, 685–696. [Google Scholar] [CrossRef]
- Luckey, J.A.; Drossman, H.; Kostichka, A.J.; Mead, D.A.; D’Cunha, J.; Norris, T.B.; Smith, L.M. High speed DNA sequencing by capillary electrophoresis. Nucleic Acids Res. 1990, 18, 4417–4421. [Google Scholar]
- Swerdlow, H.; Gesteland, R. Capillary gel electrophoresis for rapid, high resolution DNA sequencing. Nucleic Acids Res. 1990, 18, 1415–1419. [Google Scholar] [CrossRef]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar]
- Prober, J.M.; Trainor, G.L.; Dam, R.J.; Hobbs, F.W.; Robertson, C.W.; Zagursky, R.J.; Cocuzza, A.J.; Jensen, M.A.; Baumeister, K. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 1987, 238, 336–341. [Google Scholar]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Niedringhaus, T.P.; Milanova, D.; Kerby, M.B.; Snyder, M.P.; Barron, A.E. Landscape of next-generation sequencing technologies. Anal. Chem. 2011, 83, 4327–4341. [Google Scholar]
- Metzker, M.L. Sequencing technologies – the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar]
- Ronaghi, M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001, 11, 3–11. [Google Scholar] [CrossRef]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genomics 2012, 11, 3–11. [Google Scholar]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar]
- Fedurco, M.; Romieu, A.; Williams, S.; Lawrence, I.; Turcatti, G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucleic Acids Res. 2006, 34, e22. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar]
- Velasco, R.; Zharkikh, A.; Troggio, M.; Cartwright, D.A.; Cestaro, A.; Pruss, D.; Pindo, M.; Fitzgerald, L.M.; Vezzulli, S.; Reid, J.; Malacarne, G.; et al. A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS One 2007, 2, e1326. [Google Scholar]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J., Jr.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 2008, 9, 312. [Google Scholar] [CrossRef]
- Van Tassell, C.P.; Smith, T.P.; Matukumalli, L.K.; Taylor, J.F.; Schnabel, R.D.; Lawley, C.T.; Haudenschild, C.D.; Moore, S.S.; Warren, W.C.; Sonstegard, T.S. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods 2008, 5, 247–252. [Google Scholar]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef]
- Deschamps, S.; la Rota, M.; Ratashak, J.P.; Biddle, P.; Thureen, D.; Farmer, A.; Luck, S.; Beatty, M.; Nagasawa, N.; Michael, L.; et al. Rapid genome-wide single nucleotide polymorphism discovery in soybean and rice via deep resequencing of reduced representation libraries with the Illumina Genome Analyzer. Plant Genome 2010, 3, 53–68. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar]
- Wilhelm, B.; Marguerat, S.; Watt, S.; Schubert, F.; Wood, V.; Goodhead, I.; Penkett, C.; Rogers, J.; Bähler, J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single nucleotide resolution. Nature 2008, 453, 1239–1243. [Google Scholar]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; Carrington, J.C. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2007, 2, e219. [Google Scholar]
- Kasschau, K.D.; Fahlgren, N.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Carrington, J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007, 5, e57. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.K. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphate sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vitro protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Petrosino, J.F.; Highlander, S.; Luna, R.A.; Gibbs, R.A.; Versalovic, J. Metagenomic pyrosequencing and microbial identification. Clin. Chem. 2009, 55, 856–866. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 2011, 6, e19379. [Google Scholar]
- Flavell, A.J.; Pearce, S.R.; Kumar, A. Plant transposable elements and the genome. Curr. Opin. Genet. Dev. 1994, 4, 838–844. [Google Scholar] [CrossRef]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; Bennetzen, J.L. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Ma, J.; Devos, K.M. Mechanisms of recent genome size variation in flowering plants. Ann. Bot. 2005, 95, 127–135. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Clark, R.M.; Lanz, C.; Warthmann, N.; Weigel, D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008, 18, 2024–2033. [Google Scholar] [CrossRef]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; Dong, G.; Sang, T.; Han, B. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef]
- Trick, M.; Adamski, N.M.; Mugford, S.G.; Jiang, C.-C.; Febrer, M.; Uauy, C. Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploidy wheat. BMC Plant Biol. 2012, 12, 14. [Google Scholar] [CrossRef]
- Barbazuk, W.B.; Emrich, S.J.; Chen, H.D.; Li, L.; Schnable, P.S. SNP discovery via 454 transcriptome sequencing. Plant J. 2007, 51, 910–918. [Google Scholar] [CrossRef]
- Rabinowicz, P.; McCombie, W.R.; Martienssen, R.A. Gene enrichment in plant genomic shotgun libraries. Curr. Opin. Plant Biol. 2003, 6, 150–156. [Google Scholar] [CrossRef]
- Rabinowicz, P.D.; Citek, R.; Budiman, M.A.; Nunberg, A.; Bedell, J.A.; Lakey, N.; O'Shaughnessy, A.L.; Nascimento, L.U.; McCombie, W.R.; Martienssen, R.A. Differential methylation of genes and repeats in land plants. Genome Res. 2005, 15, 1431–1440. [Google Scholar] [CrossRef]
- Fellers, J.P. Genome filtering using methylation-sensitive restriction enzymes with six base pair recognition sites. The Plant Genome 2008, 1, 146–152. [Google Scholar] [CrossRef]
- Gore, M.A.; Wright, M.H.; Ersoz, E.S.; Bouffard, P.; Szekeres, E.S.; Jarvie, T.P.; Hurwitz, B.L.; Narechania, A.; Harkins, T.T.; Grills, G.S.; et al. Large-scale discovery of gene-enriched SNPs. The Plant Genome 2008, 2, 121–133. [Google Scholar]
- Tewhey, R.; Warner, J.B.; Nakano, M.; Libby, B.; Medkova, M.; David, P.H.; Kotsopoulos, S.K.; Samuels, M.L.; Hutchison, J.B.; Larson, J.W.; et al. Microdroplet-based PCR enrichment for large-scale targeted sequencing. Nat. Biotechnol. 2009, 27, 1025–1031. [Google Scholar] [CrossRef]
- Hardenbol, P.; Yu, F.; Belmont, J.; Mackenzie, J.; Bruckner, C.; Brundage, T.; Boudreau, A.; Chow, S.; Eberle, J.; Erbilgin, A.; et al. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 2005, 15, 269–275. [Google Scholar] [CrossRef]
- Hodges, E.; Xuan, Z.; Balija, V.; Kramer, M.; Molla, M.N.; Smith, S.W.; Middle, C.M.; Rodesch, M.J.; Albert, T.J.; Hannon, G.J.; McCombie, W.R. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007, 39, 1522–1527. [Google Scholar]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef]
- Li, Y.; Sidore, C.; Kang, H.M.; Boehnke, M.; Abecasis, G.R. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res. 2011, 21, 940–951. [Google Scholar] [CrossRef]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef]
- Li, Y.; Willer, C.; Sanna, S.; Abecasis, G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009, 10, 387–406. [Google Scholar] [CrossRef]
- Paran, I.; Zamir, D. Quantitative traits in plants: beyond QTL. Trends Genet. 2003, 19, 303–306. [Google Scholar] [CrossRef]
- Rahman, H.; Pekic, S.; Lazic-Jancic, V.; Quarrie, S.A.; Shah, S.M.; Pervez, A.; Shah, M.M. Molecular mapping of quantitative trait loci for drought tolerance in maize plants. Genet. Mol. Res. 2011, 10, 889–901. [Google Scholar] [CrossRef]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D.; Balint-Kurti, P.J.; Holland, J.B. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association-mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar]
- Edwards, D.; Batley, J. Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 2010, 8, 2–9. [Google Scholar] [CrossRef]
- Morrell, P.L.; Buckler, E.S.; Ross-Ibarra, J. Crop genomics: Advances and applications. Nat. Rev. Genet. 2011, 13, 85–96. [Google Scholar]
- Schneeberger, K.; Weigel, D. Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 2011, 16, 282–288. [Google Scholar] [CrossRef]
- Gore, M.A.; Chia, J.M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; Peiffer, J.A.; McMullen, M.D.; Grills, G.S.; Ross-Ibarra, J.; Ware, D.H.; Buckler, E.S. A first-generation haplotype map of maize. Science 2009, 326, 1115–1117. [Google Scholar]
- Wang, L.; Wang, A.; Huang, X.; Zhao, Q.; Dong, G.; Qian, Q.; Sang, T.; Han, B. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor. Appl. Genet. 2011, 122, 327–340. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 2008, 3, e3376. [Google Scholar]
- Chutimanitsakun, Y.; Nipper, R.W.; Cuesta-Marcos, A.; Cistué, L.; Corey, A.; Filichkina, T.; Johnson, E.A.; Hayes, P.M. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genomics 2011, 12, 4. [Google Scholar] [CrossRef]
- Szűcs, P.; Blake, V.C.; Bhat, P.R.; Chao, S.; Close, T.J.; Cuesta-Marcos, A.; Muehlbauer, G.J.; Ramsay, L.V.; Waugh, R.; Hayes, P.M. An integrated resource for barley linkage map and malting quality QTL alignment. Plant Genome 2009, 2, 134–140. [Google Scholar] [CrossRef]
- Pfender, W.F.; Saha, M.C.; Johnson, E.A.; Slabaugh, M.B. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor. Appl. Genet. 2011, 122, 1467–1480. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar]
- Saha, M.C.; Mian, R.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef]
- Saha, M.C.; Mian, R.; Zwonitzer, J.C.; Chekhovskiy, K.; Hopkins, A.S. An SSR- and AFLP-based genetic linkae map of tall fescue (Festuca arundinacea Schreb.). Theor. Appl. Genet. 2005, 110, 323–336. [Google Scholar] [CrossRef]
- Kantety, R.V.; Rota, M.L.; Matthews, D.E.; Sorrells, M.E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 2002, 48, 501–510. [Google Scholar] [CrossRef]
- Lauvergeat, V.; Barre, P.; Bonnet, M.; Ghesquiere, M. Sizty simple sequence repeat markers for use in the Festuca-Lolium complex of grasses. Mol. Ecol. 2005, 5, 401–405. [Google Scholar] [CrossRef]
- Yang, H.; Tao, Y.; Zheng, Z.; Li, C.; Sweetingham, M.; Howieson, J. Application of next-generation sequencing for rapid marker development in molecular plant breeding: A case study on antrachnose disease resistance in Lupinus angustifolius L. BMC Genomics 2012, 13, 318. [Google Scholar] [CrossRef]
- Barchi, L.; Lanteri, S.; Portis, E.; Acquadro, A.; Valè, G.; Toppino, L.; Rotino, G.L. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics 2011, 12, 304. [Google Scholar] [CrossRef]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 2012, 7, e32253. [Google Scholar]
- Iwata, H.; Ninomiya, S. AntMap: Constructing genetic linkage maps using an ant colony optimization algorithm. Breed. Sci. 2006, 56, 371–377. [Google Scholar] [CrossRef]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploidy crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef]
- Maughan, P.J.; Yourstone, S.M.; Byers, R.L.; Smith, S.M.; Udall, J.A. Single-nucleotide polymorphism genotyping in mapping populations via genomic reduction and next-generation sequencing: proof of concept. Plant Genome 2010, 3, 166–178. [Google Scholar] [CrossRef]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jørgensen, J.E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 2009, 6, 550–551. [Google Scholar]
- 1001 Genomes. A Catalog of Arabidopsis thaliana Genetic Variation. Available online: http://1001genomes.org/downloads/shore.html (accessed on 6 September 2012).
- Austin, R.S.; Vidaurre, D.; Stamatiou, G.; Breit, R.; Provart, N.J.; Bonetta, D.; Zhang, J.; Fung, P.; Gong, Y.; Wang, P.W.; McCourt, P.; Guttman, D.S. Next-generation mapping of Arabidopsis genes. Plant J. 2011, 67, 715–725. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-Sequencing in Plants. Biology 2012, 1, 460-483. https://doi.org/10.3390/biology1030460
Deschamps S, Llaca V, May GD. Genotyping-by-Sequencing in Plants. Biology. 2012; 1(3):460-483. https://doi.org/10.3390/biology1030460
Chicago/Turabian StyleDeschamps, Stéphane, Victor Llaca, and Gregory D. May. 2012. "Genotyping-by-Sequencing in Plants" Biology 1, no. 3: 460-483. https://doi.org/10.3390/biology1030460
APA StyleDeschamps, S., Llaca, V., & May, G. D. (2012). Genotyping-by-Sequencing in Plants. Biology, 1(3), 460-483. https://doi.org/10.3390/biology1030460