Facile Synthesis of Highly Hydrophobic Cellulose Nanoparticles through Post-Esterification Microfluidization



Abstract
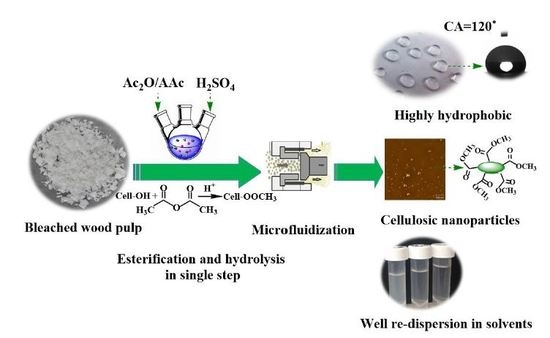
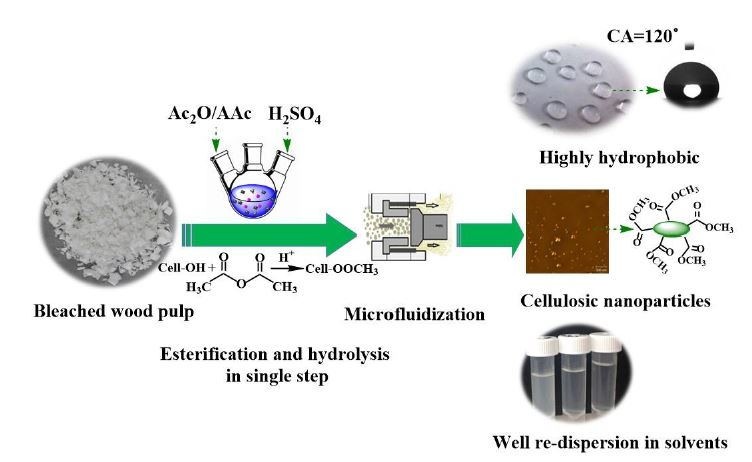
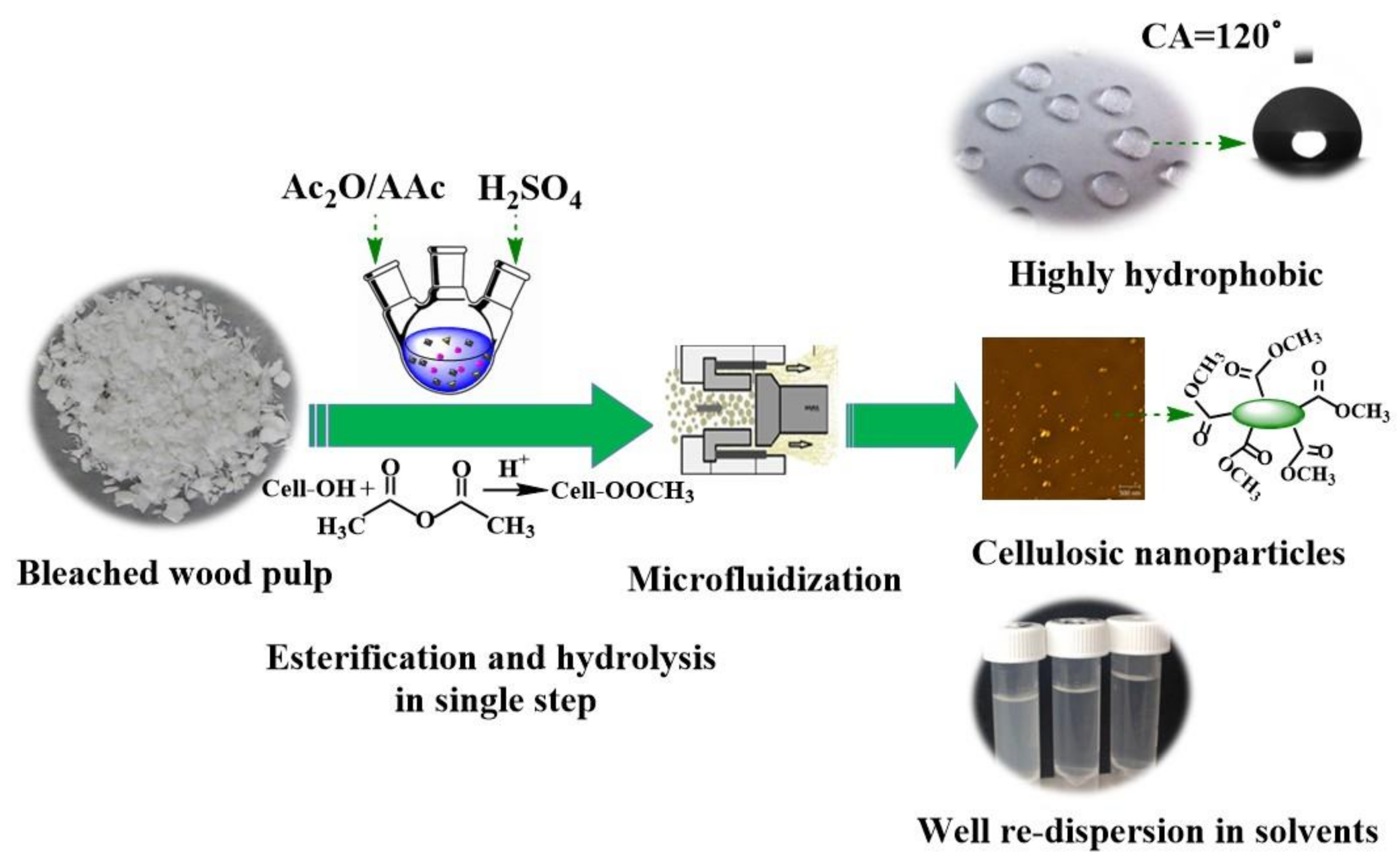
1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Synthesis of Esterified Cellulose Nanomaterials
2.3. Determination of the Degree of Substitution
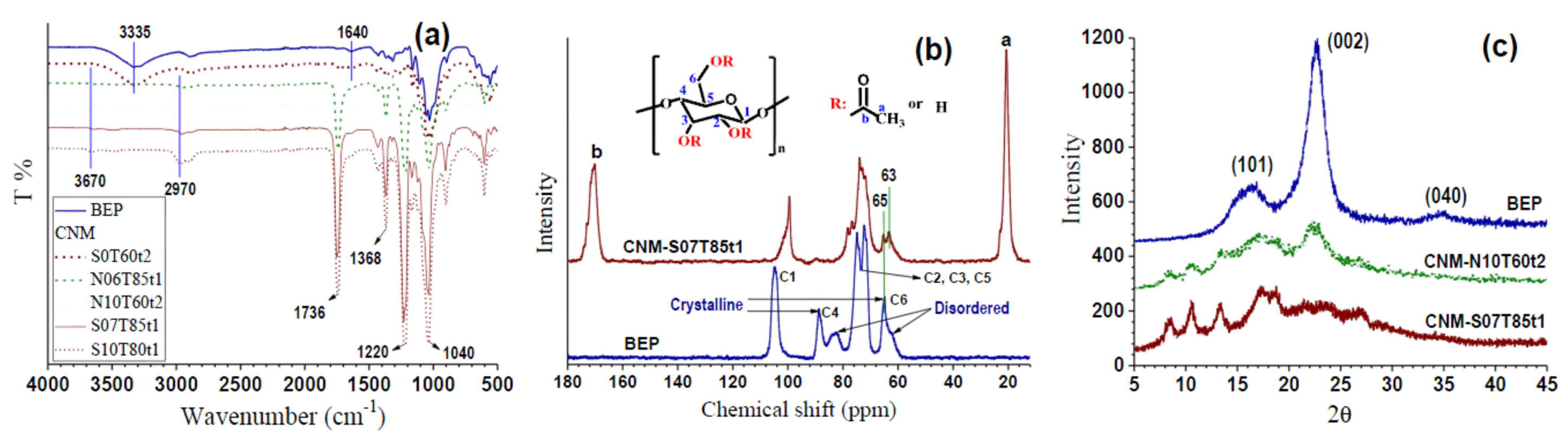
2.4. Chemical Structural Analysis
2.5. Morphology Analyses
2.6. Water Contact Angle Measurements
3. Results
3.1. Effect of Esterification Condition on Fiber DS and Morphology
3.2. Morphologies and Chemical Structures of PeM-CNPs
3.3. Degree of Substitution of PeM-CNP
3.4. Thermal and Interfacial Properties of PeM-CNP
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eichhorn, S.J.; Dufresne, A.; Aranguren, M.; Marcovich, N.E.; Capadona, J.R.; Rowan, S.J.; Weder, C.; Thielemans, W.; Roman, M.; Renneckar, S.; et al. Review: Current international research into cellulose nanofibres and nanocomposites. J. Mater. Sci. 2009, 45. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, W.; Ciesielski, P.N.; Fang, Z.; Zhu, J.Y.; Henriksson, G.; Himmel, M.E.; Hu, L. Wood-derived materials for green electronics, biological devices, and energy applications. Chem. Rev. 2016, 116, 9305–9374. [Google Scholar] [CrossRef] [PubMed]
- Carrick, C.; Wågberg, L.; Larsson, P.A. Immunoselective cellulose nanospheres: A versatile platform for nanotheranostics. ACS Macro Lett. 2014, 3, 1117–1120. [Google Scholar] [CrossRef]
- Katz, E.; Willner, I. Integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.R.; Varma, A.J. Functional nanoparticles obtained from cellulose: Engineering the shape and size of 6-carboxycellulose. Chem. Commun. 2013, 49, 8818–8820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Elder, T.J.; Pu, Y.; Ragauskas, A.J. Facile synthesis of spherical cellulose nanoparticles. Carbohydr. Polym. 2007, 69, 607–611. [Google Scholar] [CrossRef]
- Beaumont, M.; Nypelö, T.; König, J.; Zirbs, R.; Opietnik, M.; Potthast, A.; Rosenau, T. Synthesis of redispersible spherical cellulose II nanoparticles decorated with carboxylate groups. Green Chem. 2016, 18, 1465–1468. [Google Scholar]
- Nikolajski, M.; Wotschadlo, J.; Clement, J.H.; Heinze, T. Amino-Functionalized Cellulose Nanoparticles: Preparation, Characterization, and Interactions with Living Cells. Macromol. Biosci. 2012, 12, 920–925. [Google Scholar] [PubMed]
- Thorvaldsson, A.; Edvinsson, P.; Glantz, A.; Rodriguez, K.; Walkenström, P.; Gatenholm, P. Superhydrophobic behaviour of plasma modified electrospun cellulose nanofiber-coated microfibers. Cellulose 2012, 19, 1743–1748. [Google Scholar]
- Heux, L.; Chauve, G.; Bonini, C. Nonflocculating and Chiral-Nematic Self-ordering of Cellulose Microcrystals Suspensions in Nonpolar Solvents. Langmuir 2000, 16, 8210–8212. [Google Scholar]
- Petersson, L.; Kvien, I.; Oksman, K. Structure and thermal properties of poly(lactic acid)/cellulose whiskers nanocomposite materials. Compos. Sci. Technol. 2007, 67, 2535–2544. [Google Scholar] [CrossRef]
- Goussé, C.; Chanzy, H.; Excoffier, G.; Soubeyrand, L.; Fleury, E. Stable suspensions of partially silylated cellulose whiskers dispersed in organic solvents. Polymer 2002, 43, 2645–2651. [Google Scholar] [CrossRef]
- Yin, Y.; Tian, X.; Jiang, X.; Wang, H.; Gao, W. Modification of cellulose nanocrystal via SI-ATRP of styrene and the mechanism of its reinforcement of polymethylmethacrylate. Carbohydr. Polym. 2016, 142, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Berlioz, S.; Molina-Boisseau, S.; Nishiyama, Y.; Heux, L. Gas-Phase Surface Esterification of Cellulose Microfibrils and Whiskers. Biomacromolecules 2009, 10, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.X.; Chang, H.; Kumar, S.; Moon, R.J.; Youngblood, J.P. A comparative guide to controlled hydrophobization of cellulose nanocrystals via surface esterification. Cellulose 2016, 23, 1825–1846. [Google Scholar] [CrossRef]
- Sèbe, G.; Ham-Pichavant, F.; Pecastaings, G. Dispersibility and Emulsion-Stabilizing Effect of Cellulose Nanowhiskers Esterified by Vinyl Acetate and Vinyl Cinnamate. Biomacromolecules 2013, 14, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Habibi, Y. Key advances in the chemical modification of nanocelluloses. Chem. Soc. Rev. 2014, 43, 1519–1542. [Google Scholar] [CrossRef] [PubMed]
- Fahma, F.; Takemura, A.; Saito, Y. Acetylation and stepwise solvent-exchange to modify hydrophilic cellulose whiskers to polychloroprene-compatible nanofiller. Cellulose 2014, 21, 2519–2527. [Google Scholar] [CrossRef]
- Junior de Menezes, A.; Siqueira, G.; Curvelo, A.A.S.; Dufresne, A. Extrusion and characterization of functionalized cellulose whiskers reinforced polyethylene nanocomposites. Polymer 2009, 50, 4552–4563. [Google Scholar] [CrossRef]
- Kato, H.; Nakatsubo, F.; Abe, K.; Yano, H. Crosslinking via sulfur vulcanization of natural rubber and cellulose nanofibers incorporating unsaturated fatty acids. RSC Adv. 2015, 5, 29814–29819. [Google Scholar] [CrossRef]
- Tingaut, P.; Zimmermann, T.; Lopez-Suevos, F. Synthesis and Characterization of Bionanocomposites with Tunable Properties from Poly(lactic acid) and Acetylated Microfibrillated Cellulose. Biomacromolecules 2010, 11, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Uschanov, P.; Johansson, L.-S.; Maunu, S.L.; Laine, J. Heterogeneous modification of various celluloses with fatty acids. Cellulose 2011, 18, 393–404. [Google Scholar] [CrossRef]
- Vuoti, S.; Talja, R.; Johansson, L.-S.; Heikkinen, H.; Tammelin, T. Solvent impact on esterification and film formation ability of nanofibrillated cellulose. Cellulose 2013, 20, 2359–2370. [Google Scholar] [CrossRef]
- Abraham, E.; Nevo, Y.; Slattegard, R.; Attias, N.; Sharon, S.; Lapidot, S.; Shoseyov, O. Highly Hydrophobic Thermally Stable Liquid Crystalline Cellulosic Nanomaterials. ACS Sustain. Chem. Eng. 2016, 4, 1338–1346. [Google Scholar] [CrossRef]
- Espino-Pérez, E.; Domenek, S.; Belgacem, N.; Sillard, C.; Bras, J. Green Process for Chemical Functionalization of Nanocellulose with Carboxylic Acids. Biomacromolecules 2014, 15, 4551–4560. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Yu, Y.; Jiang, Z.; Zhang, L. One-pot preparation of hydrophobic cellulose nanocrystals in an ionic liquid. Cellulose 2016, 23, 1209–1219. [Google Scholar] [CrossRef]
- Braun, B.; Dorgan, J.R. Single-Step Method for the Isolation and Surface Functionalization of Cellulosic Nanowhiskers. Biomacromolecules 2009, 10, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Sehaqui, H.; Kulasinski, K.; Pfenninger, N.; Zimmermann, T.; Tingaut, P. Highly carboxylated cellulose nanofibers via succinic anhydride esterification of wheat fibers and facile mechanical disintegration. Biomacmolecules 2017, 18, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhu, J.Y.; Baez, C.; Kitin, P.; Elder, T. Highly thermal-stable and functional cellulose nanocrystals and nanofibrils produced using fully recyclable organic acids. Green Chem. 2016, 18, 3835–3843. [Google Scholar] [CrossRef]
- Bian, H.; Chen, L.; Dai, H.; Zhu, J.Y. Integrated production of lignin containing cellulose nanocrystals (LCNC) and nanofibrils (LCNF) using an easily recyclable di-carboxylic acid. Carbohydr. Polym. 2017, 167, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Huang, B.; Lu, Q.; Wang, S.; Ou, W.; Lin, W.; Chen, X. Ultrasonication-assisted manufacture of cellulose nanocrystals esterified with acetic acid. Bioresour. Technol. 2013, 127, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Sassi, J.-F.; Chanzy, H. Ultrastructural aspects of the acetylation of cellulose. Cellulose 1995, 2, 111–127. [Google Scholar] [CrossRef]
- Wang, W.; Sabo, R.C.; Mozuch, M.D.; Kersten, P.; Zhu, J.Y.; Jin, Y. Physical and mechanical properties of cellulose nanofibril films from bleached eucalyptus pulp by endoglucanase treatment and microfluidization. J. Polym. Environ. 2015, 23, 551–558. [Google Scholar] [CrossRef]
- Nutan, M.T.H. Starch Acetate as a Film Forming Excipient in Controlled Drug Delivery. Ph.D. Thesis, Texas Tech University, Lubbock, TX, USA, 2004. [Google Scholar]
- Adebajo, M.O.; Frost, R.L.; Kloprogge, J.T.; Kokot, S. Raman spectroscopic investigation of acetylation of raw cotton. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 64, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Crofton, D.J.; Moncrieff, D.; Pethrick, R.A. Dielectric studies of cellulose and its derivatives: 1. Acetylation of cellulose. Polymer 1982, 23, 1605–1608. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F. Crystallinity changes in lyocell and viscose-type fibres by caustic treatment. Eur. Polym. J. 2002, 38, 2225–2230. [Google Scholar] [CrossRef]
- Stipanovic, A.J.; Sarko, A. Packing Analysis of Carbohydrates and Polysaccharides. 6. Molecular and Crystal Structure of Regenerated Cellulose II. Macromolecules 1976, 9, 851–857. [Google Scholar] [CrossRef]
- Abraham, E.; Kam, D.; Nevo, Y.; Slattegard, R.; Rivkin, A.; Lapidot, S.; Shoseyov, O. Highly Modified Cellulose Nanocrystals and Formation of Epoxy-Nanocrystalline Cellulose (CNC) Nanocomposites. ACS Appl. Mater. Interfaces 2016, 8, 28086–28095. [Google Scholar] [CrossRef] [PubMed]
- Liitiä, T.; Maunu, S.L.; Hortling, B.; Tamminen, T.; Pekkala, O.; Varhimo, A. Cellulose crystallinity and ordering of hemicelluloses in pine and birch pulps as revealed by solid-state NMR spectroscopic methods. Cellulose 2003, 10, 307–316. [Google Scholar] [CrossRef]
- Isogai, A.; Usuda, M.; Kato, T.; Uryu, T.; Atalla, R.H. Solid-state CP/MAS carbon-13 NMR study of cellulose polymorphs. Macromolecules 1989, 22, 3168–3172. [Google Scholar] [CrossRef]
- Yamamoto, H.; Horii, F.; Hirai, A. Structural studies of bacterial cellulose through the solid-phase nitration and acetylation by CP/MAS 13C NMR spectroscopy. Cellulose 2006, 13, 327. [Google Scholar] [CrossRef]
- Takahashi, M.; Takenaka, H. Transition from Cellulose I Family to Cellulose II Family. Polym. J. 1987, 19, 855–861. [Google Scholar] [CrossRef]
- Wada, M.; Ike, M.; Tokuyasu, K. Enzymatic hydrolysis of cellulose I is greatly accelerated via its conversion to the cellulose II hydrate form. Polym. Degrad. Stab. 2010, 95, 543–548. [Google Scholar] [CrossRef]
- Dinand, E.; Vignon, M.; Chanzy, H.; Heux, L. Mercerization of primary wall cellulose and its implication for the conversion of cellulose I→cellulose II. Cellulose 2002, 9, 7–18. [Google Scholar] [CrossRef]
- Zafeiropoulos, N.E.; Vickers, P.E.; Baillie, C.A.; Watts, J.F. An experimental investigation of modified and unmodified flax fibres with XPS, ToF-SIMS and ATR-FTIR. J. Mater. Sci. 2003, 38, 3903–3914. [Google Scholar] [CrossRef]
- Dorris, G.; Gray, D. Surface analysis of paper and wood fibres by ESCA. II. Surface composition of mechanical pulps. Cellul. Chem. Technol. 1978, 12, 721–734. [Google Scholar]
- Kargarzadeh, H.; Ahmad, I.; Abdullah, I.; Dufresne, A.; Zainudin, S.Y.; Sheltami, R.M. Effects of hydrolysis conditions on the morphology, crystallinity, and thermal stability of cellulose nanocrystals extracted from kenaf bast fibers. Cellulose 2012, 19, 855–866. [Google Scholar] [CrossRef]
- Roman, M.; Winter, W.T. Effect of Sulfate Groups from Sulfuric Acid Hydrolysis on the Thermal Degradation Behavior of Bacterial Cellulose. Biomacromolecules 2004, 5, 1671–1677. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Reaction Conditions | DS of Fiber | ||
|---|---|---|---|---|
| Ac2O/HAc (mL/mL) | T (°C); Time (h) | H2SO4 (mL) | ||
| S10T80t1 | 50/50 | 80; 1 | 1.0 | 2.2 |
| S07T85t1 | 50/50 | 85; 1 | 0.7 | 1.8 |
| S05T80t2 | 50/50 | 80; 2 | 0.5 | 1.6 |
| S01T80t2 | 50/50 | 80; 2 | 0.1 | 1.2 |
| S0T60t2 | 50/50 | 60; 2 | 0 | 0.1 |
| N10T80t1 | 100/0 | 80; 1 | 1.0 | 1.5 |
| N10T60t2 | 100/0 | 60; 2 | 1.0 | 1.3 |
| N06T85t1 | 100/0 | 85; 1 | 0.6 | 1.1 |
| Sample ID | Fresh Samples | Redispersed in Ethanol | Redispersed in Water | |||
|---|---|---|---|---|---|---|
| D (nm) | PDI 1 | D (nm) | PDI 1 | D (nm) | PDI 1 | |
| S10T80t1 | 266 | 0.15 | 280 | 0.19 | 1193 | 0.36 |
| S07T85t1 | 284 | 0.10 | 326 | 0.20 | 866 | 0.33 |
| S05T80t2 | 776 | 0.32 | 698 | 0.20 | 1397 | 0.35 |
| S01T80t2 | 760 | 0.30 | 758 | 0.26 | 1048 | 0.30 |
| N10T80t1 | 352 | 0.19 | 354 | 0.20 | 666 | 0.29 |
| N10T60t2 | 432 | 0.23 | 434 | 0.24 | 1899 | 0.40 |
| N06T85t1 | 298 | 0.20 | 341 | 0.17 | 654 | 0.25 |
| Samples | C (wt %) | O (wt %) | O/C | DSCNP |
|---|---|---|---|---|
| BEP | 43.61 | 50.16 | 1.15 | - |
| S07T85t1 | 48.36 | 46.19 | 0.955 | 1.72 |
| N10T60t2 | 47.80 | 46.61 | 0.975 | 1.38 |
| Samples | Atomic Concentration (mol) | Deconvolution of the Peak C1s | DSS | CA (°) of PeM-CNP | |||||
|---|---|---|---|---|---|---|---|---|---|
| % C | % O | O/C | % C1 | % C2 | % C3 | % C4 | |||
| BEP | 57.7 | 37.9 | 0.66 | 23.0 | 59.4 | 16.7 | 0.95 | -- | 122 |
| S10T80t1 | 63.5 | 36.5 | 0.57 | 24.2 | 50.1 | 15.0 | 10.6 | 2.3 | 113 |
| S07T85t1 | 60.4 | 37.1 | 0.62 | 26.2 | 47.6 | 16.0 | 10.1 | 2.1 | 90 |
| S05T80t2 | 62.6 | 37.4 | 0.60 | 27.9 | 44.6 | 18.6 | 8.9 | 1.8 | 70 |
| S01T80t2 | 61.7 | 38.3 | 0.62 | 26.1 | 45.8 | 20.6 | 7.5 | 1.4 | 0 |
| N10T80t1 | 63.9 | 36.1 | 0.57 | 26.9 | 49.2 | 15.1 | 8.8 | 1.7 | 105 |
| N10T60t2 | 63.1 | 35.4 | 0.56 | 27.2 | 51.0 | 12.9 | 8.9 | 1.8 | 120 |
| N06T85t1 | 62.9 | 37.1 | 0.59 | 30.7 | 46.0 | 15.3 | 8.0 | 1.5 | 90 |
| Tonset | Weight Loss at Tonset (%) | Tmax | T95 | |
|---|---|---|---|---|
| BEP | 311 | 7.7 | 390.8 | 328.7 |
| S07T85t1 | 316 | 4.5 | 304.3; 380.6 | 320.3 |
| S05T80t2 | 316 | 9.5 | 304.8; 382.7 | 258.9 |
| N06T85t1 | 252 | 4.8 | 378.4 | 253.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.; Ma, Q.; Su, Q.; Bian, H.; Zhu, J.Y. Facile Synthesis of Highly Hydrophobic Cellulose Nanoparticles through Post-Esterification Microfluidization. Fibers 2018, 6, 22. https://doi.org/10.3390/fib6020022
Lin C, Ma Q, Su Q, Bian H, Zhu JY. Facile Synthesis of Highly Hydrophobic Cellulose Nanoparticles through Post-Esterification Microfluidization. Fibers. 2018; 6(2):22. https://doi.org/10.3390/fib6020022
Chicago/Turabian StyleLin, Chunxiang, Qianli Ma, Qiaoquan Su, Huiyang Bian, and J. Y. Zhu. 2018. "Facile Synthesis of Highly Hydrophobic Cellulose Nanoparticles through Post-Esterification Microfluidization" Fibers 6, no. 2: 22. https://doi.org/10.3390/fib6020022
APA StyleLin, C., Ma, Q., Su, Q., Bian, H., & Zhu, J. Y. (2018). Facile Synthesis of Highly Hydrophobic Cellulose Nanoparticles through Post-Esterification Microfluidization. Fibers, 6(2), 22. https://doi.org/10.3390/fib6020022