TE-Locate: A Tool to Locate and Group Transposable Element Occurrences Using Paired-End Next-Generation Sequencing Data

Abstract
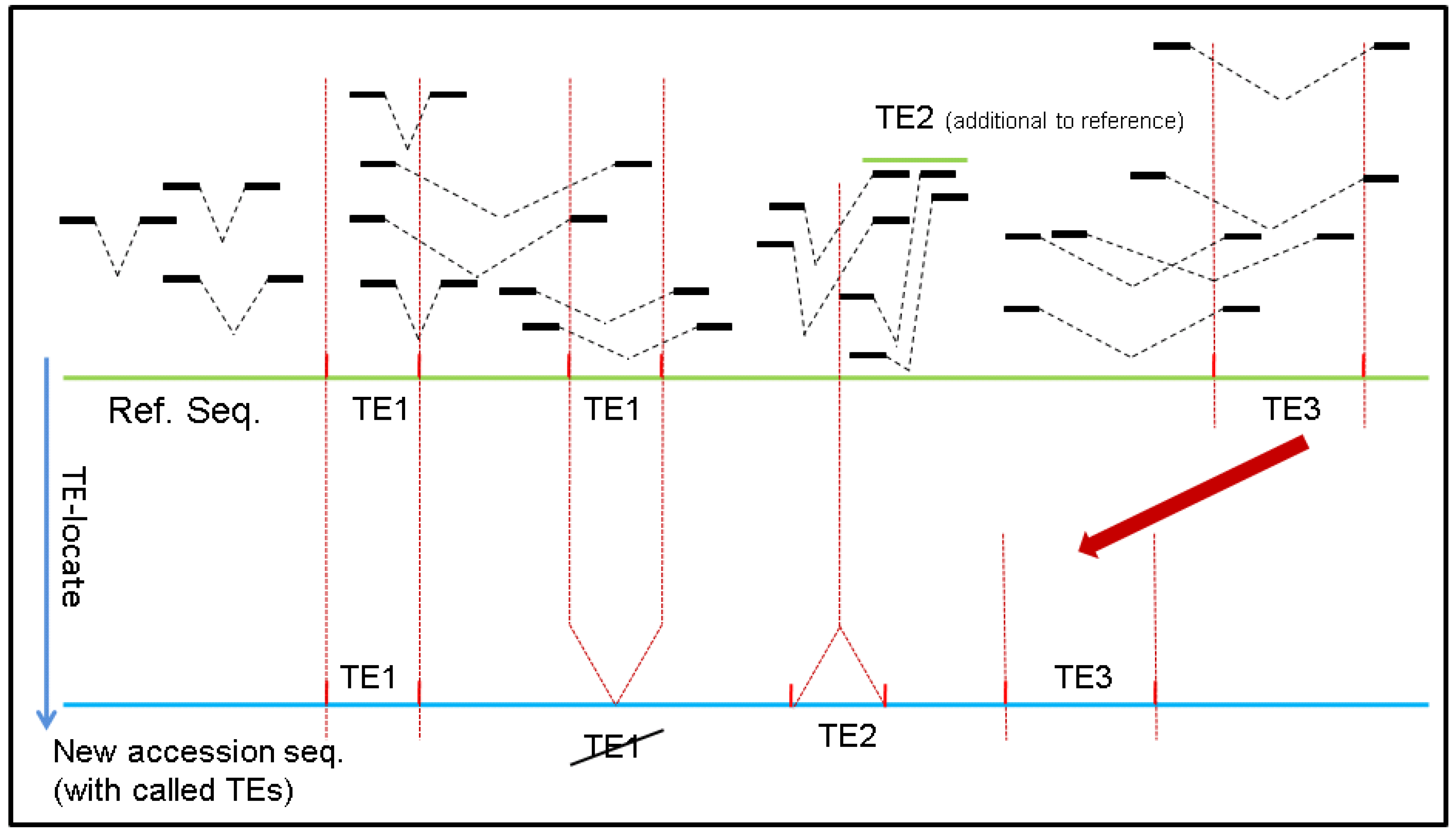
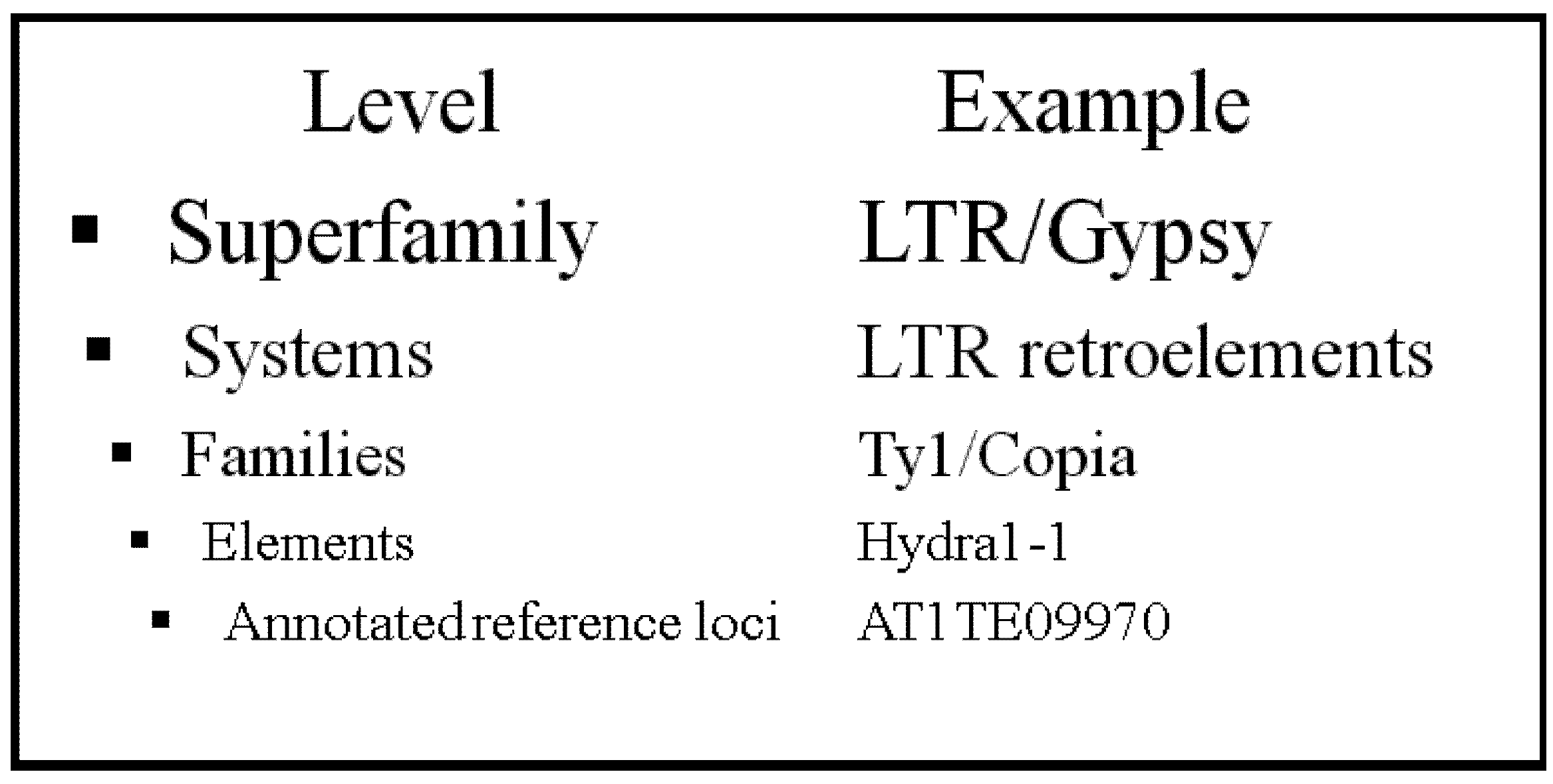
:1. Introduction
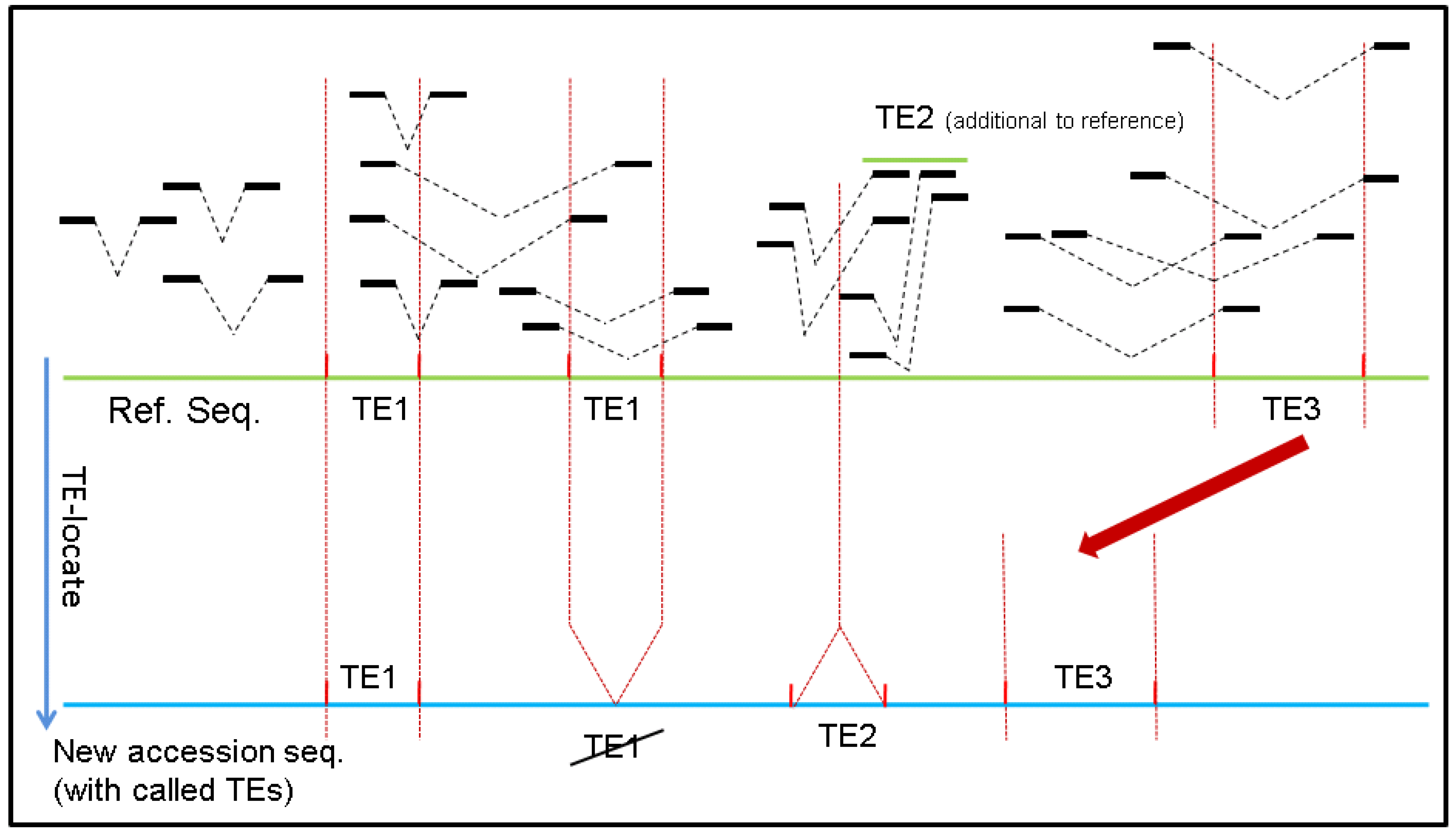
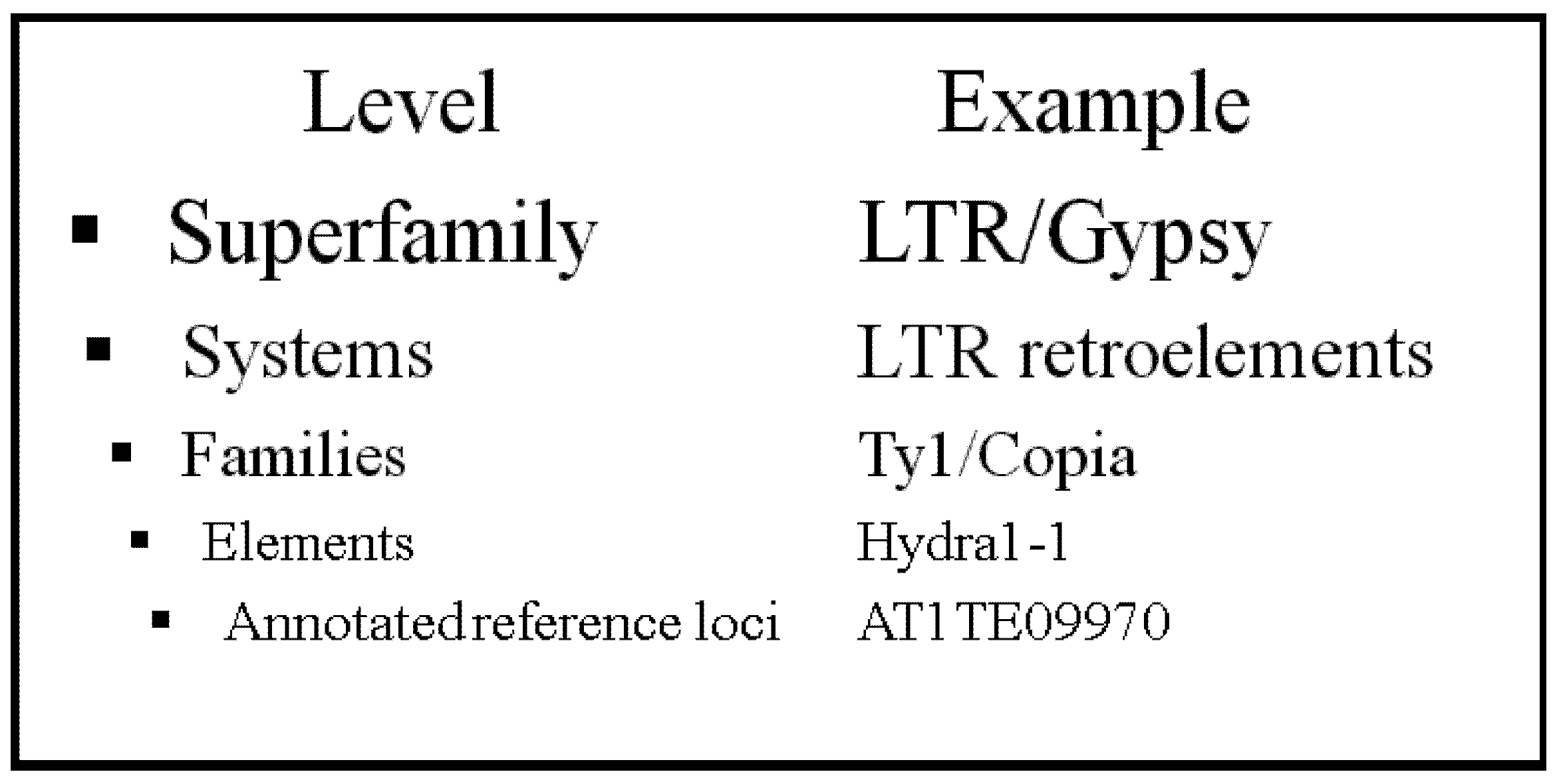
2. Results
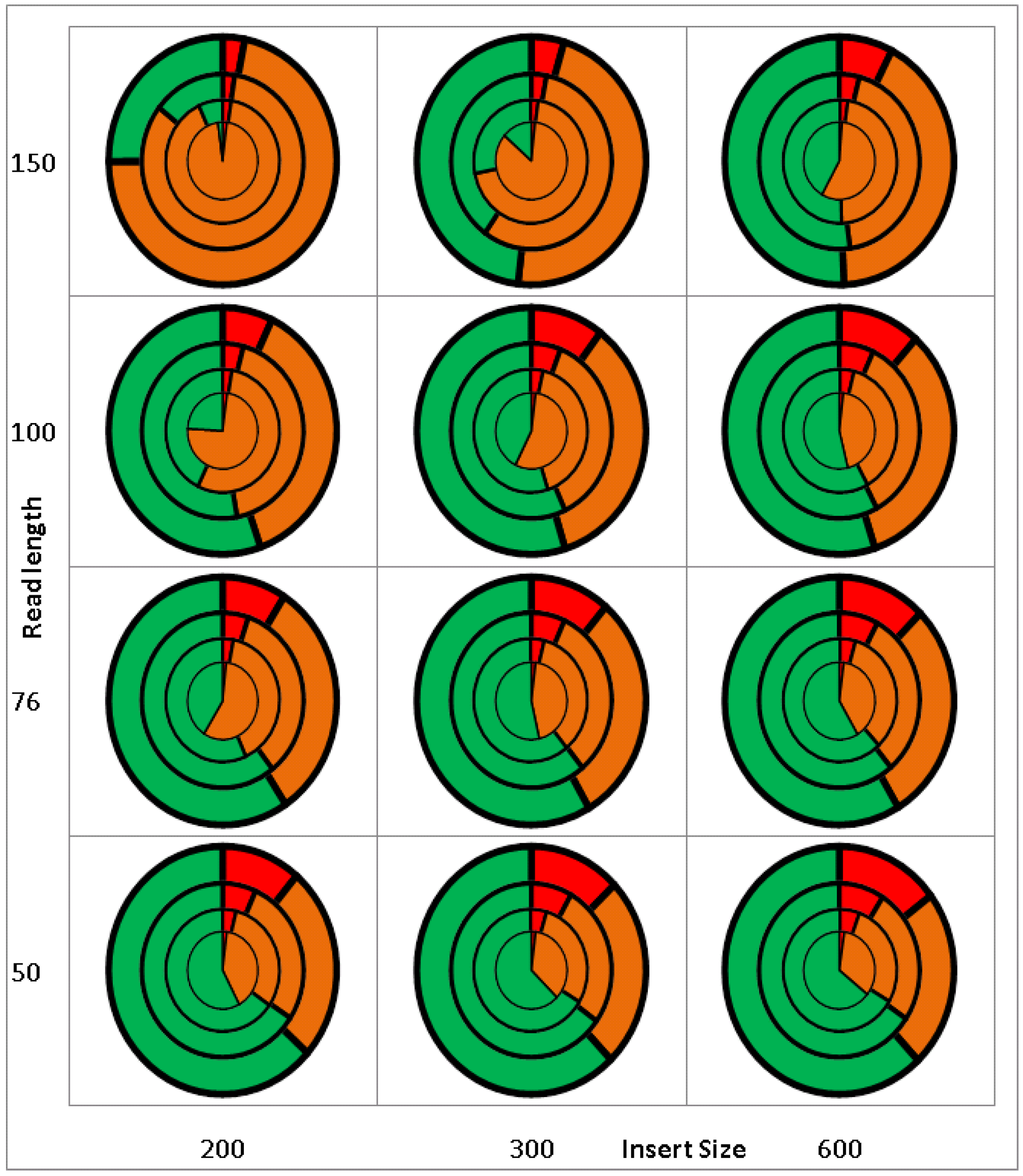
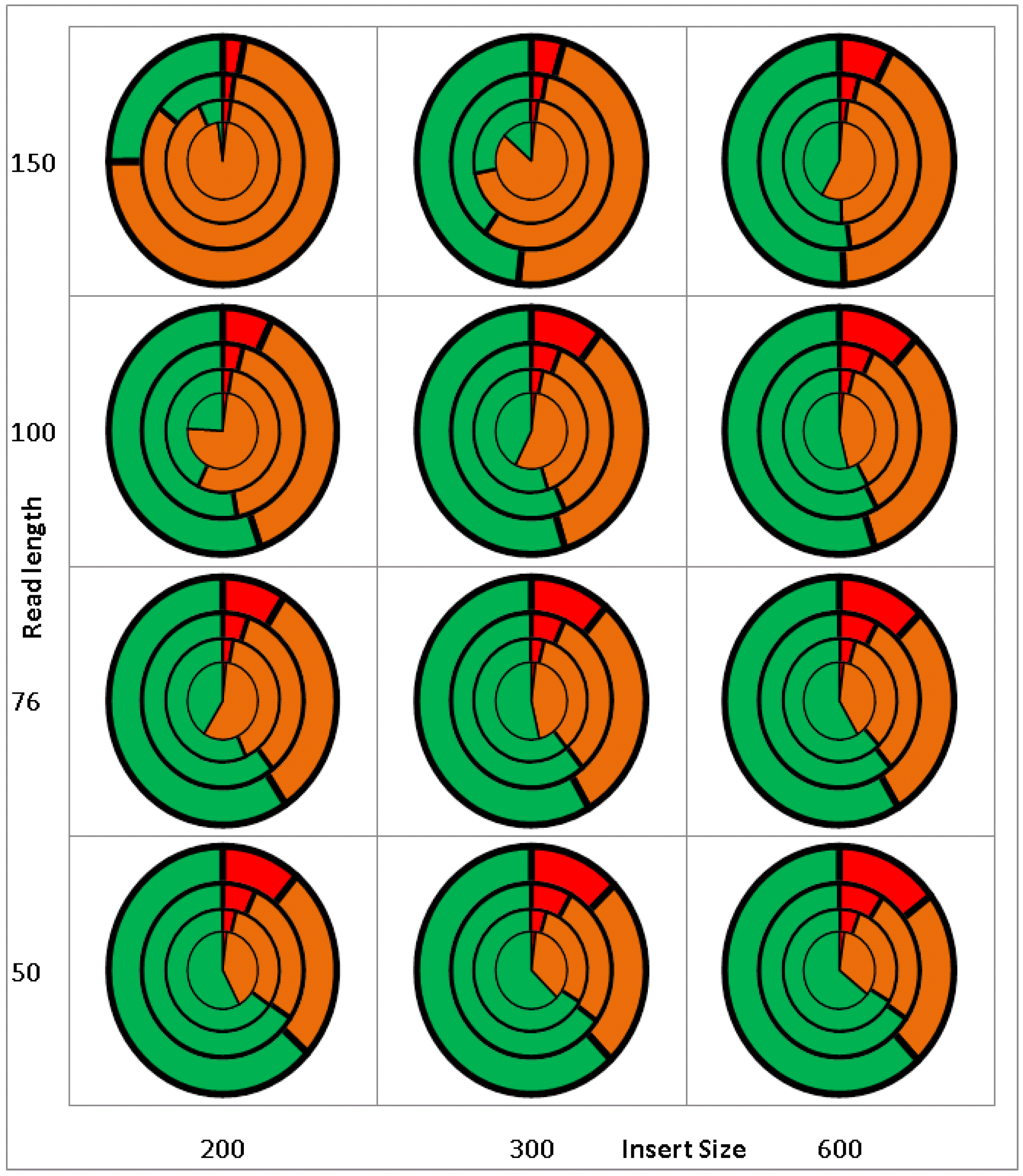
2.1. Validation/Simulation
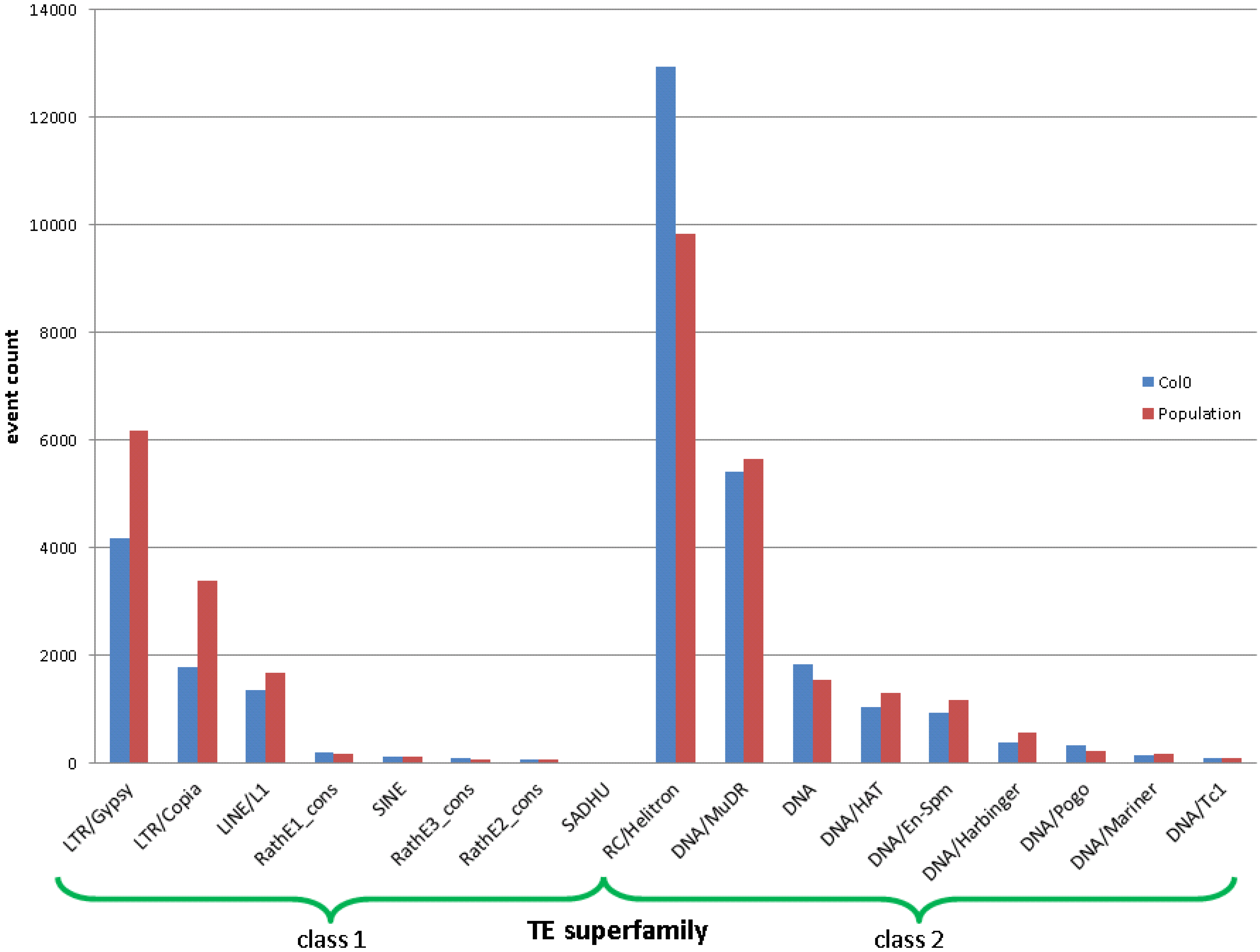
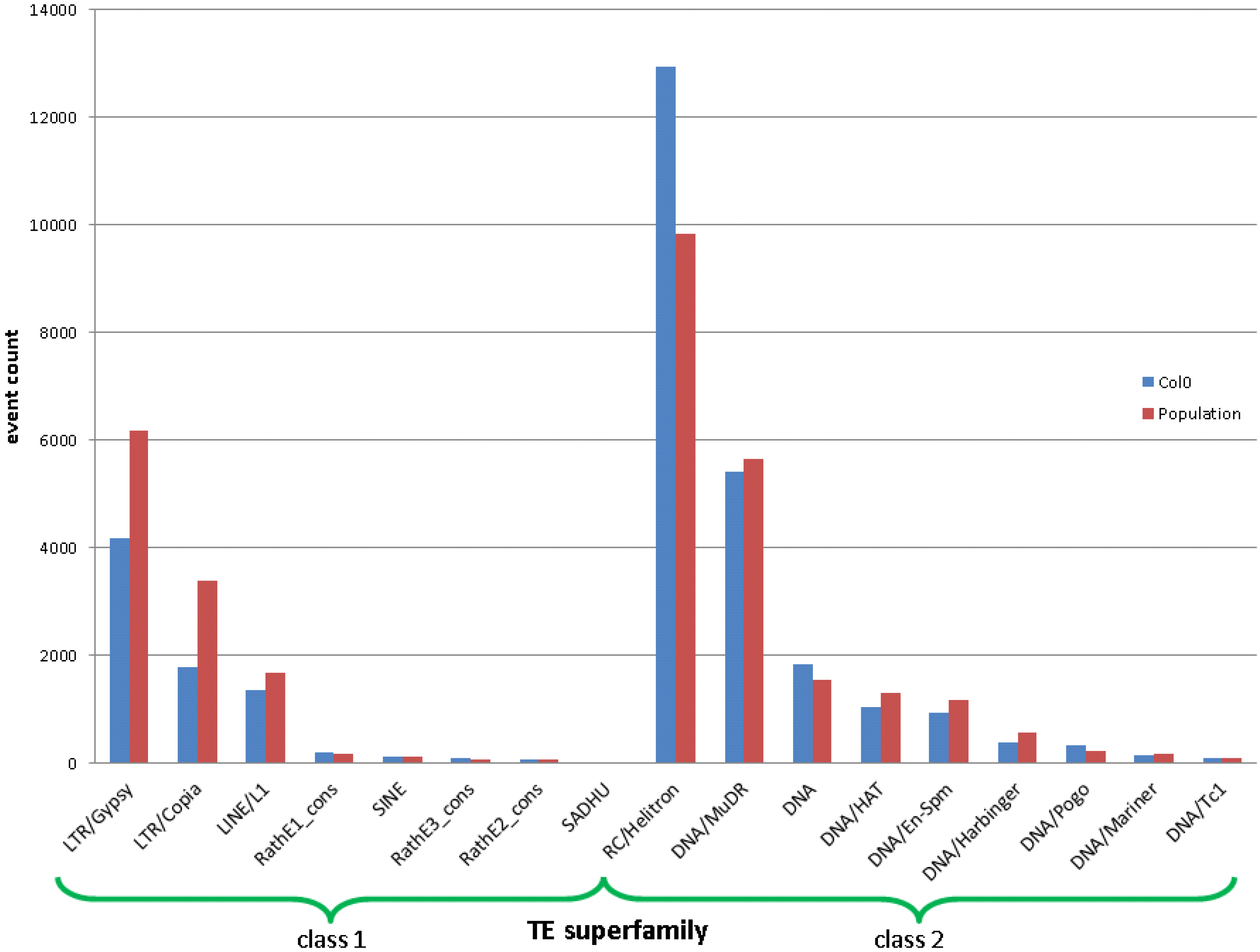
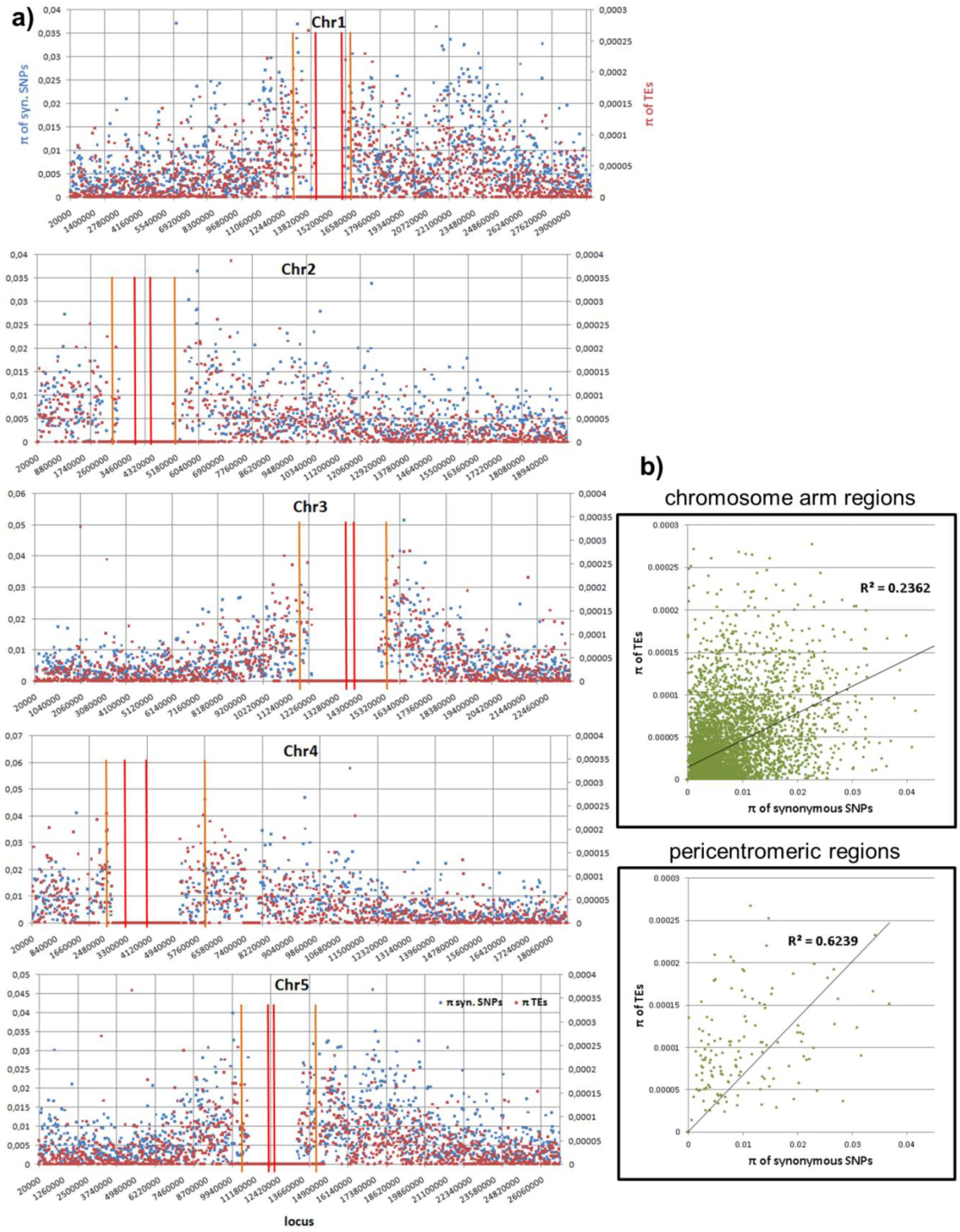
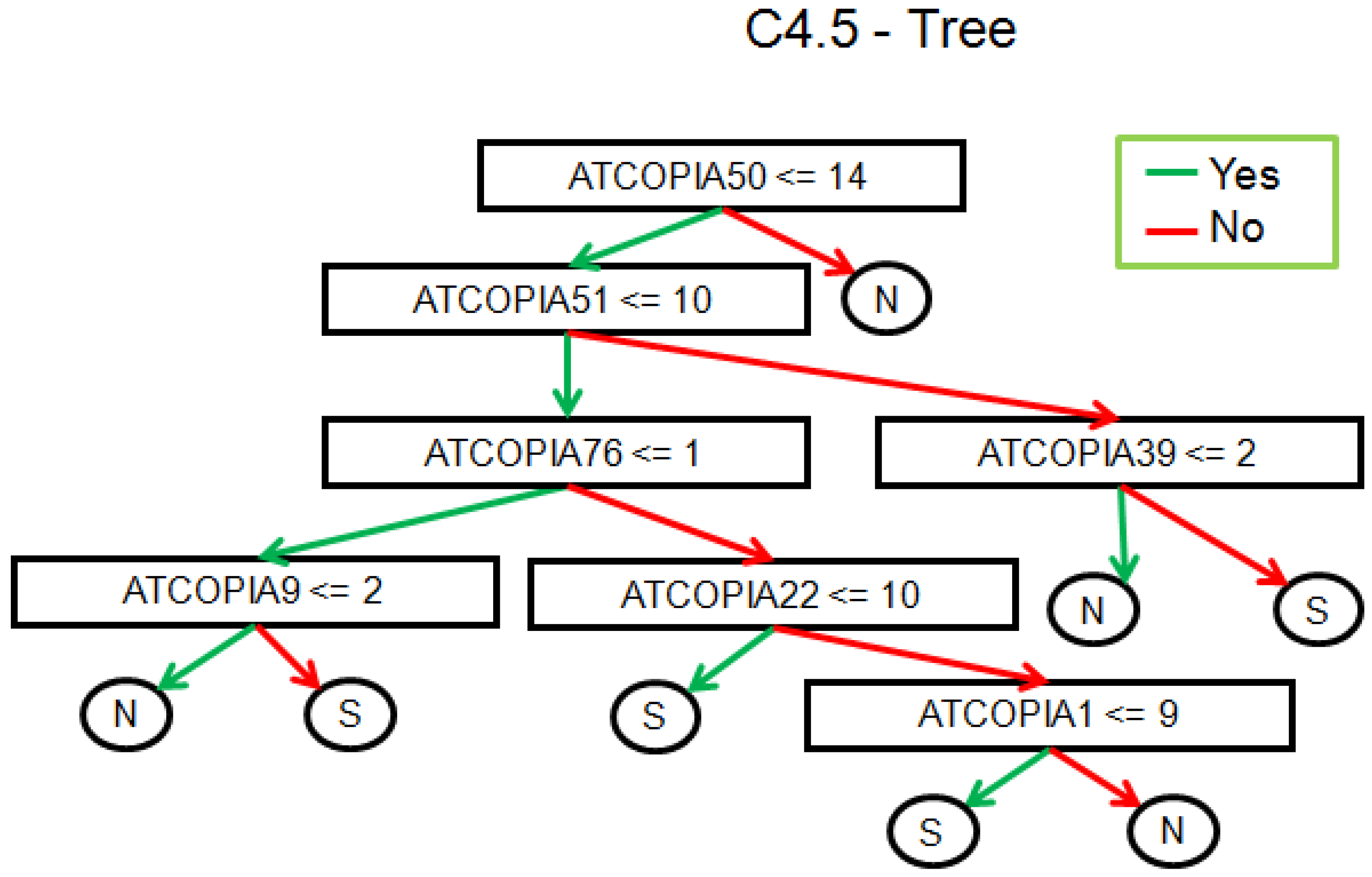
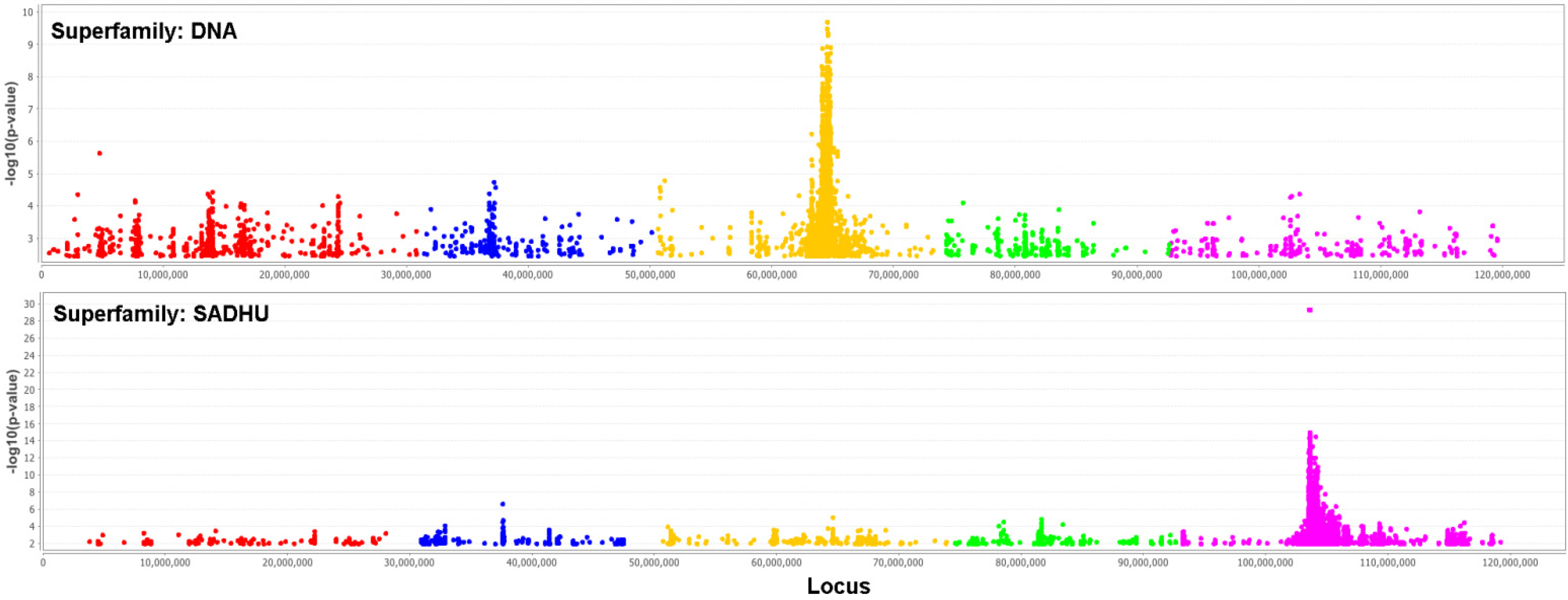
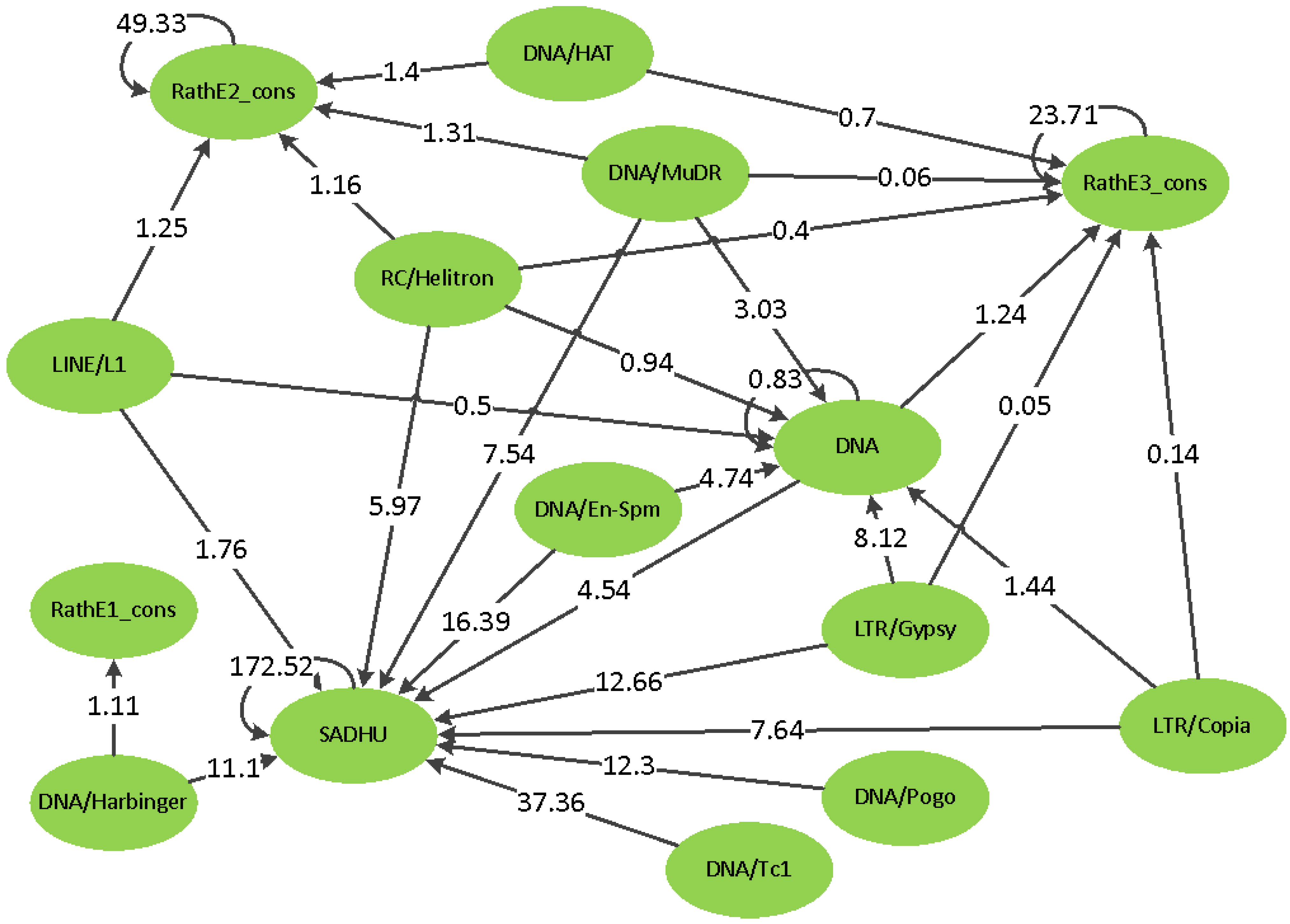
2.2. Real Data
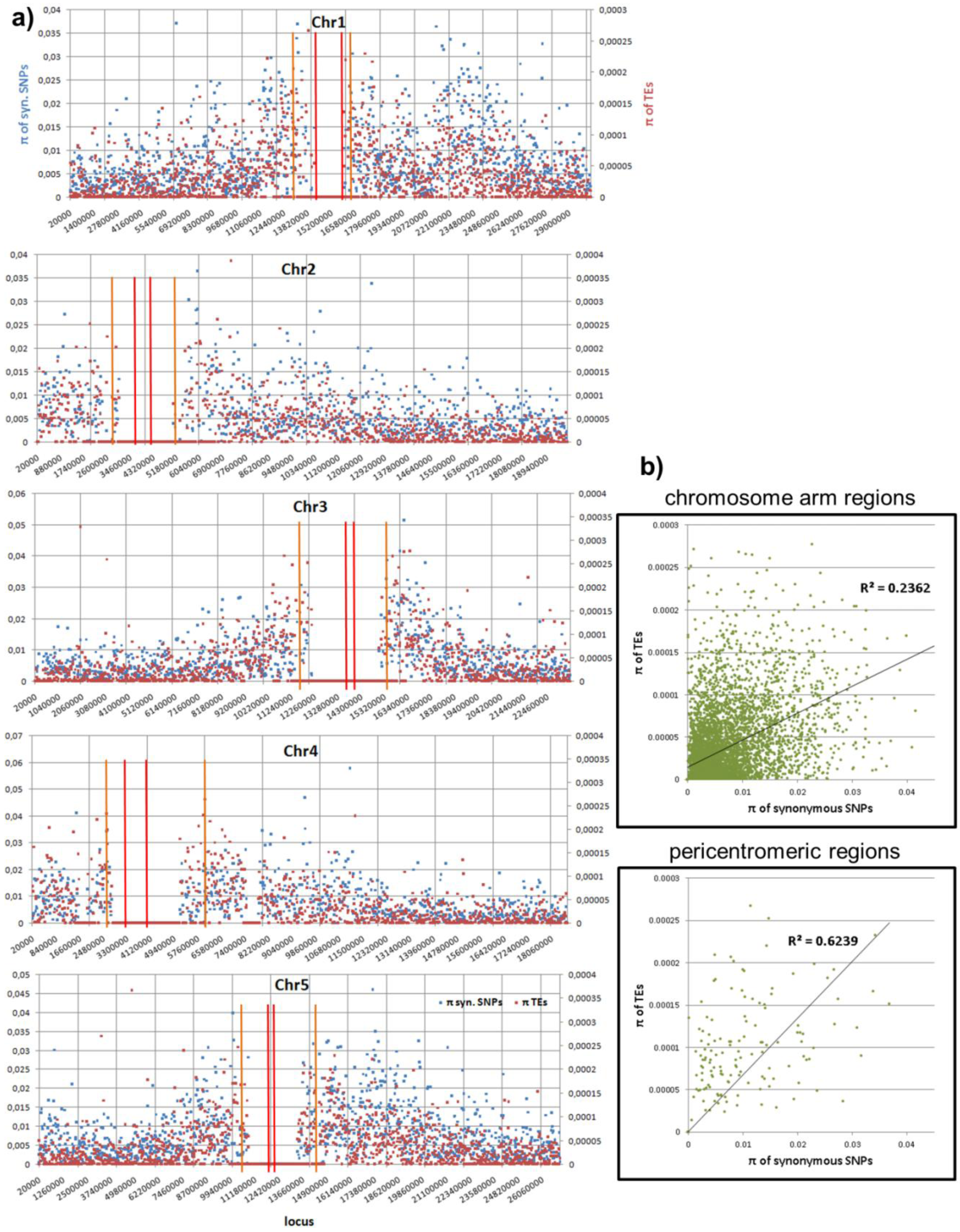
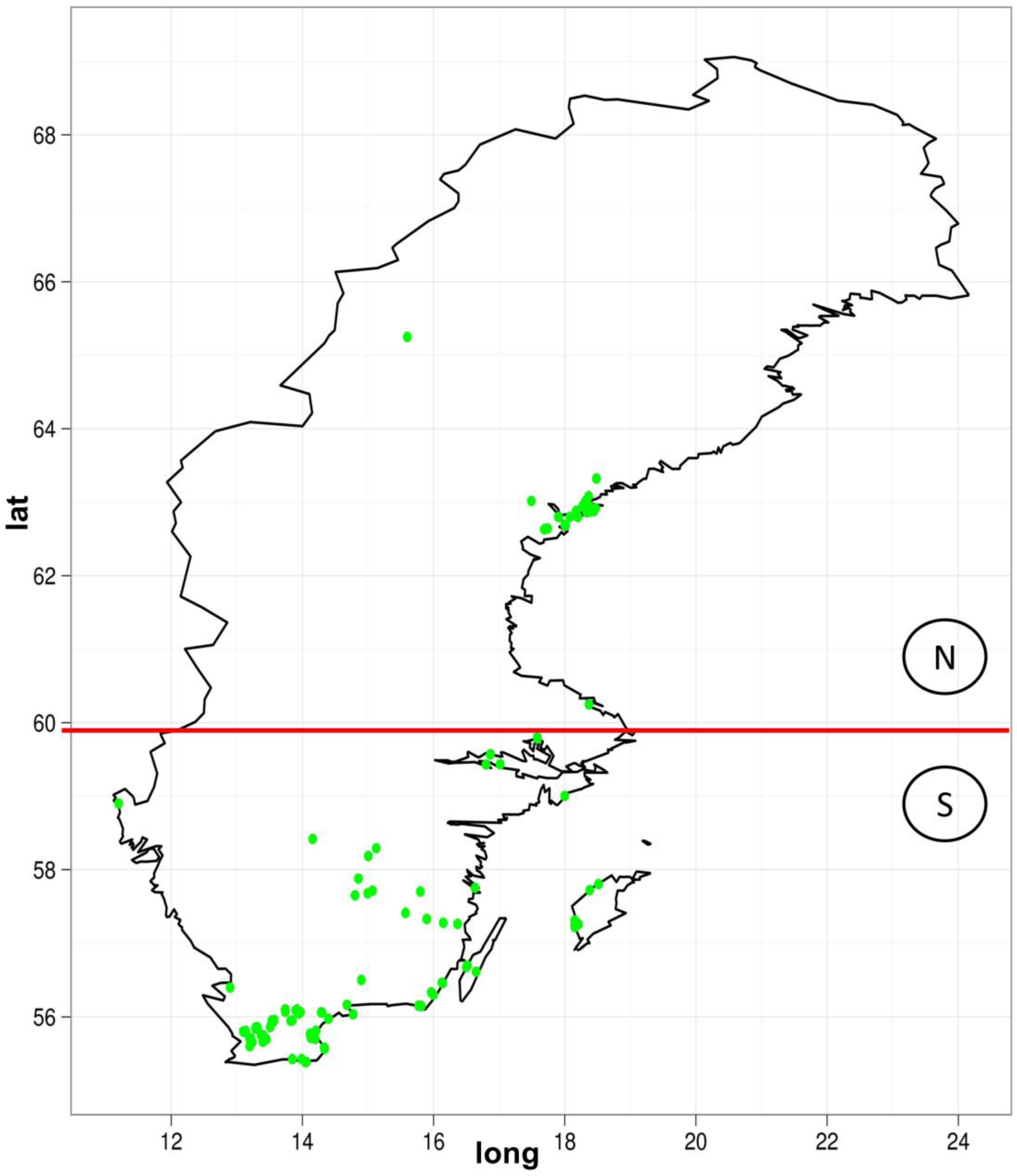
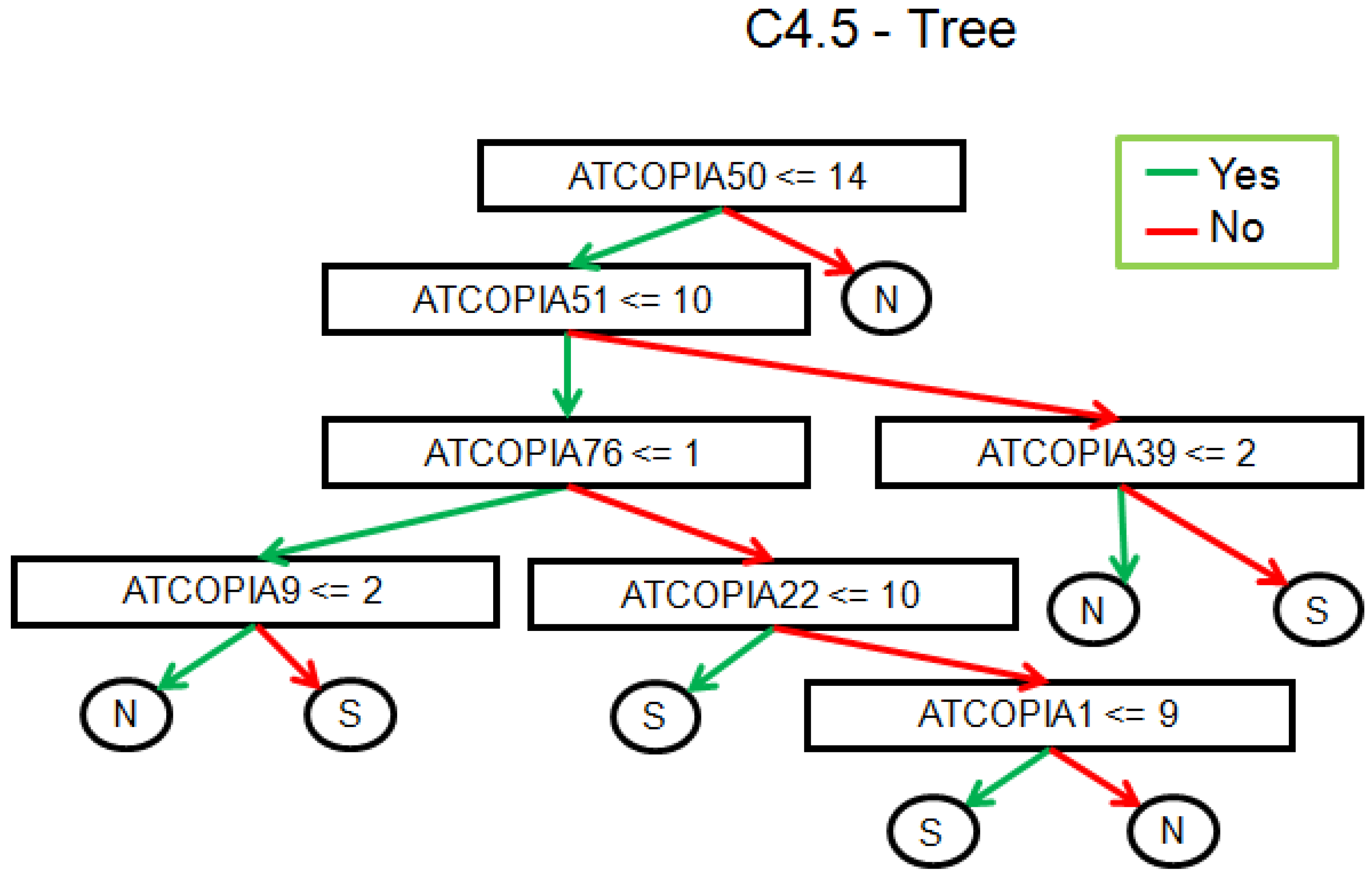
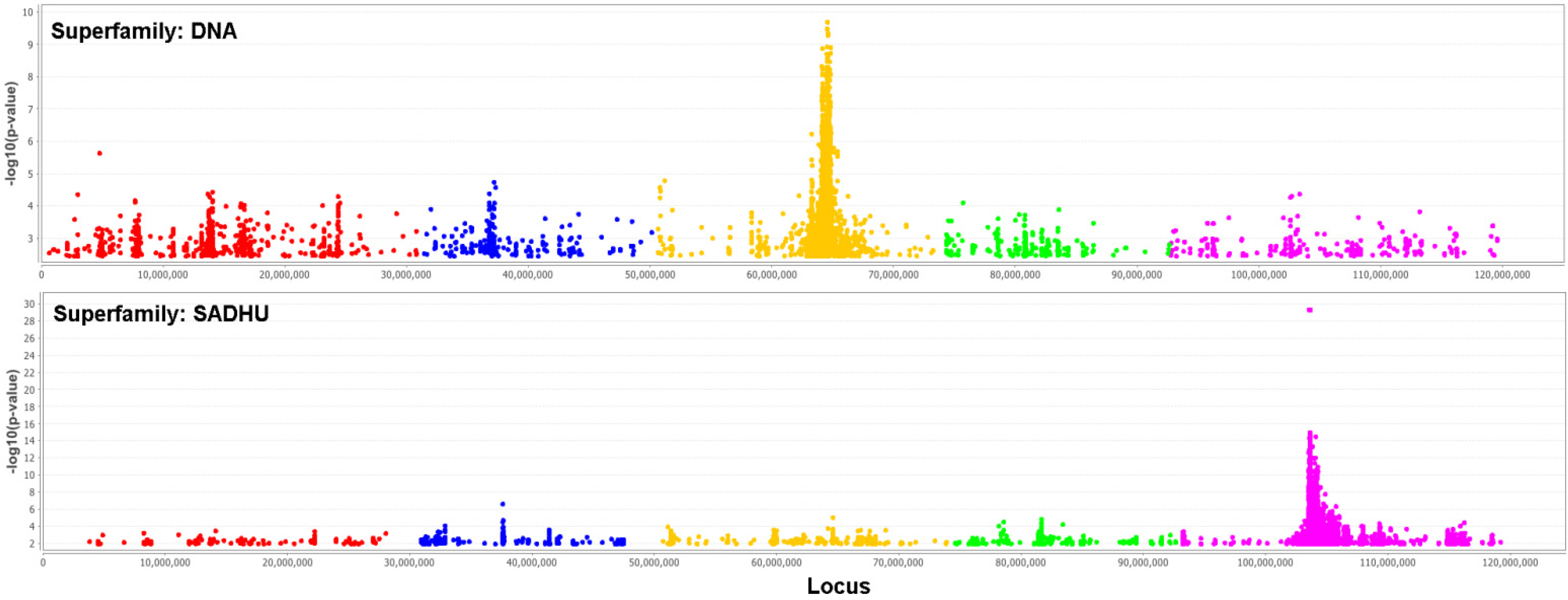

3. Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| chr | loc | len | event_type_ref | non_ref_counts | anc_status | read_pair_support | <unused>... | call_method | Orientation | #pPairs | #iPairs | new/old |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5421 | 7679 | TE+DNA/MuDR/DNA/MuDR | 5 | N | 15 | | | PairEndTE | inverse | 4 | 11 | new |
| 1 | 16726 | 3890 | TE+RC/Helitron/RC/Helitron | 171 | N | 900 | | | PairEndTE | uncertain | old | ||
| 1 | 20843 | 1292 | TE+RC/Helitron/RC/Helitron | 3 | N | 63 | | | PairEndTE | inverse | 20 | 43 | new |
| 1 | 11897 | 79 | TE+LTR/Copia/LTR/Copia | 55 | N | 69 | | | PairEndTE | uncertain | old | ||
| 1 | 22277 | 1736 | TE+DNA/MuDR/DNA/MuDR | 7 | N | 15 | | | PairEndTE | inverse | 6 | 9 | new |
| 1 | 42355 | 10046 | TE+RC/Helitron/RC/Helitron | 4 | N | 11 | | | PairEndTE | parallel | 10 | 1 | new |
| 1 | 42210 | 4671 | TE+DNA/MuDR/DNA/MuDR | 5 | N | 11 | | | PairEndTE | inverse | 1 | 10 | new |
| 1 | 50968 | 651 | TE+LTR/Gypsy/LTR/Gypsy | 6 | N | 10 | | | PairEndTE | parallel | 9 | 1 | new |
| 1 | 52425 | 382 | TE+LTR/Copia/LTR/Copia | 2 | N | 26 | | | PairEndTE | inverse | 1 | 25 | new |
| 1 | 70064 | 4814 | TE+LTR/Copia/LTR/Copia | 1 | N | 19 | | | PairEndTE | inverse | 0 | 19 | new |
| 1 | 71152 | 799 | TE+LTR/Copia/LTR/Copia | 1 | N | 31 | | | PairEndTE | parallel | 31 | 0 | new |
| 1 | 55676 | 900 | TE+DNA/HAT/DNA/HAT | 174 | N | 2133 | | | PairEndTE | uncertain | old | ||
| 1 | 77569 | 831 | TE+RC/Helitron/RC/Helitron | 178 | N | 1661 | | | PairEndTE | uncertain | old | ||
| 1 | 76844 | 656 | TE+LINE/L1/LINE/L1 | 75 | N | 753 | | | PairEndTE | uncertain | old | ||
| 1 | 84679 | 12225 | TE+LTR/Gypsy/LTR/Gypsy | 7 | N | 12 | | | PairEndTE | parallel | 10 | 2 | new |
| 1 | 91443 | 7263 | TE+LTR/Gypsy/LTR/Gypsy | 6 | N | 13 | | | PairEndTE | parallel | 11 | 2 | new |
| 1 | 116237 | 2941 | TE+LTR/Copia/LTR/Copia | 1 | N | 57 | | | PairEndTE | parallel | 47 | 10 | new |
| 1 | 129878 | 5185 | TE+LTR/Copia/LTR/Copia | 4 | N | 23 | | | PairEndTE | parallel | 23 | 0 | new |
| 1 | 154331 | 87 | TE+LINE/L1/LINE/L1 | 89 | N | 138 | | | PairEndTE | uncertain | old | ||
| 1 | 192934 | 593 | TE+RC/Helitron/RC/Helitron | 177 | N | 1915 | | | PairEndTE | uncertain | old |
| Column | Description |
|---|---|
| chr | Locus |
| loc | |
| len | The length of the corresponding reference event. |
| event_type_ref | The class of this event annotated (resp. the item/TE) |
| non_ref_counts | The number of individuals sharing this event. |
| anc_status | Unused |
| read_pair_support | The total number of all supporting read pairs of all individuals. |
| bp_range1 | Unused... |
| bp_range2 | |
| four_gamete_left | |
| four_gamete_right | |
| call_method | For TE-Locate, here is written ‘PairEndTE’, used if merged with other data in this format. |
| Orientation | ‘parallel’, ‘inverse’ or ‘uncertain’: The orientation according to the reference sequence. |
| #pPairs | The number of read pairs supporting parallel orientation. Not used if the orientation is ‘uncertain’. |
| #iPairs | The number of read pairs supporting inverse orientation. Not used if the orientation is ‘uncertain’. |
| new/old | ‘new’ or ‘old’. ‘old’ if the item is called at the locus in the reference; ‘new’ otherwise. Note that at higher hierarchical levels, all locations of this item are meant, e.g., any Copia called at a Copia locus in the reference is called ‘old’ as the item’s name is the only distinction. |
4. Discussion and Conclusions
Supplementary Files
Acknowledgments
References and Notes
- Castillo-Davis, C.I. The evolution of noncoding DNA: How much junk, how much func? Trends Genet. 2005, 21, 533–536. [Google Scholar] [CrossRef]
- McClintock, B. The Discovery and Characterization of Transposable Elements: The Collected Papers of Barbara McClintock; Garland Publishing, Inc.: New York, NY, USA, 1987. [Google Scholar]
- Nowacki, M.; Higgins, B.P.; Maquilan, G.M.; Swart, E.C.; Doak, T.G.; Landweber, L.F. A functional role for transposases in a large eukaryotic genome. Science 2009, 324, 935–938. [Google Scholar]
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile elements and disease. Curr. Opin. Genet. Dev. 1998, 8, 343–350. [Google Scholar] [CrossRef]
- Kazazian, H.H., Jr. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.L.; Ruan, Y.; Wei, C.L.; Ng, H.H.; et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef]
- Belancio, V.P.; Hedges, D.J.; Deininger, P. Mammalian non-LTR retrotransposons: For better or worse, in sickness and in health. Genome Res. 2008, 18, 343–358. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Alvarado, C.; Trifiro, M.A. Selection and mutation in the “new” genetics: An emerging hypothesis. Hum. Genet. 2010, 127, 491–501. [Google Scholar] [CrossRef]
- Gupta, S.; Gallavotti, A.; Stryker, G.A.; Schmidt, R.J.; Lal, S.K. A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudogenes. Plant Mol. Biol. 2005, 57, 115–127. [Google Scholar] [CrossRef]
- Jiang, N.; Bao, Z.; Zhang, X.; Eddy, S.R.; Wessler, S.R. Pack-MULE transposable elements mediate gene evolution in plants. Nature 2004, 431, 569–573. [Google Scholar]
- Kordis, D. Transposable elements in reptilian and avian (sauropsida) genomes. Cytogenet. Genome Res. 2009, 127, 94–111. [Google Scholar] [CrossRef]
- Lai, J.; Li, Y.; Messing, J.; Dooner, H.K. Gene movement by Helitron transposons contributes to the haplotype variability of maize. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 9068–9073. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Conconi, A.; Sogo, J.M.; Ryan, C.A. Ribosomal gene clusters are uniquely proportioned between open and closed chromatin structures in both tomato leaf cells and exponentially growing suspension cultures. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 5256–5260. [Google Scholar]
- Lamesch, P.; Dreher, K.; Swarbreck, D.; Sasidharan, R.; Reiser, L.; Huala, E. Using the Arabidopsis information resource (TAIR) to find information about Arabidopsis genes. Curr. Protoc. Bioinformatics 2010, Chapter 1. Unit1 11. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Weigel, D.; Mott, R. The 1001 genomes project for Arabidopsis thaliana. Genome Biol. 2009, 10, 107. [Google Scholar] [CrossRef]
- The 1001 Genomes Project Website. Available online: http://www.1001genomes.org (accessed on 1 July 2012).
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar]
- Long, Q.; Rabanal, F.A.; Meng, D.; Huber, C.D.; Farlow, A.; Platzer, A.; Zhang, Q.; Vilhjálmsson, B.J.; Korte, A.; Nizhynska, V.; et al. Massive genomic variation and strong selection in Swedish Arabidopsis thaliana. Gregor Mendel Institute, 2012; Unpublished work. [Google Scholar]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef]
- Quinlan, J.R. C4.5: Programs for Machine Learning; Morgan Kaufmann Publishers: Burlington, MA, USA, 1993. [Google Scholar]
- Platt, J.C. A fast algorithm for training support vector machines. Technical Reportfor Microsoft Research: Redmond, WA, USA, 21 April 1998; MSR-TR-98-14. [Google Scholar]
- Turner, A.K.; Delacruz, F.; Grinsted, J. Temperature sensitivity of transposition of class-Ii transposons. J. Gen. Microbiol. 1990, 136, 65–67. [Google Scholar] [CrossRef]
- Paquin, C.E.; Williamson, V.M. Temperature effects on the rate of ty transposition. Science 1984, 226, 53–55. [Google Scholar]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Ponstingl, H. SMALT; Wellcome Trust Sanger Institute: Cambridge, UK, 2011. [Google Scholar]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermuller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef]
- Handsaker, R.E.; Korn, J.M.; Nemesh, J.; McCarroll, S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011, 43, 269–276. [Google Scholar] [CrossRef]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar]
- Abyzov, A.; Gerstein, M. AGE: Defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics 2011, 27, 595–603. [Google Scholar] [CrossRef]
- Bergman, C.M.; Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief Bioinform. 2007, 8, 382–392. [Google Scholar] [CrossRef]
- Fiston-Lavier, A.S.; Carrigan, M.; Petrov, D.A.; Gonzalez, J. T-lex: A program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 2011, 39, e36. [Google Scholar] [CrossRef]
- Flutre, T.; Inizan, O.; Hoede, C.; Quesneville, H. REPET: Pipelines for the identification and annotation of transposable elements in genomic sequences. In Proceedings of the Plant & Animal Genome (PAG) XVIII Conference, San Diego, CA, USA, 9–13 January 2010.
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Kennedy, R.C.; Unger, M.F.; Christley, S.; Collins, F.H.; Madey, G.R. An automated homology-based approach for identifying transposable elements. BMC Bioinformatics 2011, 12, 130. [Google Scholar] [CrossRef]
- Andrieu, O.; Fiston, A.S.; Anxolabehere, D.; Quesneville, H. Detection of transposable elements by their compositional bias. BMC Bioinformatics 2004, 5. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, P.; Stanciu, M.; Brudno, M. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods 2009, 6, S13–S20. [Google Scholar] [CrossRef]
- TE-Locate Website. Available online: http://zendto.gmi.oeaw.ac.at/pickup.php?claimID=Y3tZVfN5xipYyBDN&claimPasscode=NArXMbTjmkorWjSM&emailAddr=te_locate%40gmx.at (accessed on 1 July 2012, will be long-term maintained).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Platzer, A.; Nizhynska, V.; Long, Q. TE-Locate: A Tool to Locate and Group Transposable Element Occurrences Using Paired-End Next-Generation Sequencing Data. Biology 2012, 1, 395-410. https://doi.org/10.3390/biology1020395
Platzer A, Nizhynska V, Long Q. TE-Locate: A Tool to Locate and Group Transposable Element Occurrences Using Paired-End Next-Generation Sequencing Data. Biology. 2012; 1(2):395-410. https://doi.org/10.3390/biology1020395
Chicago/Turabian StylePlatzer, Alexander, Viktoria Nizhynska, and Quan Long. 2012. "TE-Locate: A Tool to Locate and Group Transposable Element Occurrences Using Paired-End Next-Generation Sequencing Data" Biology 1, no. 2: 395-410. https://doi.org/10.3390/biology1020395