
Glass Fibre Strength—A Review with Relation to Composite Recycling


Department of Mechanical and Aerospace Engineering, University of Strathclyde, 75 Montrose Street, Glasgow G1 1XJ, UK
*
Author to whom correspondence should be addressed.
Fibers 2016, 4(2), 18; https://doi.org/10.3390/fib4020018
Submission received: 5 February 2016
/
Accepted: 8 May 2016
/
Published: 26 May 2016
(This article belongs to the Special Issue Glass Fibers)
Abstract
:The recovery and reuse of glass fibres from manufacturing waste and end-of-life composites in an environmentally-friendly, cost-effective manner is one of the most important challenges facing the thermosetting polymer composites industry. A number of processes for recycling fibres from such materials are available or under development. However, nearly all options deliver recycled glass fibres that are not cost-performance competitive due to the huge drop in strength of recycled glass fibre compared to its original state. A breakthrough in the regeneration of recycled glass fibre performance has the potential to totally transform the economics of recycling such composites. This paper reviews the available knowledge of the thermally-induced strength loss in glass fibres, discusses some of the phenomena that are potentially related and presents the status of research into processes to regenerate the strength and value of such weak recycled glass fibres.
1. Introduction
The disposal of composite manufacturing waste and end-of-life composite products in an environmentally-friendly manner is one of the most important challenges currently facing the industrial and academic composites community. The 2015 global production of fibre-reinforced composite materials significantly exceeded 10 million tons. Glass fibre (GF) is the reinforcement of choice in more than 90% of all of these composites, and more than five million tons of reinforcement grade GF was consumed in 2015 [1]. In fact, some available data indicate that E-glass consumption could already be as high as seven million tons when yarns are included [2]. Associated with this global GF consumption is the production of 0.5–1 million tons of GF manufacturing waste [3], most of which is landfilled. Furthermore, approximately 70% of reinforcement GF is used to manufacture thermoset-based Glass Reinforced Polymer composites (GRP), which also produces approximately 15% manufacturing waste [3]. Such GRP materials (both end-of-life and manufacturing waste) are difficult to recycle in an efficient manner and have historically also been disposed of in landfills. Such landfilling is rapidly becoming untenable due to legislative and landfill pricing developments. The perspectives on this issue have been recently highlighted due to the accelerating growth in the use of such composite materials in transportation and wind energy sectors. This increasing awareness of this issue is also reflected in the increasing number of composite recycling review articles appearing in the technical literature [4,5,6,7,8,9,10,11,12,13,14,15,16]. A number of processes are available for recycling composites, and thermal recycling is probably the most technologically advanced [4,7]. However, nearly all options deliver recycled glass fibres (RGF) that suffer from a lack of performance and cost competitiveness with pristine first-pass materials.
A critical technical challenge in the development of GRP recycling technology is the 80%–90% drop in the performance (and value) of RGF in comparison to its original state (see Figure 1). Consequently, RGF have a very poor performance to cost ratio and in most cases are unsuitable for reprocessing and reuse as a valuable reinforcement for composites. A breakthrough in this field could enable such RGF to compete with pristine materials in many large volume discontinuous fibre-reinforced composite applications. The development of an economically-viable process for regenerating the properties of thermally-recycled glass fibres would have major technological, societal, economic and environmental impacts. A recent analysis by Thomason et al. indicated that there is an ongoing situation where up to 50% of the global demand for GF reinforcements could potentially be supplied by RGF from end-of-life and manufacturing waste recycling [17]. The reuse of these materials could result in a huge reduction in the environmental impact of the glass-fibre and composites industry where the replacement of 50% of current GF products by RGF products would equate to a global reduction in CO2 production of two million tons per annum from reduced melting energy requirements alone. Furthermore, such a development would also vastly reduce the landfill requirements of the GF and GRP industry. This would clearly be in line with the growing societal and environmental pressure to reduce landfill disposal, increase the reuse of valuable raw materials and reduce CO2 release to the atmosphere.
In this article, we review the available literature addressing the strength loss of GF after thermal conditioning in the temperature range (below 700 °C) that must be employed in a composite recycling process if the physical form of the GF is to be retained, such that it can be reused as a reinforcement fibre. The review addresses GF strength loss, related physical and chemical changes in the GF microstructure, the role of water and sizing and research on the regeneration of RGF strength.
2. Strength of Glass Fibre
2.1. Flaws
The theoretical maximum strength of glass has been calculated by various researchers, as summarised by Sugarman [18], to within a range between 10 and 30 GPa. Experimentally-measured values do not approach this range even when dealing with strong glass fibres. Despite the long history of GF research and development, a full fundamental understanding of the strength performance of GF still eludes us. When discussing the strength of GF, one must primarily consider the effects of flaws, and it is important to make the distinction between intrinsic and extrinsic flaws (and strengths) [19]. Extrinsic strength is controlled by the presence of flaws and their severity. This general term “flaws” may refer to numerous features, such as surface scratches and cracks, devitrified regions or unintended inclusions in the bulk material. Unlike intrinsic strength, it is therefore dependent on both the diameter and length of a tested fibre sample. Intrinsic flaws are regarded as structural defects that result from thermal fluctuation [20]. It is known that the extrinsic flaws determine glass strength, but the effects of the intrinsic flaws on the glass strength are still unclear. In addition to intrinsic and extrinsic strengths, the difference between GF inert and fatigue strength must also be addressed. Gupta [19] describes inert strength as that measured in the absence of fatigue, for instance by testing at very low temperature (−196 °C), where the rate of the fatigue reaction may be neglected. Conversely, fatigue strength is measured at a higher temperature (room temperature for example) and at some known level of humidity. A constant and moderate strain rate should be used. For composite reinforcement purposes, the term glass fibre strength commonly refers to the extrinsic fatigue strength.
Hence, flaws, and more specifically, cracks, are features of key importance in the discussion of extrinsic failure of GF. A theoretical crack is a two-dimensional flaw across whose boundary the atomic bonds of the material are broken. Around this flaw, stress will be concentrated at the crack tip, which is assumed to be infinitely sharp (although in reality, it must have atomistic dimensions). Cracks are considered the most important type of flaw, as they can only grow under the application of tensile stress [19]. However, when considering a highly brittle material, like glass, where no crack tip blunting occurs, the issue with an infinitely sharp crack tip is that any externally-applied stress immediately translates to an infinitely concentrated stress at the crack tip. In reality, this makes no sense, as it would suggest the material would fail almost immediately under any load. The seminal work of Griffith [21] addressed this mathematical issue by considering the problem using an energy balance approach to obtain an equation relating fibre strength (σf), modulus (E), surface energy (γ) and the maximum dimension (a) of a surface flaw.
Although deceptively simple, this equation is generally accepted to describe the brittle failure behaviour of glass. In materials with greater ductility, a proportion of the released strain energy is dissipated around the crack tip due to plastic flow in the material. A further mathematical modification to this equation comes in the form of the Griffith–Orowan–Irwin equation:
This equation may be used to calculate the dimension of some surface or volume flaw with a circular or elliptical shape. However, proper use of the equation requires knowledge of the general dimensions of this flaw, so that the correct value of the geometric constant Y is utilised. Some values of parameter Y may be obtained in ASTM C1322-05b [22].
It is worthy of comment that, despite the extensive discussion in the literature of their role in determining glass fibre strength, these micro- or nano-dimensioned flaws have never been visualised in a glass fibre prior to fracture. This is an area where much work still remains to be done to confirm the physical existence of these strength-defining flaws.
2.2. Strength Loss of Heat-Treated Glass Fibre
2.2.1. Temperature and Time Effects
Investigations into the combined effect of temperature and time on the room temperature strength of GF began a number of decades ago. Sakka [23] was one of the first to report results on a laboratory-produced fibre with high silicon, boron, sodium and potassium oxides content. Fibres were heated for one hour, cooled in air to room temperature and then tensile tested. The results showed that for this glass composition, thermal conditioning at temperatures in the range of 200–650 °C led to a reduction in fibre strength whose magnitude was linearly dependent on the conditioning temperature. Thomas [24] took a systematic approach to both the manufacture and testing of E-glass fibres. In one experiment, he measured the effect of a 4-h thermal conditioning procedure at a selection of temperatures between 100 and 600 °C. A fairly consistent decrease in strength was shown, with a suggestion that the slope becomes steeper beyond a conditioning temperature of 300 °C. Compared to the strength of virgin fibre, a 65% loss in fibre strength was found when heat treating above 400 °C. Additionally, it was observed that strength loss proceeds with the length of the heat conditioning time until a constant minimum value is reached. At higher conditioning temperatures, this minimum value is reached within a very short time. Small sample sizes in these experiments prevented statistically-significant conclusions from being drawn, but these trends were further researched by others [25,26,27,28,29].
Cameron investigated E-glass fibres and presented data showing the strength of heat-treated fibres asymptotically approaching a minimum value with time [25]. Similar to the results shown by Thomas, this minimum value of retained strength also decreased with increasing treatment temperature. The maximum strength loss was 60% after 65 h of conditioning at 273 °C, and the strength loss after 4 h of conditioning was approximately 40%, which was also very similar to that reported by Thomas [24]. In further work [26] Cameron showed that when working with commercially-produced E-glass fibres (rather than laboratory-spun pristine fibre), the effect of thermal conditioning on room temperature strength was equally significant. Using heating times of 40–60 min, he measured a 50% drop in strength after conditioning at 300 °C. Other early research, while not directly comparable to that mentioned above, added to the body of evidence regarding time and temperature effects. Brearley and Holloway [27] studied commercial soda-lime-silica 3 mm-diameter rods. Their results showed a steep initial decrease in strength with heat treatment (for 1 h) levelling out above 200 °C and reaching a maximum loss of 70% at 530 °C. Piggott and Yokom [28] studied laboratory-produced 50 micron-diameter silica fibres and found a linear strength decrease with increasing conditioning temperature (of 1 h) with a maximum loss of 65% at 600 °C. Similarly, the work of Aslanova [29] showed the weakening effect of conditioning time and temperatures of up to 600 °C on several types of GF.
More recently, Dorzhiev et al. [30] conducted heat treatment of high-strength magnesium aluminosilicate fibre for 5 h in the temperature range 100–700 °C. Upon tensile testing, they found very little loss of strength in samples treated at 350 °C and below. However, above this temperature, some “threshold” was crossed, and a rapid deterioration in strength was observed, progressing to a maximum loss of 75% of the initial untreated value. Similar experimental data on E-glass fibre have also been reported by Feih et al. [31], who observed coupled temperature and time trends in E-glass fibre strength loss very similar to those produced by Thomas [24]. The differences in material used between these two investigations are, however, significant. Although both E-glass, in Thomas’ research, the fibres were essentially pristine and uncoated, as laboratory-drawn single fibres, and exhibited an unconditioned strength of approximately 3.7 GPa, typical for pristine single E-glass fibres. In the investigation by Feih et al., the samples were commercial 300 tex fibre rovings containing fibres coated with an industrial silane-based sizing and a much lower average single fibre strength of 2.3 GPa, which is a typical value for single fibres extracted from commercially-produced rovings.
Lund and Yue also reported results on [32] laboratory-drawn single E-glass fibres that had not been sized. The pristine nature of these samples meant that the room temperature strength of untreated fibre was very high at approximately 3 GPa. Interestingly and despite performing long 3-h heat treatments, no significant strength loss was reported until a temperature in excess of 300 °C was used. Rigorous explanations for the physical change(s) in GF that can account for the strength loss measured are often absent. The same might be said about this work, but some interesting observations may be made based on the measurements of enthalpy and anisotropy relaxation that are reported alongside the tensile strength data. Firstly, the critical temperature above which strength loss and relaxation of enthalpy both begin to occur is around 300 °C. Secondly, some links might be also drawn between heat-treated fibre strength and anisotropy relaxation. The same E-glass formulation was used to produce both standard drawn fibres and a spun wool fibre (SWF), the production of which is described in [33]: without fully detailing the SWF production process, it is important to note that it is less ordered than fibre drawing, which could allow greater fibre surface damage to occur. These E-glass spun wool fibres produced with minimal axial stress, hence very low anisotropy, had a measured tensile strength of approximately 1.5 GPa. The anisotropy of continuous E-glass fibres was found to decay to approaching zero after treating at 500 °C for 3 h; this treatment also caused a decrease in strength from around 3 down to 1.5 GPa. Taken individually, either of these correlations could be used to postulate that either: strength loss is related to enthalpy relaxation or strength loss is related to anisotropy. The authors themselves do not claim causation with respect to either of these observations of fibre relaxation. Heat-treated fibres are still brittle materials, and as such, their fracture is tied to the existence of critical surface flaws or cracks. Whether relaxation of either enthalpy or anisotropy could affect this fracture behaviour is not understood.
In a recent paper from researchers at Owens Corning, the effect of thermal condition on the room temperature strength of a range of fibre reinforcements was examined. Korwin-Edison et al. [34] studied the room temperature strength of S-glass fibres, E-glass fibres, silica fibres and basalt fibres, after 1 h of heat treatment in air over a range of temperatures of 100–800 °C. All fibres exhibited a strength decrease with temperature treatments above 200 °C. A direct comparison of the results after 650 °C heat conditioning showed that the S-glass fibres had the highest absolute strength and that the basalt fibres had the poorest level of strength retention. However, when compared to the original values, all fibres showed a strength loss of 70%–80% after treatment at 650 °C, although the basalt fibre was still ranked lowest with a 97% relative strength loss. Militly et al. also reported loss in the strength of basalt fibres after a 1-h thermal conditioning in air [35]. At conditioning temperatures up to 200 °C, the fibre strength remained approximately constant; however, when heated above 200 °C, the room temperature strength of the fibres dropped precipitously with a 90% reduction in strength after treatment at 500 °C. Jenkins et al. compared the thermally-induced fibre strength loss of commercial, epoxy-compatible, basalt and boron-free E-glass fibres [36]. Both fibre types exhibited a threshold value for strength loss of approximately 300 °C. Above this temperature, both fibre types also exhibited an approximately linear strength loss with temperature. After conditioning at 600 °C (for 25 min), the basalt had lost 80% of its original strength compared to the boron-free E-glass, which lost 65%. Sabet et al. studied the effect of thermal conditioning on the bundle strength and single fibre strength of basalt fibres [37]. Heat treatment of the basalt rovings was carried out over a 300–500 °C temperature range for times from 5 to 20 min. Significant strength loss was recorded at all conditioning temperatures, with the magnitude of the loss increasing with increasing conditioning temperature. Furthermore, the basalt single fibre strength loss was relatively constant for 5–15 min treatment times, but increased significantly when the time was increased to 20 min. Interestingly, the strength dependence on treatment temperature exhibited a threshold behaviour for 5-min treatments, but was an approximately linear relationship for 20-min treatments. The basalt fibres exhibited a strength loss of 90% after 20 min at 500 °C.
Figure 1 and Table 1 show an overview of data selected from some of the papers reviewed here and illustrate the level of the loss in room temperature fibre strength as a function of heat conditioning temperature. Figure 1 illustrates the typical steep loss in the strength as a function of conditioning temperature for E-glass and basalt fibres. Table 1 shows the maximum fibre strength loss reported for a wide range of different silica-based fibres. The scale of the challenge for the reuse of recycled reinforcement fibres is clearly illustrated by the data in Figure 1 and Table 1.
2.2.2. The Effect of Heating Atmosphere
The effect of the atmosphere in which the fibre heat conditioning occurs has also been considered as a variable, and this was studied in detail by Cameron during the 1960s. Some of his early thesis work [38] tentatively suggested that no difference in retained strength of E-glass occurred when heating in either air or argon. However, he later reported data comparing heating in an oven with vents closed (to air particle contamination) or open [25]. Some of these results appeared to show that after lengthy heating times (>10 h), more strength was retained in fibres heated in the “vents closed” furnace. A possible explanation of dust particle contamination being a strength-reducing factor, also postulated by Brearley and Holloway [27], was later shown to be unlikely by Cameron himself [25] when he performed shorter duration heat treatments on E-glass under either laboratory air or argon with a very high purity (and exceptional dryness). Up to a temperature of 300 °C, no significant differences in retained strength were measured between the two atmospheres.
More recent investigation of the heating atmosphere effect has generally yielded similar negative conclusions. In work on spun basaltic wool fibres (discontinuous-at-formation vitreous fibre), Lund and Yue [32] found no difference in the retained strength of fibres thermally conditioned for 3 h in either air or nitrogen. Similarly, Feih et al. [31], in work using silane-coated E-glass, reported no difference between treatment in ambient air or nitrogen in most cases. However, when thermally conditioning fibre at 450 °C for only 30 min, they observed greater strength retention when using a nitrogen atmosphere compared to either dry or ambient air. It appears from this body of research that, for uncoated fibres, neither dust contamination nor atmospheric moisture attack during thermal conditioning can provide an explanation to the strength loss measured in GF. What is less certain is what may occur when the normal commercial organic surface coatings have been applied to the fibres. The data of Feih et al. [31] suggest that, over the shorter time-scales of interest when considering recycling applications, an unreactive nitrogen atmosphere may lead to greater fibre strength retention. One of the possible mechanisms suggested is a reduction of the degradation of the organic sizing material, and indeed in the same work, a thermo-gravimetric analysis (TGA) comparison using both nitrogen and air shows a small, but measureable difference, with less mass lost at above 300 °C under the nitrogen atmosphere.
2.2.3. The Effect of Heat Treatment under Stress
Bartenev and Motorina [43] published some early results about the effect of applied stress on fine glass fibres of both alkaline and non-alkaline compositions during thermal conditioning. Of particular interest are the outcomes of the tests on the alkaline fibres as these were benchmarked against values for fibres that were thermally conditioned in a stress-free state. With a fairly low initial strength of little more than 1 GPa, the authors presented a roughly linear decrease in fibre strength with stress-free heating, reaching a maximum 70% loss after conditioning at 500 °C. On the other hand, fibres heated under stress showed improved strength retention. With an application of a load equalling 2% of the average ultimate fibre strength, a moderately higher strength was obtained throughout the temperature range. Application of 70% of ultimate pre-stress produced significantly improved results with only minor strength loss at 400 °C and up to a 15% strength increase above the initial non-heat treated value in the temperature range 100–350 °C.
Cameron also conducted some experiments on the heating of tensioned fibres working with largely flaw-free E-glass [25]. This difference in fibre condition, pristine versus the aged fibres of Bartenev and Motorina, has a significant effect on the strength values obtained. Cameron thermally conditioned his fibres for 2 h, either stress-free or under a pre-load, which he estimated to equal between 2% and 20% of the fibre room temperature strength of around 3.8 GPa. All heat treatment led to a decrease in retained room temperature strength. However, the conditioning temperature above which significant strength loss began was approximately 250 °C for the stressed fibres, but as low as 150 °C for the fibres conditioned stress-free. Furthermore, the magnitude of the maximum fibre strength loss at 450 °C was smaller (37%) for the stressed fibres than for the stress-free condition fibres (54%). Consequently, the tensioned fibres retained significantly more of their strength during the thermal conditioning. In the discussion of the results of both of these papers, the reduced weakening effect was attributed to alterations to the geometry around cracks or flaws on the fibre surface. Bartenev and Motorina discussed elastic and plastic deformation, and Cameron “inelastic flow”, of material in the vicinity of cracks. In both cases, they assume that this stress-forced flow leads to a less critical crack geometry, and hence, reduced stress concentrations develop when tensile testing is conducted.
More recently, Lezzi et al. [44] reported on work using silica glass fibre, of an approximate diameter of 125 µm. Single fibres were loaded in tension at stresses of up to 60% of the breaking stress and treated for 60 s with a hot gas stream at temperatures between 100 and 500 °C while under tension. After heat treatment, the fibres were unloaded and tensile tested at room temperature and humidity. They reported that fibres heat treated while under tension retained more strength than those treated in a stress-free state. The degree of improvement of strength retention was related to the applied stress during the heat treatment. For example, after 60 s at 500 °C in the stress-free state, the fibre strength was approximately 1.3 GPa, but when a 1-, 2- or 3-GPa applied stress was applied, the retained strengths were 1.8, 2.5 and 3.5 GPa, respectively. The explanation given for this phenomenon was that a thin residual compressive stress layer forms on the surface of fibres when they are heated under tension while exposed to water vapour. The investigation was also extended to 100 µm-diameter E-glass fibres [45], which were conditioned under stress and humidity for various times. Stress was induced by placing fibres in a two-point bending configuration inside silica tubes, and testing was correspondingly carried out using a two-point bending method. Holding fibres under bending loads of 1 and 2 GPa led to an increase in the failure strain of fibres beyond that of the as-received value. Again, the explanation given for this observed strength increase was surface residual stress formation by surface stress relaxation.
The full implications that this work may have for RGF obtained from composite recycling are open to discussion. These experiments were all carried out using unsized fibres, whereas in practical applications, it is almost certain that some silane-based sizing will have been applied to the fibre surface. Although an accepted plausible hypothesis, it is not absolutely certain that physical cracks or flaws in the GF surface are always the source of critical stress concentrations leading to the failure of thermally-conditioned GF. For example, agglomerations of sizing or matrix material or foreign particles that have become bonded to the fibre surface could act as sufficient stress raisers to cause fibre failure. Whether or not this further level of complexity regarding the surface coating would affect the ameliorating conditioning-under-tension effect described is unknown at this time.
2.3. Physical Changes Following Heat Treatment
The precise mechanism, or mechanisms, that contribute to the strength loss of heat-treated GF are not yet fully established in the literature. There are, however, a number of changes that occur during thermal conditioning that have been reported. In some cases, these phenomena are well understood, and in others, observations are reported, but the discussion and hypotheses presented may still require further verification.
2.3.1. Thermal Compaction or Enthalpy Relaxation
Physical changes occurring in heat-treated GF tend to be separated into two discussions; either of surface or bulk phenomena. One phenomenon that may be considered a bulk change is thermal compaction. A volume of early work, concerning bulk changes in E-glass fibre, was conducted by Otto [46]. The phenomenon “thermal compaction” was first noted by Otto and Preston [47] in 1950. Later, using very fine fibres (less than 10 µm), Otto applied various heating regimes and measured changes in the properties. The main conclusion was that the previously-observed densification process, which he called thermal compaction, occurred when heating was carried out using suitably high temperatures or long time scales. As might be expected, thermal expansion occurred at lower temperatures (between room temperature and 300 °C); however, beyond this, the phenomenon of thermal compaction dominated over expansion. Subsequently holding the fibre at around 500 °C caused a contraction to below the starting fibre length. Furthermore, isothermal heating temperatures as low as 300 °C led to measureable contraction in fibre length given sufficient time; the process occurring much faster at higher temperatures. Alongside this compaction, Otto also showed other bulk changes, such as an increase in density and, indirectly, in Young’s modulus. These property changes also related directly to heating temperature. Although other experimental results were not presented, the author noted that the phenomenon was not unique to this glass formulation, and similar exploratory results were obtained for other glasses.
Aslanova et al. [48] published experimental data on GF of numerous compositions and at many diameters, which were tested using a low frequency torsion pendulum under vacuum and over a range of temperatures. Changes with temperature in both internal friction and shear modulus were reported, and numerous explanations were suggested for various observed effects. Particularly interesting comments on the behaviour of an aluminoborosilicate glass were made: with increasing temperature, its relative shear modulus initially decreased, but at around 300 °C, a turning point occurred, and it started to increase again. Similarly to Otto [46], they explained this effect as being due to compaction of the glass structure. They further stated that this can occur because of the relaxation of deformations within the fibre structure that are formed during drawing. Results showing contraction of fibres in both the longitudinal and radial directions have also been reported [49].
In some more recent studies [32,50,51,52,53], the relaxation of GF was studied using differential scanning calorimetry (DSC). The release of energy from fibre samples during temperature scans was measured over a wide range of times and temperatures. Different types of fibre were analysed within these various works, including a boron-containing E-glass composition. Results for these, as well as calcium metaphosphate and basaltic fibres demonstrated that heating below the glass transition temperature (Tg) allows the release of what the authors term excess enthalpy from the fibres. The phenomenon is therefore called enthalpy relaxation. It was demonstrated [52] that the enthalpy relaxation is related to the quenching of the glass during its production; a larger amount of energy (excess enthalpy) is stored within the glass structure when quenching is performed at a higher cooling rate. The explanation offered for this phenomenon is a “cooperative rearrangement of the frozen-in isotropic network”. With respect to E-glass, this rearrangement occurs only once the heat treatment temperature exceeds around 300 °C and its rate increases significantly at higher temperatures.
Recent results published by Yang and Thomason [54] provided further evidence of this fibre structural relaxation phenomenon by investigation of the thermo-mechanical properties of single boron-free E-glass fibres. Using a thermo-mechanical analyser, fibre length changes were monitored, and in situ measurement of the fibres Young’s modulus was performed. The results agree with those discussed above [32,46,50]. A small contraction in fibre length was found at an isothermal treatment of 300 °C, but the effect was far more significant at higher temperatures (400–500 °C). Furthermore, the room temperature Young’s modulus of fibres was tested after isothermal heat treatments; an increase of almost 20% compared to the original value was measured when treating to 600 °C. Once again, this effect initiated when a treatment temperature of approximately 300 °C or greater was used.
From the results of Otto [46] and Yang and Thomason [54], it is quite clear that thermal compaction of GF occurs when they are heated above approximately 300 °C, and this effect correlated with a corresponding increase in the room temperature Young’s modulus of the fibre. The temperature range over which this phenomenon acts corresponds closely with that over which enthalpy relaxation of GF by DSC has been shown in numerous works [32,50,51,52,53]. It seems reasonable to conclude that these works demonstrate related phenomena; the conclusions that long-range relaxation or reorganisation of the glass network is responsible appear plausible. Although this is the case, an explanation of exactly what is happening within the fibre structure during the process has yet to be reported. A link between thermal compaction, enthalpy relaxation of fibres and the decrease in fibre tensile strength of heat-treated fibre is also as yet unreported.
2.3.2. Anisotropy (Birefringence) Relaxation
A reasonable volume of evidence, largely collected using mechanical methods, suggests that the structure of E-glass fibres is isotropic. There is, however, a separate discussion regarding the axial anisotropy imparted to GF due to straining during the fibre drawing process [54], which results in optical birefringence. In many of the studies that have reported measurements of the birefringence of GF, experiments were also conducted to analyse the effect of heat treatment [32,49,50,55]. Lu et al. [55] suggested that anisotropy relaxation was temperature dependent; complete relaxation occurred if samples were heated to Tg, but lower temperature annealing led to incomplete relaxation. Ya et al. [50] carried out birefringence measurements on E-glass after heat treating for various times over the range 300–550 °C. It was shown that a relatively low temperature treatment of 300 °C for 3 h was sufficient to reduce the measured anisotropy by approximately 30%. Full anisotropy relaxation was achieved when heating at 550 °C for 3 h or at a lower temperature of 400 °C for around 10 h. Lund and Yue [32] reported further experiments using an identical glass composition. Their results showed some anisotropy relaxation for a 3-h treatment at temperatures as low as 200 °C, and 100% anisotropy relaxation occurred well below 500 °C. The differences between the two works may well be explained by the differences in fibre drawing temperature.
2.3.3. Crystallisation
Crystallisation of GF has been proposed as the source of strength loss following heat treatment. Aslanova [29] reported on an electronographic study conducted using a non-alkaline GF. In its standard form, electronograms gave a weakly diffracted picture, but after heating to 400–600 °C, rings or points appeared in the pictures obtained. These were attributed to the growth of a modified form of cristobalite. The levels of GF crystallisation reported on in this work do not seem to have been replicated since. However, a recent study of stone wool fibres [56] has shown evidence of a surface nano-crystallisation phenomenon involving periclase (MgO) crystals. Whether these findings have relevance to the thermal recycling of E-glass fibres is uncertain. There are reasons to suppose that this may not be the case. Firstly, nano-crystallisation was only observed after treatment close to the Tg. Secondly, and more crucially, the mechanism by which the process occurred relates to the high iron oxide content of stone wool fibres. In these fibres, oxidisation of ferrous to ferric ions and the movement of electron holes caused a movement of divalent MgO towards the fibre surface. As E-glass contains only trace quantities of iron oxide and small amounts of MgO, the occurrence of the same process would seem improbable. Crystallisation of other (non-reinforcement grade) glass systems has also been demonstrated [57,58], but once again, these findings followed heating of the samples for relatively long periods of around a day. Potential crystallisation of the E-glass system as an explanation of heat-treated fibre strength loss remains relatively unexplored.
2.3.4. Ion Exchange
Gy [59] and Varshneya [60] discussed ion exchange as a method by which glasses may be strengthened, in a similar way to tempering. However, unlike tempering, which is applicable to massive glass with large internal stresses and potentially flaws on the millimetre scale, ion exchange can potentially deliver a strengthening effect to fine glass fibres. When considering the movement of ions through the glass network, one must have an idea of the appearance of the network structure and its chemical makeup. In general terms, by its nature, the structure of E-glass is vitreous and disordered, and the main component is silicon dioxide. However, there are also significant concentrations of compounds whose metallic ions have valences of two or three [61]. In the glass network, the Si-O bonds have the highest bond energy; hence, they are the strongest. Kistler [62] suggested that the bonding of trivalent atoms, such as aluminium, will be less strong, but that those ions that are di- or mono-valent are the most likely to be able diffuse through the glass network. The findings of Yue et al. [56] regarding magnesium oxide migration in stone wool fibres would appear to be concurrent with this. Changes in surface concentrations of Mg, Al and Si following sub-Tg treatments of magnesium aluminosilicate fibres have been reported [30]. Some evidence suggests that a change in surface concentration of Ca occurs when E-glass fibre is heated close to its Tg. Nichols et al. investigated the effect of heat treating E-glass fibre samples for 3 h at temperatures up to 720 °C. Using X-ray photoelectron spectroscopy (XPS), they found that the surface concentration of Ca increased throughout, suggesting that diffusion to the surface occurred during thermal treatment [63].
In the context of this work the source of potential allowing diffusion is heat. In that case, the critical temperature is the annealing temperature (Tann), above which tempering of massive glass to remove internal stresses is carried out. Molecular diffusion may occur below this temperature assuming the straining temperature (Tstrain) at least has been surpassed, but the time taken will be greater. For boron-free E-glass, these two temperatures can be given as approximately Tann = 740 °C and Tstrain = 690 °C [61]. Pugh et al. [64] reported that only monovalent alkali metal ions (K and Na) possessed any significant mobility through silica-based optical fibres that were held at high temperatures for relatively long lengths of time. These heat treatments were far longer than both those typically employed in any composite recycling process and the heat treatments used when studying E-glass fibre strength. The case for ion migration in glass is clear; however, it appears to be limited primarily to monovalent ions, and long heating times at temperatures approaching Tg appear to be required. In the context of the strength loss of E-glass fibre, with its very low content of monovalent ions, diffusion would seem a less likely candidate to play a significant role.
The surface of E-glass has been studied by numerous researchers, and in a few studies, it has been shown that there are differences between the surface and bulk of GF in terms of composition at an atomic level. Wong [65] used Auger electron spectroscopy (AES) to compare the elemental compositions of E-glass fibre surfaces with the bulk (obtained by analysis of fracture surfaces). His results suggested that the fibre surface is rich in silicon, a result also reported by Thomason and Dwight [66]. Wong also reported an enrichment of aluminium at the surface, but to a lesser extent compared to silicon. Conversely, the surface of the fibres studied appeared to have a significantly lower concentration of calcium and magnesium compared to the fibre bulk. The conclusion of changes in the atomic composition of the surface of E-glass fibres was also reported by Liu et al. [67]. Using XPS, they measured the surface and bulk composition of E-glass and reported calcium and magnesium depletion and aluminium enrichment in the fibre surface as compared to the bulk value. Similar results, of a silicon- and oxygen-rich and calcium-depleted surface, have also been reported by Wang et al. [68], but using thin glass slides of E-glass composition rather than fibres directly. The results of Wesson et al. [69] conflict somewhat with the aforementioned studies, as they suggest an E-glass fibre surface that has a lower concentration of oxygen than the bulk and is comparatively enriched with calcium and aluminium. Nonetheless, the suggested aluminium surface enrichment is in agreement with the finding by Wong and Liu [65,67].
3. The Role of Water and Hydroxyls
3.1. Hydroxyl Groups
In reviewing the strength on bare (unsized) glass fibre, it is of utmost importance to discuss the fibre surface, as this is considered to be directly linked to fracture behaviour [21]. Some research into the surface of E-glass has been published, but a greater volume on pure silica exists in the literature. This is a useful place to begin the discussion, as the complexities introduced by adding or altering even one component in a silica or glass system can significantly alter its state and complicate analysis of the surface [70,71]. The study of pure silica systems began many decades ago [72] and has continued since [71,73,74].
Unless it has been treated under certain specific conditions, the surface of silica can be assumed to be covered to some extent with hydroxyl (OH) groups; when these hydroxyls are bonded to a silicon atom, they are known as silanols. These silanols exist in one of two states: they are either free (isolated) or they are hydrogen bonded with a neighbouring silanol [72]. The overall coverage of hydroxyl groups on a surface is expressed by the hydroxyl number (αOH). This value was calculated for many types of silica with a range of specific surfaces by Zhuravlev [75]; for a fully-hydroxylated silica, he produced the so-called Kiselev–Zhuravlev constant, αOH = 4.6 nm−2. A similar value of between 4.2 and 5.4 nm−2 was also reported by Bakaev and Pantano [74] using a hydrogen/deuterium exchange method. The Kiselev–Zhuravlev constant appears to be generally accepted; however, differing values for fully-hydroxylated silica have been reported; for example, αOH = 1.3 nm−2 [71] and αOH = 8.2–9.8 nm−2 [73]. Significant differences in the materials and methods for which these values were obtained may explain the disagreement in αOH values.
The silanol coverage on a silica surface has been shown to be changed by the application of heat and/or vacuum treatments. Zhuravlev [75] presented perhaps the most extensive data regarding dehydration of physically-adsorbed water, the dehydroxylation of surface silanols and the rehydroxylation of heat-treated samples. Mass thermograms of a standard silica were presented, showing that the intensity of water evolved against sample temperature using a heating rate of approximately 5 °C/min. Prior to the measurement, however, samples were pre-treated simultaneously under vacuum and at various temperatures. A key temperature of 200 °C was identified with respect to surface water; after vacuum treatment below 200 °C, physically-adsorbed water remained on the silica surface. However, following treatment at 200 °C or above, the surface was fully dehydrated. Further heating led to further evolution of water, due to dehydroxylation of the surface. Two regions of dehydroxylation were found, one between approximately 200 and 450 °C and the second above 450 °C. The lower range was attributed to the removal of hydrogen bonded hydroxyls, the coverage of which halved by 450 °C. Above 450 °C, only free hydroxyls remained; a temperature in excess of 1000 °C was required to fully dehydroxylate the surface.
E-glass, like silica, is considered to be an amorphous material whose surface is populated with hydroxyl groups. However, differences between the surface hydroxyl coverage of silica and more complex glass systems are perhaps not surprising given the changes to the molecular network caused by the additions of various network formers and modifiers in glasses. It has been shown [70] that the addition of one extra glass-forming component, in this case boron, to silica significantly alters the surface state, as demonstrated by a change in the interaction with adsorbate molecules; its effect on surface hydroxyl coverage has also been demonstrated [76]. The surface of E-glass fibre has been shown using angle-resolved XPS [69] to be populated with silanol groups whose concentration is greater than in the bulk of the fibre. Values for the surface hydroxyl coverage of glass samples, obtained using contact angle measurements, have also been published. The hydroxyl number of heat-cleaned laboratory glass slides was reported as αOH = 2.5 nm−2 [77]. For slides of carefully-cleaned and fully-hydrolysed boron-free E-glass, a value of approximately αOH = 2.4 nm−2 was obtained [78]. In both cases, it is clear that hydroxyl coverage is significantly lower than that of the idealised fully-hydroxylated silica system.
3.2. Interaction with Water
Any discussion of the strength of glass must include reference to the role of water, which is generally viewed as deleterious [79,80,81,82,83]. The reduction in strength of glasses exposed to water has been a field of research in and of itself for a long time, primarily because the effect is so clear, but the exact explanation of the mechanism responsible is a challenging research question. Water can be present in significant quantities on glass surfaces and in the glass bulk. Glasses manufactured using standard melt technologies always contain residual water [84,85]. Water can exist in, and diffuse through, glass as both molecular water (H2O) and hydroxyl (OH) [86,87]. The mobile species, molecular water, can diffuse into and react with the silica to produce immobile hydroxyl by the reaction H2O + ≡Si–O–Si≡ ⇔ 2≡Si–OH. It is noteworthy that this reaction breaks structural bonds, which must have consequences on the response of the glass structure to applied stress. At high enough temperatures, the reaction is fast, and reaction equilibrium is established with an equilibrium constant K = [OH]2/[H2O], where [OH] and [H2O] are the concentrations of hydroxyl and molecular water, respectively [88]. The concentration of dissolved molecular water, [H2O], in the glass is proportional to atmospheric water vapour pressure. Mechanical loss increases and the modulus decreases with increasing water content in glass [89,90]. Interestingly, this would imply that exposure of glass to elevated temperatures could presumably lower the water content and increase the measured modulus (as has been experimentally observed by Yang and Thomason [54]). It has been suggested that water enters into glasses, accelerated by tensile stress and causes swelling, which reduces modulus and strength in a manner similar to the plasticization effect in polymers [88,89]. Furthermore, the magnitude of K has been shown to increase in the temperature range 200–550 °C in silica glass [91], implying the likelihood of increasing Si–O–Si structural bond breakage with increasing conditioning temperature in the temperature range, which causes significant strength loss in GF.
Much of the experimental research on the interaction of water with GF has been focussed on the lowering of the performance of the strong, often freshly drawn, relatively undamaged and free of severe flaws, fibres by stress corrosion [79]. However, a number of researchers have commented on the strength of relatively damaged or deliberately abraded glass or GF being increased by interaction with water. As early as 1960, Mould [92] studied the liquid nitrogen strength of abraded glass microscope slides after storage in either water or a 100% RH atmosphere. A one-day storage in water increased strength by 30%–60% depending on the method of abrasion. Ito and Tomozawa reported [93] a strength increase of abraded high-silica 3 mm-diameter glass rods measured after immersion in water and silicic acid solution at 88 °C. There was no significant strength increase observed when the immersion was at room temperature. Li and Tomozawa [94] extended this study on silica rods to high temperatures and pressures. They reported a strength increase of more than 200% in the abraded rods after a four-day water vapour treatment at 250 °C. In both of these studies, it was concluded that the mechanism behind this strength increase was one of crack blunting caused by dissolution and subsequent precipitation of the glass in the region of crack tips. This hypothesis was further discussed by Hirao and Tomozawa [95].
Recently, Weiderhorn et al. revisited Tomozawa’s results on hot water conditioning and proposed another hypothesis for the strengthening effect [96,97]. They proposed that water diffuses from the environment into surface cracks to generate a zone of swelling around the crack tip. Because the swollen material is constrained from expanding by the surrounding glass, they suggest that a zone of compressive stress is generated at the fracture surface around the crack tip. The volume expansion of the material around the crack tip consequently causes a fracture mechanics shielding of cracks present in the glass surface and an increase in the toughness. The swelling is time-dependent and so, the toughening of the glass increases with time. This hypothesis was strengthened by a very recent publication from the same group of researchers. By measuring the curvature of thin disks of vitreous silica that were penetrated by water vapour at either 90 °C or 200 °C from one side only, Weiderhorn et al. [98] also determined the volume expansion of the silica and the effect of this volume expansion on its strength. They showed that the water strengthening process depended on crack size, temperature and the amount of swelling of the silica. They further suggest that their results show that swelling and the consequent bending of the disks was caused by silanol group formation in the silica structure and that molecular water played little of a role in the swelling process.
It is interesting to speculate how the above discussion can relate to strength loss in heat-conditioned fibres. Firstly, it has to be noted that none of the water treatment effects discussed in the above works have been (yet) investigated and verified in E-glass or in fibres of similar dimensions to that of reinforcement-grade fibres (approximate diameter of 9–17 µm). Nevertheless, it is a relatively small step to extend the idea that (hot) water exposure can increase the strength of relatively weak fibres to the inverse hypothesis that heating GF causes dehydroxylation and dehydration, which leads to strength loss through shrinkage and loss of compressive stress around crack tips.
As discussed above, Zhuravlev [75] has extensively reviewed work on the role of OH groups on the surface of silica. It was shown that the bonded OH groups could be removed (as water molecules) from the silica surface by heat treatment in the temperature range from 200 to 500 °C. In addition, Zhuravlev also investigated the rehydroxylation of silica surface. It was found that the complete rehydroxylation of the silica surface can be achieved easily for those samples of silica that were subjected to heat treatment at temperatures below 400 °C. After the treatment at a higher temperature, only partial rehydroxylation was achieved. Since glass surfaces are also silica-rich, they have an analogously hydrated structure. Nishioka and Schramke [99,100] compared the thermal desorption of water from E-glass fibres and silica. They concluded that surface silanol (Si–OH) group condensation occurred at temperatures above 200 °C and that sub-surface water desorbed at 300 °C, and at this temperature, silica skeletal bonds are reported to be hydrolysable, so that the surface composition of the heat-treated fibres may be highly variable. The quantity of water desorbed between 500 °C and 800 °C was suggested to result from the diffusion of bulk water from the inner structure. Trens et al. [101] reported a step increase in the weight loss of E-glass fibres during heating at around 450 °C. This was attributed to the loss of surface OH groups and discussed in association with the diffusion of metal ions from the bulk towards the fibre surface. Despite this available information on the temperature dependence of the amount, and state, of water in GF, it is notable that there has been no associated research of how these phenomena relate to thermally-conditioned GF strength loss. It is clear that there appears to be a number of water-related phenomena that are associated with temperature ranges similar to those associated with the GF strength loss phenomenon and that investigation of this relationship could lead to a new understanding of this critical effect.
4. The Role of Silanes and Sizings
Reinforcement-grade GF are always coated (or sized) with a mixture of chemicals during their manufacture. GF sizings are generally designed and applied with the intention of improving GF processability and ability to transfer stress across the interphase between fibre and matrix in the final composite. However, the capability of the sizing to protect the surface of the GF so that it maintains the highest possible level of strength is also of great importance. At the heart of all GF sizings is the organosilane coupling agent, which plays a critical role in all of the important functions of GF sizing [3,102]. In addition to protecting against the weakening effects of moisture and mechanical damage, it has been proposed that silanes have the ability to “heal” fibre surface flaws, alleviating their negative effect on the fibre strength [102,103,104,105]. Many of the important functions of these silane coupling agents depend on the action of the organic fraction of the molecules, such as the aminopropyl group of the commonly-used aminopropylsilane (APS) coupling agent. In any GRP composite thermal recycling process (or GF thermal conditioning experiment), relatively high temperatures will be involved so that the polymer may be removed. It seems probable that these high temperatures will also degrade any silane molecules on the GF surface.
Tiefenthaler and Urban [106] coated Nextel ceramic fibres with various different silanes. Mats of heat-cleaned cloth were coated and dried then exposed to one hour of furnace heating at different temperatures. Their photoacoustic infrared spectroscopy study showed that, following a 200 °C treatment, almost 50% of the silane propyl chains were removed, and by 300 °C, none at all were detected. Feih et al. [31] presented data for E-glass fibres sized with a thermoset compatible sizing. Using TGA in air at 10 °C/min, they measured a total mass loss of 0.5% by 500 °C with the majority of mass loss occurring between 250 and 450 °C. Their results also showed that TGA under a pure nitrogen atmosphere caused slightly less weight loss than air. When analysing the strength loss of heat-treated or recycled glass fibres upon which a silane coupling agent or full sizing was present, it is necessary to consider the effect of the degradation of this coating, as well as any changes in the GF itself that may contribute to strength loss. A study by Zinck et al. [104] also suggested the possibility that adsorption of silane coupling agents on surfaces of already processed fibres could increase their strength, by a mechanism in which flaws of a critical size were healed.
Thomason et al. [39] studied the bundle strength and single fibre strength of boron-free E-glass fibres that had been sized with only APS or only water (bare fibre) during their production on a commercial GF pilot facility. The bare fibres were significantly weaker that the APS sized fibres, and their strength was further reduced by thermal conditioning up to 400 °C. The silane sized GF exhibited relative stability in average tensile strength up to 300 °C. However, conditioning above this temperature resulted in a precipitous drop in room temperature strength. The origins of these observations were attributed, in part, to the thermal degradation of the organic part of the silane coating and the consequential increase in sensitivity to fibre surface mechanical and environmental damage. However, the parallel changes in the performance of the uncoated fibres with conditioning temperature may imply that there are also other changes taking place in the glass fibres at these relatively low processing temperatures. In a later paper, the same group extended their study of these fibres to higher conditioning temperatures and confirmed similar trends [40]. It was shown that, although the bare GF samples were initially much weaker than the APS-coated GF, after thermal conditioning above about 450 °C, the results converged, and both fibre types exhibited very low levels of single fibre strength. The authors also presented results showing that both GF samples showed an approximately linear increase in room temperature modulus with conditioning temperature above approximately 200 °C. Furthermore, although both samples started with approximately the same level of tensile modulus, the modulus of the water sized fibres was found to be significantly higher (at 95% confidence level) than that of the silane-coated fibres after elevated temperature conditioning. Hence, it appeared that the silane coating impeded the mechanism that led to an increase in modulus of the fibres during the hot conditioning.
Jenkins et al. [41] also confirmed the effect of thermal conditioning on the single fibre strength of these same APS sized and bare GF. They observed that many researchers studying this strength loss phenomenon carried out the heat conditioning process on the “as-received” fibre roving or mats. They pointed out that the degradation and removal of the protective organic silane/sizing on such fibre bundles could lead to an increased level of fibre-fibre flaw generation and obfuscate the analysis and explanations of the observed strength loss. This hypothesis was validated by the use of a novel experimental design where fibres were isolated from the GF rovings and heat conditioned individually, which removed any possibility of extra fibre damage from surface contact after the sizing degradation. The results for average fibre strength versus conditioning temperature showed a significantly different trend than for (the more commonly used) single fibres, which were removed from fibre rovings after heat conditioning. Using single fibre thermal conditioning, it was possible to achieve significantly greater retained strengths for the bare fibres. Furthermore, the starting temperature for the precipitous drop in the strength of the APS fibres was raised from 300 °C up to 450 °C, above which temperature the bare fibre also showed a drop in strength. They interpreted these results as showing that the critical temperature for GF strength loss due to silane sizing layer degradation was approximately 300 °C. This was further confirmed by a combination of thermal and chemical analyses. However, the single GF thermal conditioning results indicated that there was also some other glass structure-related mechanism that led to significant strength loss when the conditioning temperature was raised above 450 °C.
In their paper on thermally-induced GF strength loss, Thomason et al. [40] also reported investigation of the use of hydrolysed silane coupling agents to regenerate the strength of weakened fibre according to the silane flaw-healing hypothesis. Their results showed that a range of common silane coupling agents (individually and blended) had little significant effect in raising the strength of E-glass fibres, which had been heat treated at 450 °C for 15 min. It was noted that although statistically significant (at the 95% confidence level), increases of up to 25% were observed, complete strength regeneration would require a 300% increase. The authors suggested that one of the possibilities for the failure of direct silanisation in strength regeneration of the heat-treated GF may be the deactivation of glass surface, as it has been shown with both amorphous silica and glass fibre that severe dehydration can occur when subjected to thermal treatment above 400 °C in air [75,99,100,107].
It was notable that the silane flaw healing hypothesis is based on experimental data obtained on new fibres that have been sized directly during production, are relatively strong and have not experienced a thermal conditioning, which seriously weakens the fibres. It can be assumed that for very weak heat-treated/recycled fibres, it will be necessary to reactivate the fibre surface in order for silane coupling agent molecules to react with the fibre surface and enable a flaw healing process to take place. González et al. studied the reactivation of the surface of calcinated E-glass fibres that had been heat cleaned at 450 °C for 1 h [107]. They concluded that calcination created a generally homogeneous surface layer of amorphous silica with a low surface silanol concentration and that regeneration of silanol groups (necessary for reaction with silanes) on this silica is not an easy process. They further concluded that activation with boiling water for 1 h may hydrate the fibre surface, but without any appreciable silanol regeneration. However, refluxing the fibres with 10% aqueous HCl solution for 1–3 h generated “a great number” of silanol groups and enabled a much greater degree of silane coating to be achieved. Unfortunately, the authors did not investigate the effects of these treatments on the strength on the heat-treated fibres. Nevertheless, Thomason et al. did study this effect in their paper [40]. Heat-treated fibres were also subjected to a 1–2 h pre-treatment in 10% HCl solution before treatment with various silane solutions. The use of this HCl pre-treatment combined with a silane coating had little significant effect on raising the strength of the weakened GF. Results were also presented showing that application of silane coating (with and without HCl pre-treatment) had little significant effect on the strength of the bare GF. Although these GF had not been thermally conditioned, they were still significantly weaker that the APS sized GF due to the accumulation of mechanical and environmental damage during handling and storage. The authors concluded that these results raise significant doubts on the validity of the silane flaw healing theory, but more importantly, indicate that further work is needed to improve the understanding of the role of post-silanisation in the strength of glass fibres.
5. Glass Fibre Recycling and Reuse as Reinforcements
A number of researchers have reported the investigation of recycled glass fibres (RGF) from composites and on the reuse of such fibres as reinforcements in composite applications. These reports all show that using such RGF without any attempt to regenerate the GF strength produces composites with significantly reduced performance. This is obviously a significant disadvantage in any attempt to use RGF as a replacement for pristine reinforcement products. Kennerley et al. [108] presented tentative results on the strength of single GF recovered from both heat-conditioned GF bundles and composites processed in their fluidized bed combustion (FBC) rig. Although some of the average values reported were based on far smaller sample sizes than preferable for these brittle fibres, the inverse relationship between retained fibre strength and temperature was observed. Crucially in this work, fibres were exposed to elevated temperatures for short times (20 min or less), yet significant strength loss was still measured. In the case of fibres that were FBC processed at 650 °C, only 5% of the original fibre strength remained. Extreme mechanical damage was reported as the most likely cause for this very low strength retention. In a further report, Kennerley et al. [109] confirmed significant reduction of the strength of RGF produced under different FBC conditions and temperatures. In the most severe, they recorded a 98% loss in fibre strength when recycled at 650 °C. They also went on to produce dough moulding compound (DMC) composites using RGF that had been recycled from sheet moulding compound (SMC) mouldings at 450 °C. It was found that RGF composites had only 50% of the flexural strength and (most significantly) only 35% of Charpy impact strength of similar DMC samples prepared using pristine GF. The same research group later reported further significant fibre strength losses in RGF obtained from a range of composite materials recycled in their FBC rig [110]. They also reported the production of an RGF veil, which had only 40% of the strength of a similar pristine GF veil.
Williams et al. also produced RGF from automotive SMC waste materials by pyrolysis at 450 °C. They reported a loss in fibre tensile strength of 45% [111]. They also used the RGF to prepare DMC composites [112]. In this case, the Charpy impact performance of the RGF-based composite was reduced to less than 25% of the sample containing pristine GF. Perrin et al. produced RGF on a laboratory scale by solvolysis of shredded SMC (also involving selective dissolution of the calcium carbonate filler in an acid bath) in diethylenetriamine at 200 °C [113]. The strength of their RGF was not reported. However, composite samples were produced by extrusion and injection moulding 20% RGF with a PP/PE copolymer matrix. The RGF-based samples exhibited a 30% drop in tensile strength and (surprisingly) a 50% drop in tensile modulus. It should be noted that the RGF composites also had 5% adhesion promoter added to the composite, whereas the control composite system based on pristine GF had none. This would have significantly advantaged the apparent RGF performance. More recently, Åkesson et al. isolated RGF from a scrap wind turbine blade by microwave pyrolysis under nitrogen at 440 °C [114]. The RGF was used to produce non-woven fibre mats, which were further processed into composite laminates using polyester resin, hand lay-up and compression moulding. The RGF-based laminate exhibited a 70% reduction in flexural strength and a 60% reduction in modulus in comparison to those made with pristine GF. Oliveux et al. investigated RGF obtained on the laboratory scale by subcritical hydrolysis of a GF-polyester composite [115]. Conditions were varied with a 250–350 °C temperature range and a range of pressures from 45–180 bar. RGF obtained at 350 °C exhibited a reduction of approximately 60% in fibre strength. Kao et al. [116] reported similar strength losses from RGF, which appear to have been produced from the same composites using the same hydrolysis rig under similar conditions. In a different project, Kao et al. produced composites using silane-coated boron-free E-glass fibres combined with a polyester resin [117]. RGF was obtained from these composites by thermal decomposition in the range of 500–600 °C for 45 min. The RGF exhibited a strength loss of 70%. Beauson et al. obtained RGF from an end-of-life wind turbine blade by pyrolysis at 400 °C or glycolysis at 180 °C [118]. In both processes, the RGF exhibited approximately a 50% strength loss. The strength for the RGF obtained by pyrolysis at 450 °C fits well with the general picture, but the poor performance of the RGF obtained by glycolysis is surprising given the relatively low processing temperature. It may be that there are other factors involved with this chemical-based process.
It should be noted that, to fully appreciate the reduction in the reinforcement value of the RGF in these experiments, a significant fraction of the reported composite performance is contributed by the matrix material. This matrix contribution can be assumed to be approximately the same in both cases. Consequently, the loss in reinforcement performance of the RGF compared to pristine GF is actually proportionally much greater than the measured loss in composite performance. This has been confirmed in recent work by Thomason et al. [17,119] and Yang et al. [120], where heat-conditioned fibres were used to produce RGF-PP composites and RGF-epoxy composites. In both cases, reference values for the polymer matrix were produced, and in both cases, the tensile strength of the RGF composites was a little better than the unreinforced polymer matrix.
A number of authors have also produced composites using blends of RGF and pristine GF. In all cases, this also leads to a degradation of composite performance, although obviously to a lesser (but still highly) significant degree in comparison with 100% RGF reinforced composites [109,110,111,112,113,114,121]. This may sometimes give the appearance that such RGF could potentially be used to replace some fraction of pristine GF in real composite applications. However, this ignores a number of real-world commercial considerations that would weigh significantly against such a route being easily accepted by most composite end-users.
Criado et al. produced RGF from glass-reinforced polyester waste materials by pyrolysis at 550 °C. They reported an 82% reduction in the tensile strength of their RGF with respect to the values of the virgin fibres [122]. The authors proceeded to “reuse” this material as a filler in cement mortar after grinding down the RGF to a particulate. There have been a number of reviews of this “reuse” of RGF in cement and concrete. However, this technology does not “reuse” the reinforcement potential of the RGF and is in reality more a method of disposal than reuse.
6. Glass Fibre Strength Regeneration and Reuse
Donald reviewed known methods available for improving the mechanical properties of oxide glasses [123]. These were divided into surface and bulk techniques and included thermal and chemical strengthening, controlled crystallization and particle, fibre and whisker reinforcement. Many of the reviewed methods were not obviously applicable to fibres. However, methods such as surface etching, chemical strengthening (by ion exchange) and surface coating could most probably be applied to fibres. Until recently, there has been little systematic research effort focused on the challenge of reversing the strength loss observed in thermally-conditioned GF in order to regenerate their usefulness as a composite reinforcement. As discussed above, the use of silanes alone, or in combination with acid surface reactivation, has been investigated, but not proven as a significant success in the regeneration of the strength of heat-conditioned E-glass fibres.
Ion exchange is [110,123] known as a method by which glasses may be strengthened, but there has been very little investigation of the application of this technology to RGF. Karlsson et al. [124] recently reviewed the technology of the chemical strengthening of glass, but there was no mention of application to GF. Cooper and Krohn [125] described the strengthening of soda-alumina-silica fibres by ion exchange in potassium nitrate. The 0.25 mm-diameter fibres were immersed in a KNO3 salt bath at 400 °C for different times up to 16 h. At shorter times, a strong increase in fibre strength was observed, which reached a plateau level of approximately +200% after 1 h. It is assumed that potassium ions that occupy a larger volume replace the sodium ions in the fibre surface, causing an increased compressive stress in the surface layer, which delays flaw/crack opening during a tensile test. However, reinforcement fibre glass formulations such as E-glass contain only a trace amount of sodium, and the ion exchange method of fibre strengthening does not appear to have been systematically researched on E-glass fibres. The method was briefly mentioned by Kennerly in his PhD thesis [126], which reports a single experiment attempting the ion exchange method of chemical strengthening on E-glass fibre that had been heat treated at 450 °C. A 16% increase in average single fibre strength was reported.
Early studies by Sakka [23] and Aslanova [29] focussed on using hydrogen fluoride (HF) etching in order to obtain evidence that the thermally-induced fibre strength loss was related to a fibre surface phenomenon. Both researchers reported significant strength recovery in HF-etched heat-conditioned fibres. The authors explained the strength regeneration obtained with HF as being related to the removal of damaged surface layers and the modification of existing cracks. The HF effect then remained relatively unexplored until a recent conference paper by Pico and Bartl reported on their research related to recycling of end-of-life wind turbine blades [42]. They studied the effect of thermal conditioning and HF etching on the strength of a number of reinforcement fibres, including two forms of E-glass, S-glass and basalt. Heat conditioning at 400 °C for 2 h caused significant strength loss in all fibre types. The heat-treated fibres were then exposed to 1% HF (further details absent) to investigate the effect of surface etching on fibre strength. It was stated that the S-glass and standard (boron containing) E-glass fibres were too brittle to be measured after the HF treatment. However, partial recovery of the strength of the basalt and boron-free E-glass fibres was recorded.
A very recent paper by Yang et al. further explored the potential of HF treatment in glass fibre recovery [120]. They reported on a systematic study of the effects of heat treatment, HF treatment and silane treatment on the strength of boron-free E-glass fibres and RGF-epoxy composites produced by vacuum infusion moulding. Their results showed that treatment with 1% volume HF solution tripled the strength of heat-treated GF. The fibre strength was shown to be dependent on the HF treatment time, as well as the heat conditioning temperature. The authors further discussed the need to reactivate the regenerated fibre surface in order to enable an adequate level fibre-matrix adhesion required to obtain acceptable composite performance. Heat-treated fibres produced composites with very poor strength and impact performance. Heat-treated fibre with HF treatment exhibited high levels of fibre strength recovery, but only small improvements in composite performance. This was explained as being due to the poor level of interfacial strength in this system. In a similar manner, heat-treated fibres that were only silane treated exhibited full recovery of the interfacial strength, but very little recovery of fibre strength and, consequently, also showed poor composite performance recovery. However, heat-treated fibres that had received an HF treatment followed by silanisation resulted in composites with high levels of composite performance recovery. The authors stated that their results provide strong evidence of the feasibility of achieving closed-loop recycling of GRP composites through regenerating the performance of the thermally-degraded GF. However, they also noted that the use of HF in an industrial environment would be problematic due to its highly toxic nature and that it is most unlikely that the HF route could ever lead to a cost-effective process for the regeneration of the properties of RGF [127]. However, they also referred to a recently-filed patent on a technology of glass fibre recovery that does not involve any HF or its derivatives. Further details of three treatments were discussed by the same authors in a recent conference paper [119]. Significant recovery in the strength of heat-treated E-glass fibres was reported using three (non-HF) routes. Significant recovery of the performance of injection-moulded RGF-polypropylene composites was also presented.
Further details of one of these treatments was revealed in a recent journal paper [17], where Thomason et al. presented data on the effect of hot sodium hydroxide (NaOH) treatment on the strength of heat-conditioned E-glass fibres and RGF-polypropylene composites prepared by a wet deposition glass mat thermoplastic (GMT) moulding process. Following a similar protocol to that of the HF work by Yang et al. [120], the authors presented the use of a combination of thermal conditioning, hot NaOH treatment and silanisation on the average strength of single fibres, the interfacial shear strength between the fibres and PP and the performance of random in-plane GMT laminates. Results indicated that the GF lose 80% or more of their strength when exposed to temperatures typically found in GRP thermal recycling processes, which makes them unsuitable for reuse as a composite reinforcement. The single fibre tensile testing results showed that up to 75% of that strength loss can be recovered by a short treatment in hot NaOH solution. This fibre strength recovery was maintained and improved upon by a further application of a silane sizing. This silane layer also acted to recover the compatibility of the recycled fibre surface with a polymer composite matrix and to regenerate a high level of fibre-matrix interfacial stress transfer capability. The regeneration of the RGF performance by these simple chemical methods resulted in GF-PP GMT composites, which exhibited a 74% strength recovery compared to GMT based on thermally-recycled fibres, which had received no further treatment. It was proposed that further optimisation of these chemical treatments could lead to further improvements in the fibre performance, enabling them to be used to replace the pristine first-pass fibre products used in many discontinuous glass fibre-reinforced composite applications. The authors noted that there are many non-technical factors that will play a role in the eventual commercial profitability of any GRP recycling process. Nevertheless, the development of a non-HF-based glass fibre strength regeneration treatment is an exciting development in the progress towards a cost-effective closed-loop GRP recycling technology.
Acknowledgments
The authors would like to acknowledge financial support from the Engineering and Physical Sciences Research Council under the grants EP/I037970/1 and EP/I038616/1.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| GMT | Glass mat thermoplastic |
| APS | Aminopropylsilane |
| DMC | Dough moulding compound |
| DSC | Differential scanning calorimetry |
| FBC | Fluidized bed combustion |
| GF | Glass fibre |
| RGF | Recycled glass fibre |
| SMC | Sheet moulding compound |
| Tg | Glass transition temperature |
| TGA | Thermo-gravimetric analysis |
References
- Owens Corning Investor Presentation (Slide 22). Available online: http://s1.q4cdn.com/942908807/files/doc_presentations/2015/Q3/Q3-Presentation-v9.pdf (accessed on 12 January 2016).
- Li, H.; Watson, J.C. Continuous glass fibers for reinforcement. In Encyclopaedia of Glass Science, Technology, History and Culture; John Wiley: New York, NY, USA, 2016. [Google Scholar]
- Thomason, J.L. Glass Fibre Sizing: A Review of Size Formulation Patents. Blurb Incorporated 2015. Available online: http://www.blurb.co.uk/b/6244662-glass-fibre-sizing (accessed on 3 February 2016).
- Derosa, R.; Telfeyan, E.; Mayes, J.S. Current state of recycling sheet molding compounds and related materials. J. Thermoplast. Compos. Mater. 2005, 18, 219–240. [Google Scholar] [CrossRef]
- Pickering, S.J. Recycling technologies for thermoset composite materials-current status. Compos. A Appl. Sci. Manuf. 2006, 37, 1206–1215. [Google Scholar] [CrossRef]
- Cuesta, J.L.; Perrin, D.; Sonnier, R. Waste management, recycling and regeneration of filled polymers. In Handbook of Multiphase Polymer Systems; Boudenne, A., Ibos, L., Candau, Y., Thomas, S., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2011; pp. 921–957. [Google Scholar]
- Goodship, V. Recycling issues in polymer matrix composites. In Failure Mechanisms in Polymer Matrix Composites: Criteria, Testing and Industrial Applications; Robinson, P., Greenhalgh, E., Pinho, S., Eds.; Woodhead Publishing Ltd.: Cambridge, UK, 2012; pp. 336–366. [Google Scholar]
- Yang, Y.; Boom, R.; Irion, B.; van Heerden, D.J.; Kuiper, P.; de Wit, H. Recycling of composite materials. Chem. Eng. Process. Process Intensif. 2012, 51, 53–68. [Google Scholar] [CrossRef]
- Job, S. Recycling glass fibre reinforced composites—History and progress. Reinf. Plast. 2013, 57, 19–23. [Google Scholar] [CrossRef]
- Job, S. Recycling composites commercially. Reinf. Plast. 2014, 58, 32–38. [Google Scholar] [CrossRef]
- Yazdanbakhsh, A.; Bank, L.C. A critical review of research on reuse of mechanically recycled FRP production and end-of-life waste for construction. Polymers 2014, 6, 1810–1826. [Google Scholar] [CrossRef]
- Miller, L.; Soulliere, K.; Sawyer-Beaulieu, S.; Tseng, S.; Tam, E. Challenges and alternatives to plastics recycling in the automotive sector. Materials 2014, 7, 5883–5902. [Google Scholar] [CrossRef]
- Asmatulu, E.; Twomey, J.; Overcash, M. Recycling of fiber-reinforced composites and direct structural composite recycling concept. J. Compos. Mater. 2014, 48, 593–608. [Google Scholar] [CrossRef]
- Beauson, J.; Lilholt, H.; Brøndsted, P. Recycling solid residues recovered from glass fibre-reinforced composites—A review applied to wind turbine blade materials. J. Reinf. Plast. Compos. 2014, 33, 1542–1556. [Google Scholar] [CrossRef]
- Oliveux, G.; Dandy, L.O.; Leeke, G.A. Current status of recycling of fibre reinforced polymers: Review of technologies, reuse and resulting properties. Prog. Mater. Sci. 2015, 72, 61–99. [Google Scholar] [CrossRef]
- Rybicka, J.; Tiwari, A.; Leeke, G.A. Technology readiness level assessment of composites recycling technologies. J. Clean. Prod. 2016, 112, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Thomason, J.L.; Nagel, U.; Yang, L.; Sáez, E. Regenerating the strength of thermally recycled glass fibres using hot sodium hydroxide. Compos. A Appl. Sci. Manuf. 2016, in press. [Google Scholar]
- Sugarman, B. Strength of Glass (A Review). J. Mater. Sci. 1967, 2, 275–283. [Google Scholar] [CrossRef]
- Gupta, P.K. Strength of glass fibers. In Fiber Fracture; Elices, M., Llorca, J., Eds.; Elsevier Science Ltd.: Oxford, UK, 2002; pp. 127–153. [Google Scholar]
- Kurkjian, C.R.; Gupta, P.K.; Brow, R.K.; Lower, N. The intrinsic strength and fatigue of oxide glasses. J. Non-Cryst. Solids 2003, 316, 114–124. [Google Scholar] [CrossRef]
- Griffith, A.A. The Phenomena of Rupture and Flow in Solids. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1921, 221, 163–198. [Google Scholar] [CrossRef]
- Standard Practice for Fractography and Characterization of Fracture Origins in Advanced Ceramics; ASTM C1322-05b; ASTM: West Conshohocken, PA, USA, 2010.
- Sakka, S. Effects of reheating on strength of glass fibers. Bull. Inst. Chem. Res. 1957, 34, 316–320. [Google Scholar] [CrossRef]
- Thomas, W.F. An investigation of the factors likely to affect the strength and properties of glass fibres. Phys. Chem. Glasses 1960, 1, 4–18. [Google Scholar]
- Cameron, N.M. The effect of environment and temperature on the strength of E-glass fibres. Part 2. Heating and ageing. Glass Technol. 1968, 9, 121–130. [Google Scholar]
- Cameron, N.M. Effect of prior heat treatment on the strength of glass fibers measured at room temperature. J. Am. Ceram. Soc. 1965, 48. [Google Scholar] [CrossRef]
- Brearley, W.; Holloway, D.G. The effect of heat-treatment on the breaking strength of glass. Phys. Chem. Glasses 1963, 4, 69–75. [Google Scholar]
- Piggott, M.R.; Yokom, J.C. The weakening of silica fibres by heat treatment. Glass Technol. 1968, 9, 172–175. [Google Scholar]
- Aslanova, M.S. The effect of different factors on the mechanical properties of glass fibers. Steklo Keram. 1960, 17, 10–15. [Google Scholar] [CrossRef]
- Dorzhiev, D.B.; Khazanov, V.E.; Gorbachev, V.V. Some features of the structure and strength of a magnesium aluminosilicate fiber. Sov. J. Glass Phys. Chem. 1990, 15, 99–102. [Google Scholar]
- Feih, S.; Boiocchi, E.; Mathys, Z.; Gibson, A.G.; Mouritz, A.P. Mechanical properties of thermally-treated and recycled glass fibres. Compos. B Eng. 2011, 42, 350–358. [Google Scholar] [CrossRef]
- Lund, M.D.; Yue, Y. Impact of drawing stress on the tensile strength of oxide glass fibers. J. Am. Ceram. Soc. 2010, 93, 3236–3243. [Google Scholar] [CrossRef]
- Lund, M.D. Tensile Strength of Glass Fibres. Ph.D. Thesis, Aalborg University, Aalborg, Denmark, 2010. [Google Scholar]
- Korwin-Edson, M.L.; Hofmann, D.A.; McGinnis, P.B. Strength of high performance glass reinforcement fiber. Int. J. Appl. Glass Sci. 2012, 3, 107–121. [Google Scholar] [CrossRef]
- Militky, J.; Kovacic, V.; Rubnerova, J. Influence of thermal treatment on tensile failure of basalt fibers. Eng. Fract. Mech. 2002, 69, 1025–1033. [Google Scholar] [CrossRef]
- Jenkins, P.G.; Riopedre-Méndez, S.; Sáez, E.R.; Yang, L.; Thomason, J.L. Investigation of the strength of thermally conditioned basalt and E-glass fibres. In Proceedings of the 20th International Conference on Composite Materials (ICCM20), Copenhagen, Denmark, 19–24 July 2015.
- Sabet, S.M.M.; Akhlaghi, F.; Eslami-Farsani, R. The effect of thermal treatment on tensile properties of basalt fibers. J. Ceram. Sci. Tech. 2015, 6, 245–248. [Google Scholar]
- Cameron, N.M. The Effect of Annealing on the Room Temperature Strength of Glass Fibres. Ph.D. Thesis, University of Illinois at Urbana–Champaign, Champaign, IL, USA, 1962. [Google Scholar]
- Thomason, J.L.; Kao, C.C.; Ure, J.; Yang, L. The strength of glass fibre reinforcement after exposure to elevated composite processing temperatures. J. Mater. Sci. 2014, 49, 153–162. [Google Scholar] [CrossRef]
- Thomason, J.L.; Yang, L.; Meier, R. The properties of glass fibres after conditioning at composite recycling temperatures. Compos. A Appl. Sci. Manuf. 2014, 61, 201–208. [Google Scholar] [CrossRef]
- Jenkins, P.G.; Yang, L.; Liggat, J.J.; Thomason, J.L. Investigation of the strength loss of glass fibre after thermal conditioning. J. Mater. Sci. 2015, 50, 1050–1057. [Google Scholar] [CrossRef]
- Pico, D.; Bartl, A. Chemical treatment of glass fibers after composite recycling process. In Proceedings of the ISWA World Congress, Hamburg, Germany, 15–18 November 2010.
- Bartenev, G.M.; Motorina, L.I. Effect of tensile stresses on the strength of heat-treated glass fibers. Mekhanika Polim. 1965, 1, 89–92. [Google Scholar] [CrossRef]
- Lezzi, P.J.; Xiao, Q.R.; Tomozawa, M.; Blanchet, T.A.; Kurkjian, C.R. Strength increase of silica glass fibers by surface stress relaxation: A new mechanical strengthening method. J. Non-Cryst. Solids 2013, 379, 95–106. [Google Scholar] [CrossRef]
- Lezzi, P.J.; Seaman, J.H.; Tomozawa, M. Strengthening of E-glass fibers by surface stress relaxation. J. Non-Cryst. Solids 2014, 402, 116–127. [Google Scholar] [CrossRef]
- Otto, W.H. Compaction effects in glass fibers. J. Am. Ceram. Soc. 1961, 44, 68–72. [Google Scholar] [CrossRef]
- Otto, W.H.; Preston, F.W. Evidence against oriented structure in glass fibres. J. Soc. Glass Technol. 1950, 34, 63–68. [Google Scholar]
- Aslanova, M.S.; Ivanov, N.V.; Balashov, Y.S. Effect of chemical composition on the relaxation properties of thin glass fibers. Steklo Keram. 1970, 8, 21–24. [Google Scholar]
- Stockhorst, H.; Brückner, R. Structure sensitive measurements on E-glass fibers. J. Non-Cryst. Solids 1982, 49, 471–484. [Google Scholar] [CrossRef]
- Ya, M.; Deubener, J.; Yue, Y. Enthalpy and anisotropy relaxation of glass fibers. J. Am. Ceram. Soc. 2008, 91, 745–752. [Google Scholar] [CrossRef]
- Yue, Y.; Jensen, S.L.; Christiansen, J. Physical aging in a hyperquenched glass. Appl. Phys. Lett. 2002, 81, 2983–2985. [Google Scholar] [CrossRef]
- Yue, Y. Features of the relaxation in hyperquenched inorganic glasses during annealing. Phys. Chem. Glasses 2005, 46, 354–358. [Google Scholar]
- Deubener, J.; Yue, Y.; Bornhöft, H.; Ya, M. Decoupling between birefringence decay, enthalpy relaxation and viscous flow in calcium boroalumosilicate glasses. Chem. Geol. 2008, 256, 299–305. [Google Scholar] [CrossRef]
- Yang, L.; Thomason, J.L. The thermal behaviour of glass fibre investigated by thermomechanical analysis. J. Mater. Sci. 2013, 48, 5768–5775. [Google Scholar] [CrossRef]
- Lu, X.; Arruda, E.M.; Schultz, W.W. The effect of processing parameters on glass fiber birefringence development and relaxation. J. Non-Newton. Fluid Mech. 1999, 86, 89–104. [Google Scholar] [CrossRef]
- Yue, Y.; Korsgaard, M.; Kirkegaard, L.F.; Heide, G. Formation of a nanocrystalline layer on the surface of stone wool fibers. J. Am. Ceram. Soc. 2009, 92, 62–67. [Google Scholar] [CrossRef]
- Wakasugi, T.; Burgner, L.L.; Weinberg, M.C. A DTA study of crystal nucleation in Na2O-SiO2 glasses. J. Non-Cryst. Solids 1999, 244, 63–73. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Olabi, A.G. Effect of nucleation temperature and time on crystallization behaviour of zirconia/zircon added borosilicate glass. In Proceedings of the ECCM15—15th European Conference on Composite Materials, Venice, Italy, 24–28 June 2012.
- Gy, R. Ion exchange for glass strengthening. Mater. Sci. Eng. B 2008, 149, 159–165. [Google Scholar] [CrossRef]
- Varshneya, A.K. Chemical strengthening of glass: Lessons learned and yet to be learned. Int. J. Appl. Glass Sci. 2010, 1, 131–142. [Google Scholar] [CrossRef]
- Wallenberger, F.T.; Watson, J.C.; Hong, L. Glass Fibers. In ASM Handbook, Volume 21: Composites; ASM International: Novelty, OH, USA, 2001; pp. 1–8. [Google Scholar]
- Kistler, S.S. Stresses in glass produced by nonuniform exchange of monovalent ions. J. Am. Ceram. Soc. 1962, 45, 59–68. [Google Scholar] [CrossRef]
- Nichols, D.; Hercules, D.M.; Peek, R.C.; Vaughan, D.J. Application of X-ray photoelectron spectroscopy to the study of fiberglass surfaces. Appl. Spectrosc. 1974, 28, 219–222. [Google Scholar] [CrossRef]
- Pugh, A.C.; Stratton, R.P.; Lewis, D.B. Investigation of elemental diffusion during the drawing and heat treatment of glass optical fibres. J. Mater. Sci. 1994, 29, 1036–1040. [Google Scholar] [CrossRef]
- Wong, R. Recent aspects of glass fiber-resin interfaces. J. Adhes. 1972, 4, 171–179. [Google Scholar] [CrossRef]
- Thomason, J.L.; Dwight, D.W. The use of XPS for characterisation of glass fibre coatings. Compos. A Appl. Sci. Manuf. 1999, 30, 1401–1413. [Google Scholar] [CrossRef]
- Liu, X.; Thomason, J.L.; Jones, F.R. XPS and AFM study of interaction of organosilane and sizing with E-Glass fibre surface. J. Adhes. 2008, 84, 322–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jones, F.R.; Denison, P. TOF SIMS and XPS study of the interaction of hydrolysed γ-aminopropyltriethoxysilane with E-glass surfaces. J. Adhes. Sci. Technol. 1992, 6, 79–98. [Google Scholar] [CrossRef]
- Wesson, S.P.; Jen, J.S.; Nishioka, G.M. Acid-base characteristics of silane- treated E glass fiber surfaces. J. Adhes. Sci. Technol. 1992, 6, 151–169. [Google Scholar] [CrossRef]
- Hair, M.L. Hydroxyl groups on silica surface. J. Non-Cryst. Solids 1975, 19, 299–309. [Google Scholar] [CrossRef]
- Fry, R.A.; Tsomaia, N.; Pantano, C.G.; Mueller, K.T. 19F MAS NMR quantification of accessible hydroxyl sites on fiberglass surfaces. J. Am. Chem. Soc. 2003, 125, 2378–2379. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.S. Surface functionality of amorphous silica by infrared spectroscopy. J. Phys. Chem. 1956, 62, 1168–1178. [Google Scholar] [CrossRef]
- Peng, L.; Qisui, W.; Xi, L.; Chaocan, Z. Investigation of the states of water and OH groups on the surface of silica. Colloids Surfaces A Physicochem. Eng. Asp. 2009, 334, 112–115. [Google Scholar] [CrossRef]
- Bakaev, V.A.; Pantano, C.G. Inverse reaction chromatography 2. Hydrogen/Deuterium exchange with silanol groups on the surface of fumed silica. J. Phys. Chem. 2009, 113, 13894–13898. [Google Scholar] [CrossRef]
- Zhuravlev, L.T. Surface characterization of amorphous silica—A review of work from the former USSR. Colloids Surfaces A Physicochem. Eng. Asp. 1993, 74, 71–90. [Google Scholar] [CrossRef]
- Pantano, C.G. Effect of boron oxide on surface hydroxyl coverage of aluminoborosilicate glass fibres: A (19)F solid state NMR study. Phys. Chem. Glasses 2003, 44, 64–68. [Google Scholar]
- Carré, A.; Lacarrière, V.; Birch, W. Molecular interactions between DNA and an aminated glass substrate. J. Colloid Interface Sci. 2003, 260, 49–55. [Google Scholar] [CrossRef]
- Liu, X.M.; Thomason, J.L.; Jones, F.R. The concentration of hydroxyl groups on glass surfaces and their effect on the structure of silane deposits. In Proceedings of the Sixth International Symposium on Silanes and Other Coupling Agents, Cincinnati, OH, USA, 13–15 June 2007.
- Martin, D.M.; Akinc, M.; Oh, S.M. Effect of forming and aging atmospheres on E-glass strength. J. Am. Ceram. Soc. 1978, 61, 308–311. [Google Scholar] [CrossRef]
- Proctor, B.A.; Whitney, I.; Johnson, J.W. The strength of fused silica. Proc. R. Soc. A Math. Phys. Eng. Sci. 1967, 297, 534–557. [Google Scholar] [CrossRef]
- Orowan, E. Fracture and strength of solids. Rep. Prog. Phys. 1948, 12, 185–232. [Google Scholar] [CrossRef]
- Tomozawa, M. Stress corrosion reaction of silica glass and water. Phys. Chem. Glasses 1998, 39, 65–69. [Google Scholar]
- Michalske, T.A.; Freiman, S.W. A molecular interpretation of stress corrosion in silica. Nature 1982, 295, 511–512. [Google Scholar] [CrossRef]
- Naraev, V.N. The influence of water on the glass properties. Glass Phys. Chem. 2004, 30, 367–389. [Google Scholar] [CrossRef]
- Shelby, J.E. A limited review of water diffusivity and solubility in glasses and melts. J. Am. Ceram. Soc. 2008, 91, 703–708. [Google Scholar] [CrossRef]
- Davis, K.M.; Tomozawa, M. An infrared spectroscopic study of water-related species in silica glasses. J. Non-Cryst. Solids 1996, 201, 177–198. [Google Scholar] [CrossRef]
- Davis, K.M.; Tomozawa, M. Water diffusion into silica glass: Structural changes in silica glass and their effect on water solubility and diffusivity. J. Non-Cryst. Solids 1995, 185, 203–220. [Google Scholar] [CrossRef]
- Oehler, A.; Tomozawa, M. Water diffusion into silica glass at a low temperature under high water vapor pressure. J. Non-Cryst. Solids 2004, 347, 211–219. [Google Scholar] [CrossRef]
- Ito, S.; Tomozawa, M. Dynamic fatigue of sodium-silicate glasses with high water content. J. Phys. 1982, C9, 611–614. [Google Scholar] [CrossRef]
- Day, D.E.; Stevels, J.M. Effect of dissolved water on the internal friction of glass. J. Non-Cryst. Solids 1974, 14, 165–177. [Google Scholar] [CrossRef]
- Tomozawa, M.; Kim, D.-L.; Agarwal, A.; Davis, K.M. Water diffusion and surface structural relaxation of silica glasses. J. Non-Cryst. Solids 2001, 288, 73–80. [Google Scholar] [CrossRef]
- Mould, R.E. Strength and static fatigue of abraded glass under controlled ambient conditions: III, aging of fresh abrasions. J. Am. Ceram. Soc. 1960, 43, 160–167. [Google Scholar] [CrossRef]
- Ito, S.; Tomozawa, M. Crack blunting of high-silica glass. J. Am. Ceram. Soc. 1982, 65, 368–371. [Google Scholar] [CrossRef]
- Li, H.; Tomozawa, M. Mechanical strength increase of abraded silica glass by high pressure water vapor treatment. J. Non-Cryst. Solids 1994, 168, 287–292. [Google Scholar] [CrossRef]
- Hirao, K.; Tomozawa, M. Dynamic Fatigue of treated high-silica glass: Explanation by crack tip blunting. J. Am. Ceram. Soc. 1987, 70, 377–382. [Google Scholar] [CrossRef]
- Wiederhorn, S.M.; Fett, T.; Rizzi, G.; Fuenfschilling, S.; Hoffmann, M.J.; Guin, J.P. Effect of water penetration on the strength and toughness of silica glass. J. Am. Ceram. Soc. 2011, 94, S196–S203. [Google Scholar] [CrossRef]
- Fett, T.; Rizzi, G.; Hoffmann, M.J.; Wagner, S.; Wiederhorn, S.M. Effect of water on the inert strength of silica glass: Role of water penetration. J. Am. Ceram. Soc. 2012, 95, 3847–3853. [Google Scholar] [CrossRef]
- Wiederhorn, S.M.; Yi, F.; LaVan, D.; Richter, L.J.; Fett, T.; Hoffmann, M.J. Volume expansion caused by water penetration into silica glass. J. Am. Ceram. Soc. 2015, 98, 78–87. [Google Scholar] [CrossRef]
- Nishioka, G.M.; Schramke, J.A. Desorption of water from glass fibers. In Molecular Characterization of Composite Interfaces; Ishida, H., Kumar, G., Eds.; Plenum Press: New York, NY, USA, 1983; pp. 387–400. [Google Scholar]
- Nishioka, G.M.; Schramke, J.A. Thermodesorption of water from silicate surfaces. J. Colloid Interface Sci. 1985, 105, 102–111. [Google Scholar] [CrossRef]
- Trens, P.; Denoyel, R.; Guilloteau, E. Evolution of surface composition, porosity, and surface area of glass fibers in a moist atmosphere. Langmuir 1996, 12, 1245–1250. [Google Scholar] [CrossRef]
- Thomason, J.L. Glass Fibre Sizing: A Review of the Literature. Blurb Incorporated 2015. Available online: http://www.blurb.co.uk/b/6244677-glass-fibre-sizing (accessed on 3 February 2016).
- Zinck, P.; Mader, E.; Gerard, J.F. Role of silane coupling agent and polymeric film former for tailoring glass fiber sizings from tensile strength measurements. J. Mater. Sci. 2001, 36, 5245–5252. [Google Scholar] [CrossRef]
- Zinck, P.; Pays, M.F.; Rezakhanlou, R.; Gerard, J.F. Mechanical characterisation of glass fibres as an indirect analysis of the effect of surface treatment. J. Mater. Sci. 1999, 34, 2121–2133. [Google Scholar] [CrossRef]
- Yang, L.; Thomason, J.L. Effect of silane coupling agent on mechanical performance of glass fibre. J. Mater. Sci. 2012, 48, 1947–1954. [Google Scholar] [CrossRef] [Green Version]
- Tiefenthaler, A.; Urban, M.W. Thermal stability of silane coupling agents on Nextel fibres. Composites 1989, 20, 145–150. [Google Scholar] [CrossRef]
- González-Benito, J.; Baselga, J.; Aznar, A.J. Microstructural and wettability study of surface pretreated glass fibres. J. Mater. Process. Technol. 1999, 92–93, 129–134. [Google Scholar] [CrossRef]
- Kennerley, J.R.; Fenwick, N.J.; Pickering, S.J.; Rudd, C.D. The properties of glass fibers recycled from the thermal processing of scrap thermoset composites. J. Vinyl Addit. Technol. 1997, 3, 58–63. [Google Scholar] [CrossRef]
- Kennerley, J.R.; Kelly, R.M.; Fenwick, N.J.; Pickering, S.J.; Rudd, C.D. The characterisation and reuse of glass fibres recycled from scrap composites by the action of a fluidised bed process. Compos. A Appl. Sci. Manuf. 1998, 29A, 839–845. [Google Scholar] [CrossRef]
- Pickering, S.J.; Kelly, R.M.; Kennerley, J.R.; Rudd, C.D.; Fenwick, N.J. A fuidised bed process for the recovery of glass fibres from scrap thermoset composites. Compos. Sci. Technol. 2000, 60, 509–523. [Google Scholar] [CrossRef]
- Williams, P.T.; Cunliffe, A.; Jones, N. Recovery of value-added products from the pyrolytic recycling of glass-fibre-reinforced composite plastic waste. J. Energy Inst. 2005, 78, 51–61. [Google Scholar] [CrossRef]
- Cunliffe, A.M.; Williams, P.T. Characterisation of products from the recycling of glass fibre reinforced polyester waste by pyrolysis. Fuel 2003, 82, 2223–2230. [Google Scholar] [CrossRef]
- Perrin, D.; Guillermain, C.; Bergeret, A.; Lopez-Cuesta, J.-M.; Tersac, G. SMC composites waste management as reinforcing fillers in polypropylene by combination of mechanical and chemical recycling processes. J. Mater. Sci. 2006, 41, 3593–3602. [Google Scholar] [CrossRef]
- Åkesson, D.; Foltynowicz, Z.; Christéen, J.; Skrifvars, M. Microwave pyrolysis as a method of recycling glass fibre from used blades of wind turbines. J. Reinf. Plast. Compos. 2012, 31, 1136–1142. [Google Scholar] [CrossRef]
- Oliveux, G.; Bailleul, J.-L.; Salle, E.L.G.L. Chemical recycling of glass fibre reinforced composites using subcritical water. Compos. A. Appl. Sci. Manuf. 2012, 43, 1809–1818. [Google Scholar]
- Kao, C.C.; Ghita, O.R.; Hallam, K.R.; Heard, P.J.; Evans, K.E. Mechanical studies of single glass fibres recycled from hydrolysis process using sub-critical water. Compos. Part A. Appl. Sci. Manuf. 2012, 43, 398–406. [Google Scholar] [CrossRef]
- Kao, C.C.; Thomason, J.L. Regeneration of thermally recycled glass fibre for cost-effective composite recycling: Performance of fibre recyclates from thermoset composites and with subsequent ReCoVeR treatments. In Proceedings of the 16th European Conference on Composite Materials (ECCM16), Seville, Spain, 22–26 June 2014.
- Beauson, J.; Fraisse, A.; Toncelli, C.; Bech, J.I.; Brøndsted, P. Thermoset composite recycling: Properties of recovered glass fiber. In Proceedings of the 20th International Conference on Composite Materials (ICCM20), Copenhagen, Denmark, 19–24 July 2015.
- Thomason, J.L.; Sáez-Rodríguez, E.; Kao, C.C.; Nagel, U.; Yang, L. ReCoVeR: Regenerating the strength of glass fibres thermally recycled from end-of-life composites. In Proceedings of the 20th International Conference on Composite Materials (ICCM20), Copenhagen, Denmark, 19–24 July 2015.
- Yang, L.; Sáez, E.R.; Nagel, U.; Thomason, J.L. Can thermally degraded glass fibre be regenerated for closed-loop recycling of thermosetting composites? Compos. A Appl. Sci. Manuf. 2015, 72, 167–174. [Google Scholar] [CrossRef]
- Palmer, J.P.; Ghita, O.R.; Savage, L.; Evans, K.E. Successful closed-loop recycling of thermoset composites. Compos. A. Appl. Sci. Manuf. 2009, 40, 490–498. [Google Scholar] [CrossRef]
- Criado, M.; Garcia-Dáz, I.; Bastidas, J.M.; Alguacil, F.J.; López, F.A.; Monticelli, C. Effect of recycled glass fiber on the corrosion behavior of reinforced mortar. Constr. Build. Mater. 2014, 64, 261–269. [Google Scholar] [CrossRef]
- Donald, I.W. Methods for improving the mechanical properties of oxide glasses. J. Mater. Sci. 1989, 24, 4177–4208. [Google Scholar] [CrossRef]
- Karlsson, S.; Jonson, B.; Stålhandske, C. The technology of chemical glass strengthening—A review. Glass Technol. Eur. J. Glass Sci. Technol. 2010, 51, 41–54. [Google Scholar]
- Cooper, A.R.; Krohn, D.A. Strengthening of glass fibers: Part 2, ion exchange. J. Am. Ceram. Soc. 1969, 52, 665–669. [Google Scholar] [CrossRef]
- Kennerley, J. Recycling Fibres Recovered from Composite Materials Using a Fluidised Bed Process. Ph.D. Thesis, University of Nottingham, Nottingham, UK, 1998. [Google Scholar]
- Yang, L.; Jenkins, P.G.; Liggat, J.J.; Thomason, J.L. Strength of thermally conditioned glass fibre degradation, retention and regeneration. In Proceedings of the 20th International Conference on Composite Materials (ICCM20), Copenhagen, Denmark, 19–24 July 2015.
Figure 1.
Typical results from the literature on residual fibre strength after heat conditioning.
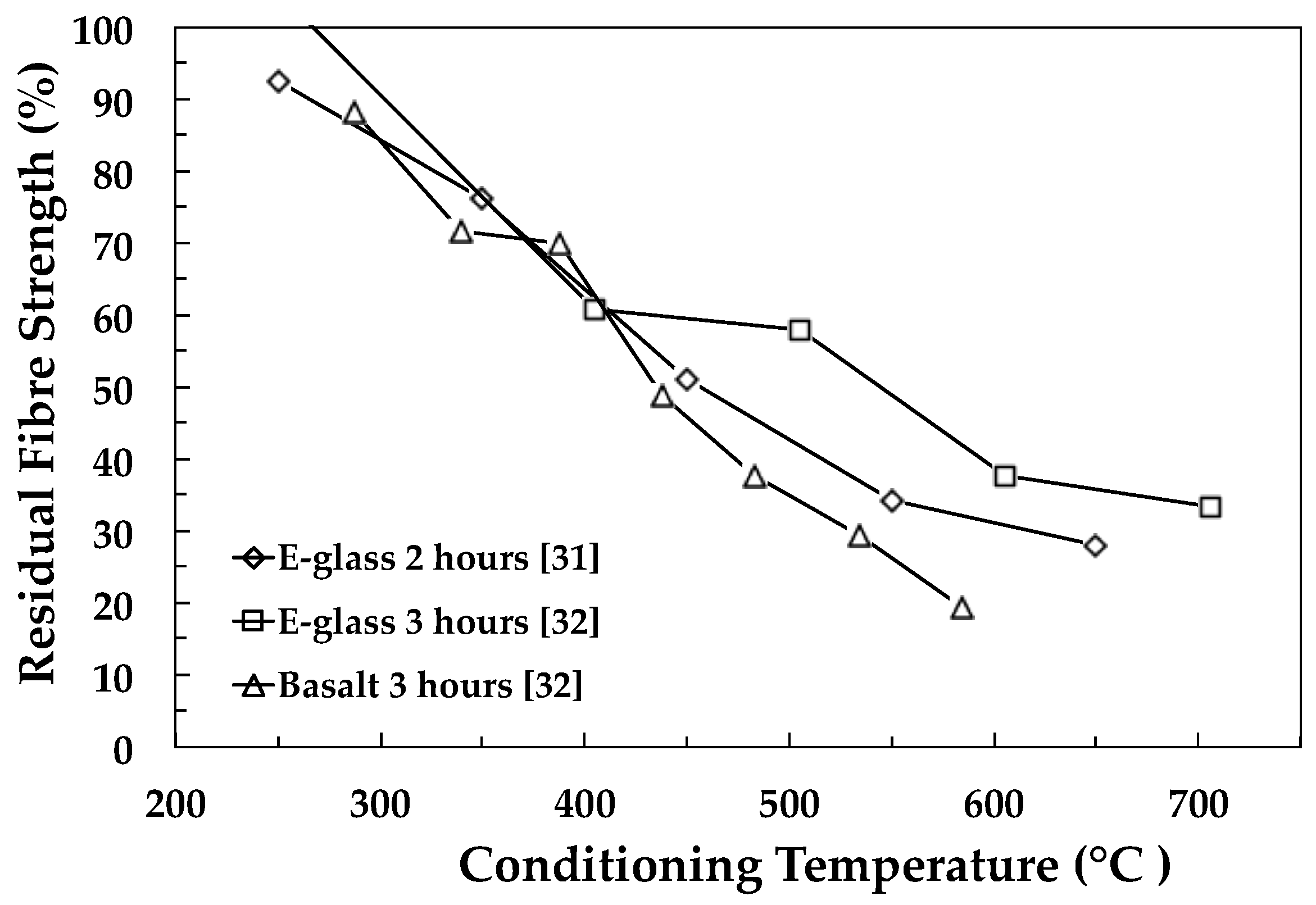

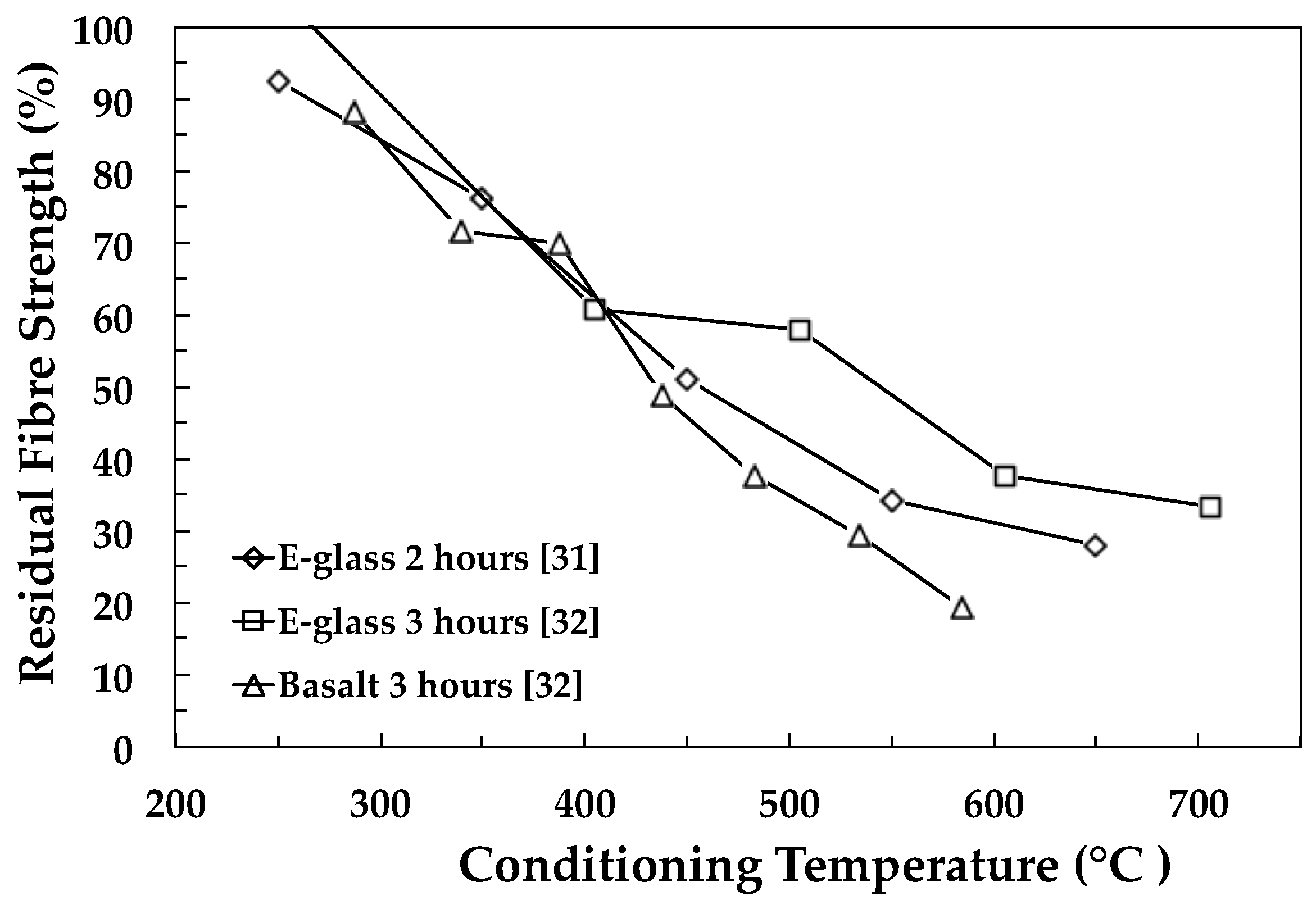{kind=link}
Table 1.
Overview of selected results from the literature on fibre strength loss after heat conditioning.
| Ref. | Fibre Type | Fibre Diameter (microns) | Coating, Sizing | Maximum Conditioning Temperature (°C) | Time (min) | Maximum Strength Loss (%) |
|---|---|---|---|---|---|---|
| [28] | Silica | 50 | unsized | 600 | 60 | 65 |
| [34] | Silica | – | unsized | 650 | 60 | 79 |
| [34] | S-glass | – | – | 650 | 60 | 71 |
| [24] | E-glass | 12 | unsized | 600 | 240 | 67 |
| [31] | E-glass | 12 | 111A | 650 | 15 | 70 |
| [32] | E-glass | 8–16 | unsized | 600 | 180 | 62 |
| [34] | E-glass | – | unsized | 650 | 60 | 80 |
| [38] | E-glass | 6–17 | unsized | 273 | 240 | 40 |
| [39] | E-glass | 17 | unsized | 400 | 25 | 45 |
| [39] | E-glass | 17 | aminosilane | 400 | 25 | 55 |
| [40] | E glass | 17 | unsized | 600 | 25 | 45 |
| [40] | E-glass | 17 | aminosilane | 600 | 25 | 73 |
| [41] | E-glass | 17 | unsized | 600 | 25 | 65 |
| [41] | E-glass | 17 | aminosilane | 600 | 25 | 75 |
| [42] | E-glass | 17 | SE1500 | 600 | 25 | 65 |
| [32] | Basalt | 9–16 | unsized | 600 | ? | 80 |
| [34] | Basalt | – | unsized | 650 | 60 | 97 |
| [35] | Basalt | 9 | – | 500 | 60 | 90 |
| [37] | Basalt | 10 | – | 500 | 5 | 77 |
| [37] | Basalt | 10 | – | 500 | 20 | 89 |
| [42] | Basalt | 15 | epoxy | 600 | ? | 80 |
| [23] | NA-ABG * | 12 | unsized | 650 | ? | 75 |
| [23] | NA-ABG * | 22 | unsized | 650 | 60 | 70 |
| [27] | Soda-lime | 3000 | unsized | 530 | 60 | 70 |
| [29] | NA-ABG * | – | – | 550 | ? | 55 |
| [43] | Alkaline | 20 | unsized | 500 | 60 | 70 |
* Non-alkaline aluminoborosilicate glass.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thomason, J.; Jenkins, P.; Yang, L. Glass Fibre Strength—A Review with Relation to Composite Recycling. Fibers 2016, 4, 18. https://doi.org/10.3390/fib4020018
AMA Style
Thomason J, Jenkins P, Yang L. Glass Fibre Strength—A Review with Relation to Composite Recycling. Fibers. 2016; 4(2):18. https://doi.org/10.3390/fib4020018
Chicago/Turabian StyleThomason, James, Peter Jenkins, and Liu Yang. 2016. "Glass Fibre Strength—A Review with Relation to Composite Recycling" Fibers 4, no. 2: 18. https://doi.org/10.3390/fib4020018
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.