Enhanced Adhesion of Continuous Nanoporous Au Layers by Thermochemical Oxidation of Glassy Carbon


Abstract
:1. Introduction
1.1. Nanoporous Gold (NPG)-Based Catalyst on Glassy Carbon (GC)
1.2. Thermochemical Oxidation for Improved Adhesion
1.3. The Paper’s Objective
2. Experimental Section
2.1. Electrode Preparation
2.2. Pd Seeding
2.3. Thermochemical Oxidation
2.4. Thin Film Alloy Deposition
2.5. The De-Alloying Process
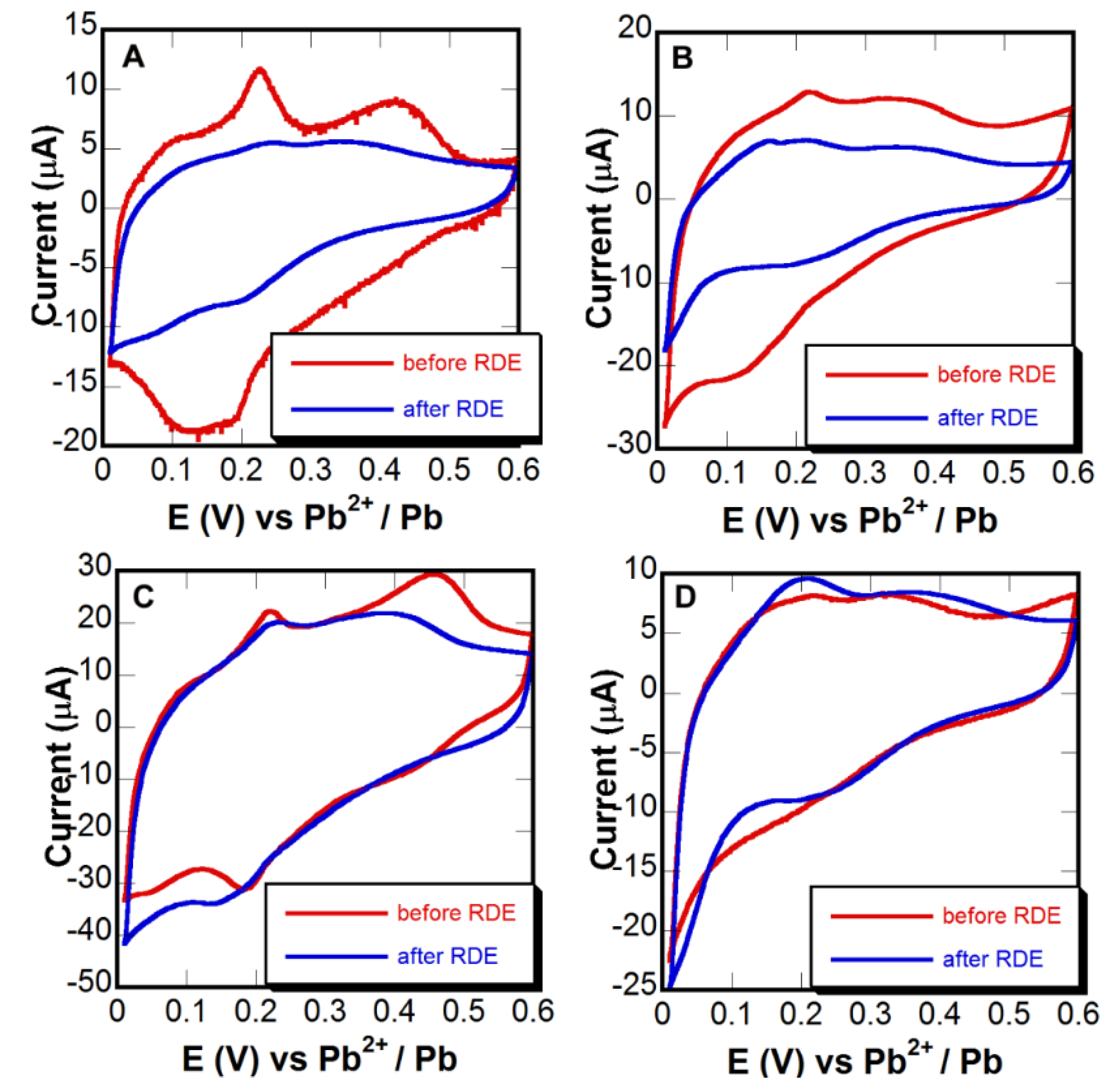
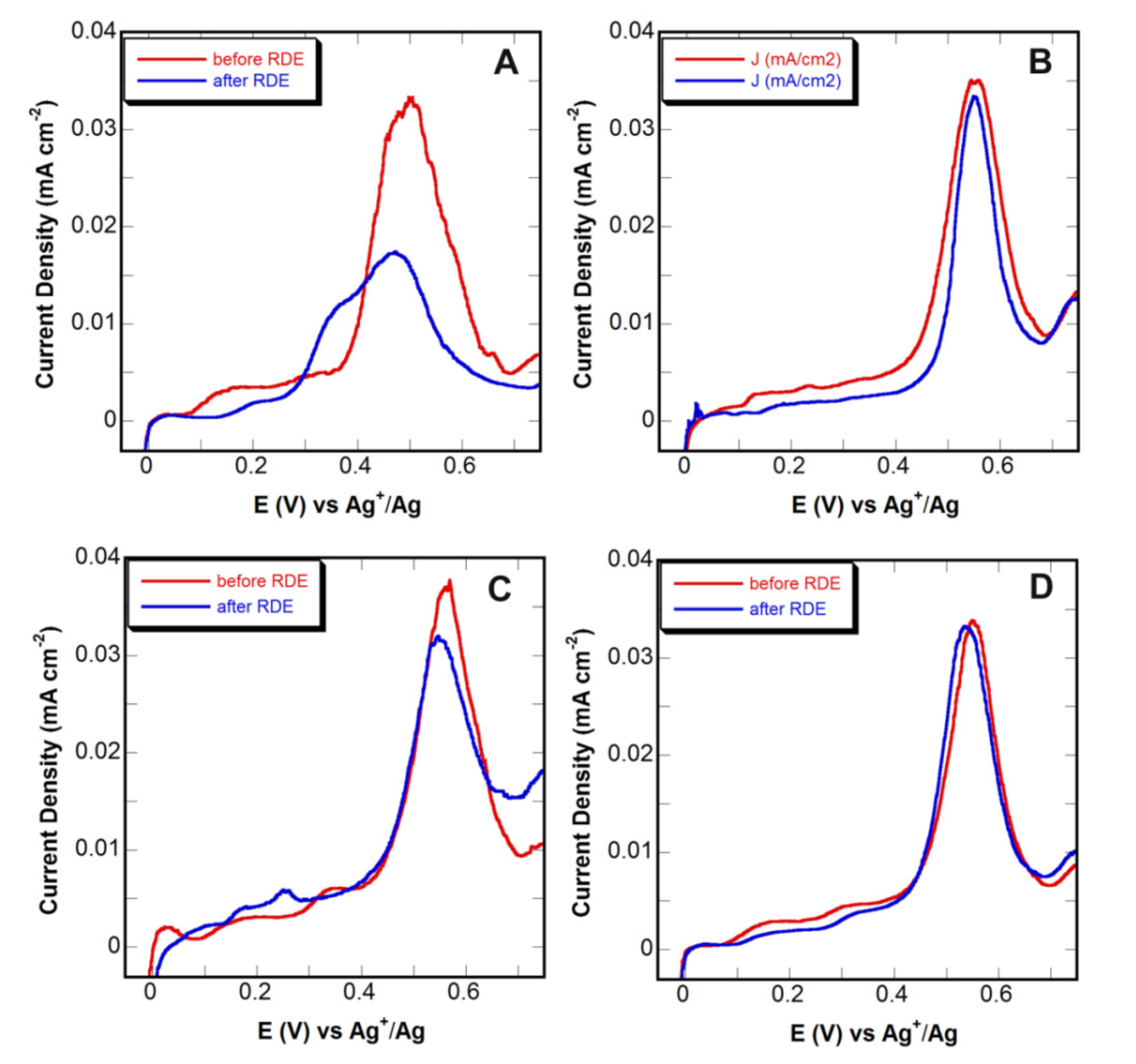
2.6. Surface Area Measurements
2.7. Adhesion Assessment
2.8. Scanning Electron Microscopy, Dual-Beam FIB/SEM and Energy Dispersive X-ray Spectroscopy
2.9. Contact Angle Measurements
3. Results and Discussion
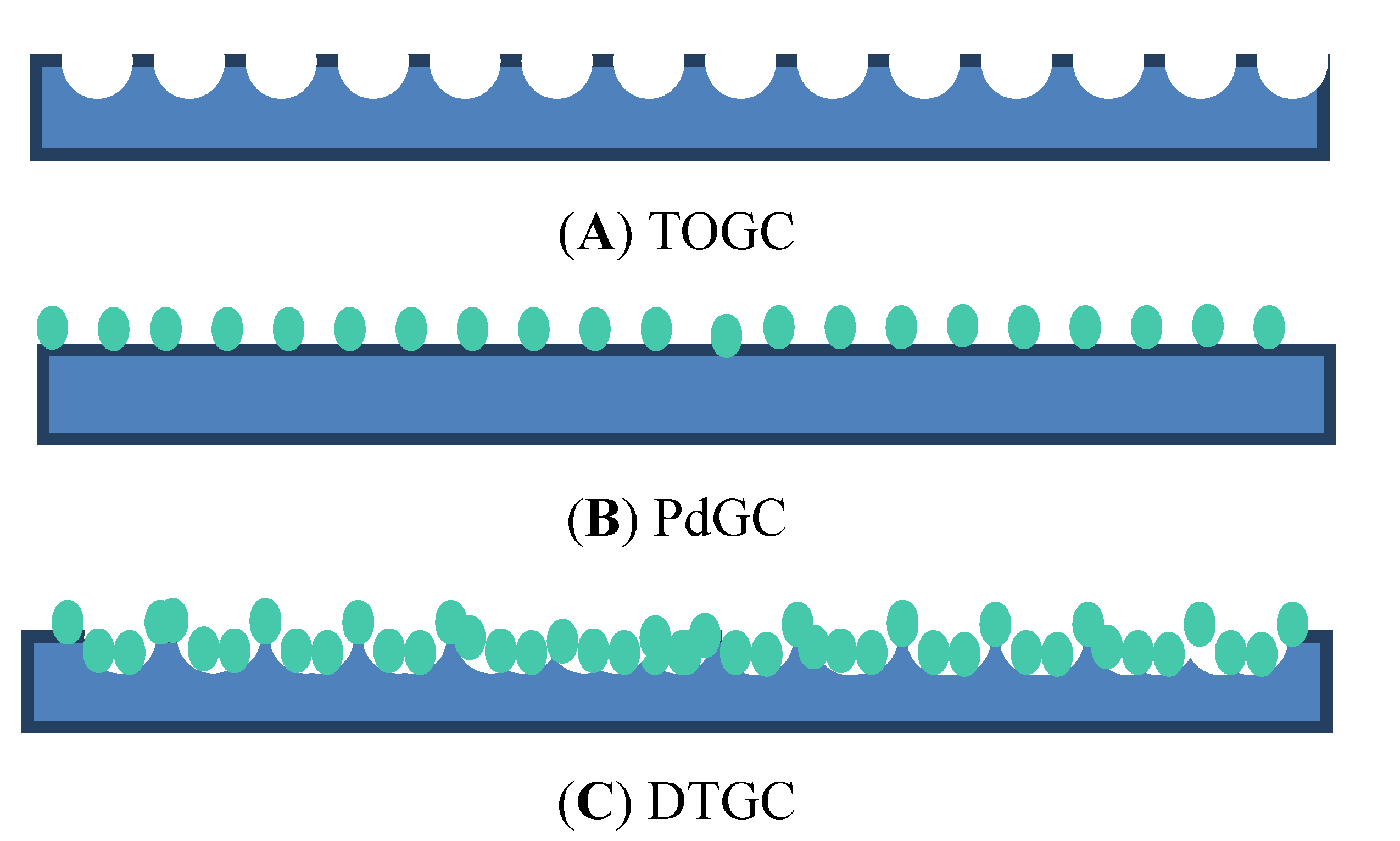
3.1. Nucleation Density





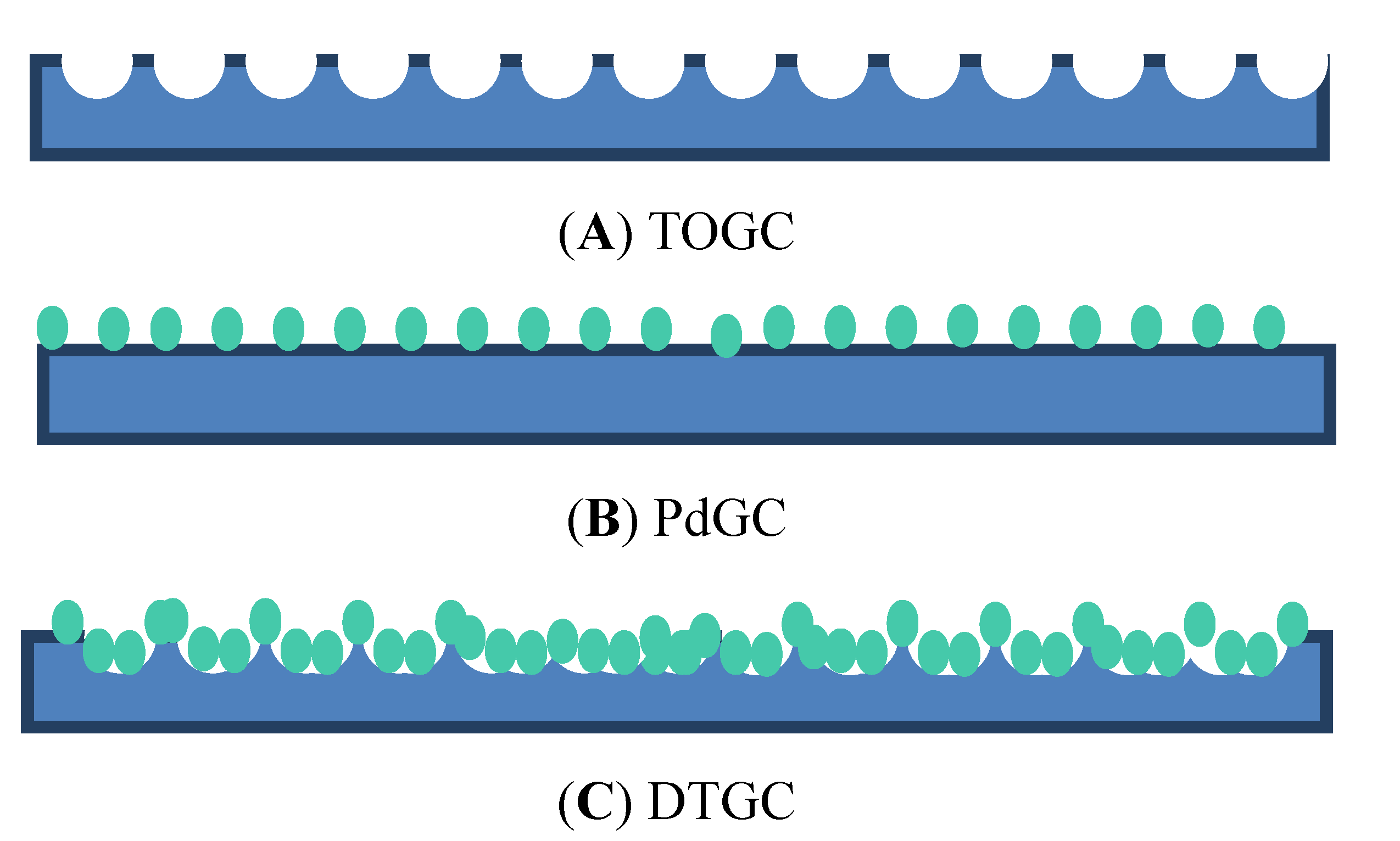
3.2. Contact Angle Measurements

3.3. Adhesion Tests



| Material | Treatment | % Surface Area Loss (based on Pb UPD) | % Surface Area Loss (based on de-alloying) |
|---|---|---|---|
| Au0.3Ag0.7 thin film | Untreated-control | 62 ± 12 | 42 ± 8 |
| Au0.3Ag0.7 thin film | Pd seed layer only | 45 ± 5 | 22 ± 5 |
| Au0.3Ag0.7 thin film | Thermochemical oxidation | 5 ± 4 | 1 ± 3 |
| Au0.3Ag0.7 thin film | Thermochemical oxidation followed by Pd seed layer | 1 ± 3 | 2 ± 3 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Erlebacher, J.; Aziz, M.J.; Karma, A.; Dimitrov, N.; Sieradzki, K. Evolution of nanoporosity in dealloying. Nature 2001, 410, 450–453. [Google Scholar] [CrossRef]
- Collinson, M.M. Nanoporous gold electrodes and their applications in analytical chemistry. ISRN Anal. Chem. 2013, 2013, 692484:1–692484:21. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.W.; Erlebacher, J. Metallic mesoporous nanocomposites for electrocatalysis. J. Am. Chem. Soc. 2004, 126, 6876–6877. [Google Scholar]
- Thotiyl, M.M.O.; Freunberger, S.A.; Peng, Z.Q.; Chen, Y.H.; Liu, Z.; Bruce, P.G. A stable cathode for the aprotic li-o-2 battery. Nat. Mater. 2013, 12, 1049–1055. [Google Scholar]
- Wittstock, A.; Biener, J.; Erlebacher, J.; Bäumer, M. Nanoporous Gold: From an Ancient Technology to a High-Tech Material; Royal Society of Chemistry: London, UK, 2012; pp. 1–252. [Google Scholar]
- Zeis, R.; Mathur, A.; Fritz, G.; Lee, J.; Erlebacher, J. Platinum-plated nanoporous gold: An efficient, low pt loading electrocatalyst for pem fuel cells. J. Power Sources 2007, 165, 65–72. [Google Scholar] [CrossRef]
- McCurry, D.A.; Kamundi, M.; Fayette, M.; Wafula, F.; Dimitrov, N. All electrochemical fabrication of a platinized nanoporous au thin-film catalyst. ACS Appl. Mater. Inter. 2011, 3, 4459–4468. [Google Scholar]
- Zeis, R.; Lei, T.; Sieradzki, K.; Snyder, J.; Erlebacher, J. Catalytic reduction of oxygen and hydrogen peroxide by nanoporous gold. J. Catal. 2008, 253, 132–138. [Google Scholar] [CrossRef]
- Seker, E.; Reed, M.L.; Begley, M.R. Nanoporous gold: Fabrication, characterization, and applications. Materials 2009, 2, 2188–2215. [Google Scholar] [CrossRef]
- Bromberg, L.; Fayette, M.; Martens, B.; Luo, Z.P.; Wang, Y.; Xu, D.; Zhang, J.; Fang, J.; Dimitrov, N. Catalytic performance comparison of shape-dependent nanocrystals and oriented ultrathin films of pt4cu alloy in the formic acid oxidation process. Electrocatalysis 2013, 4, 24–36. [Google Scholar] [CrossRef]
- Fayette, M.; Liu, Y.; Bertrand, D.; Nutariya, J.; Vasiljevic, N.; Dimitrov, N. From au to pt via surface limited redox replacement of pb upd in one-cell configuration. Langmuir 2011, 27, 5650–5658. [Google Scholar] [CrossRef]
- Ge, X.; Wang, R.; Ding, Y. Platinum-decorated nanoporous gold leaf for methanol electrooxidation. Chem. Mater. 2007, 19, 5827–5829. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Kim, J.Y.; Vairavapandian, D.; Stickney, J.L. Platinum nanofilm formation by ec-ale via redox replacement of upd copper: Studies using in-situ scanning tunneling microscopy. J. Phys. Chem. B 2006, 36, 17998–18006. [Google Scholar]
- Bicelli, L.P.; Bozzini, B.; Mele, C.; D’Urzo, L. A review of nanostructural aspects of metal electrodeposition. Int. J. Electrochem. Sci. 2008, 3, 356–408. [Google Scholar]
- Kamundi, M.; Bromberg, L.; Fey, E.; Mitchell, C.; Fayette, M.; Dimitrov, N. Impact of structure and composition on the dealloying of AuxAg(1−x) alloys on the nanoscale. J. Phys. Chem. C 2012, 116, 14123–14133. [Google Scholar]
- Kamundi, M.; Bromberg, L.; Ogutu, P.; Dimitrov, N. Seeding strategies for the deposition of high density network of nanoporous au cluster catalyst on glassy carbon electrodes. J. Appl. Electrochem. 2013, 43, 879–890. [Google Scholar] [CrossRef]
- Bromberg, L.; Xia, J.; Fayette, M.; Dimitrov, N. Synthesis of ultrathin and continuous layers of nanoporous Au on glassy carbon substrates. J. Electrochem. Soc. 2014, 161, D3001–D3010. [Google Scholar] [CrossRef]
- Miklos, J.; Mund, K.; Naschwitz, W. Siemens AG, Offenlegungsschrift DE. German Patent 30 11 701 A1, 1980.
- Hahn, M.; Bärtsch, M.; Schnyder, B.; Kotz, R.; Carlen, M.; Ohler, C.; Edvard, D. A 24 V Bipolar Electrochemical Double Layer Capacitor Based on Activated Glassy Carbon. In Power Sources for the New Millenium; Ryan, M.A., Surampudi, S., Jain, M., Eds.; The Electrochemical Society: Pennington, NJ, USA, 2001; pp. 220–228. [Google Scholar]
- Savouchkina, A.; Foelske-Schmitz, A.; Scherer, G.G.; Wokaun, A.; Kötz, R. Study of platinum deposition on untreated and thermally modified glassy carbon. J. Electrochem. Soc. 2011, 158, D420–D425. [Google Scholar] [CrossRef]
- Braun, A.; Bartsch, M.; Schnyder, B.; Kotz, R.; Haas, O.; Haubold, H.G.; Goerigk, G. X-ray scattering and adsorption studies of thermally oxidized glassy carbon. J. Non-Cryst. Solids 1999, 260, 1–14. [Google Scholar] [CrossRef]
- Lewis, J.; Redfern, B.; Cowlard, F. Vitreous carbon as a crucible material for semiconductors. Solid State Electron. 1963, 6, 251–257. [Google Scholar] [CrossRef]
- Cowlard, F.; Lewis, J. Vitreous carbon—A new form of carbon. J. Mater. Sci. 1967, 2, 507–512. [Google Scholar] [CrossRef]
- Braun, A. Development and Characterization of Glassy Carbon Electrodes for a Bipolar Electrochemical Double Layer Capacitor. Ph.D. Dissertation, Swiss Federal Institute of Technology, Zurich, Switzerland, December 1999. [Google Scholar]
- Liu, Y.; Bliznakov, S.; Dimitrov, N. Comprehensive study of the application of a pb underpotential deposition-assisted method for surface area measurement of metallic nanoporous materials. J. Phys. Chem. C 2009, 113, 12362–12372. [Google Scholar] [CrossRef]
- Maruyama, J.; Abe, I. Influence of anodic oxidation of glassy carbon surface on voltammetric behavior of Nafion (R)-coated glassy carbon electrodes. Electrochim. Acta. 2001, 46, 3381–3386. [Google Scholar] [CrossRef]
- Engstrom, R. Electrochemical pretreatment of glassy carbon electrodes. Anal. Chem. 1982, 54, 2310–2314. [Google Scholar] [CrossRef]
- Engstrom, R.; Strasser, V.P. Characterization of electrochemically pretreated glassy carbon electrodes. Anal Chem. 1984, 56, 136–141. [Google Scholar] [CrossRef]
- Harris, P.J.F. Fullerene-related structure of commercial glassy carbons. Philos. Mag. 2004, 84, 3159–3167. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Kawamura, K. Structure of glassy carbon. Nature 1971, 231, 175–176. [Google Scholar] [CrossRef]
- Braun, A.B.; Bärtsch, M.; Merlo, O.; Schnyder, B.; Schaffner, B.; Kotz, R.; Haas, O.; Wokaun, A. Exponential growth of electrochemical double layer capacitance in glassy carbon during thermal oxidation. Carbon 2003, 41, 759–765. [Google Scholar] [CrossRef]
- Ergun, S.T.; Tiensuu, V.H. Alicyclic structures in coals. Nature 1959, 183, 1668–1670. [Google Scholar] [CrossRef]
- Barbero, C.; Silber, J.J.; Soreno, L. Studies of surface-modified glassy carbon electrodes obtained by electrochemical treatment: Its effect on
![Coatings 04 00416 i001]() adsorption and the electron transfer rates of the Fe2+/Fe3+ couple. J. Electroanal. Chem. 1988, 248, 321–340. [Google Scholar] [CrossRef]
adsorption and the electron transfer rates of the Fe2+/Fe3+ couple. J. Electroanal. Chem. 1988, 248, 321–340. [Google Scholar] [CrossRef] - Sullivan, M.G.; Schnyder, B.; Bärtsch, M.; Alliata, D.; Barbero, C.; Imhof, R.; Kötz, R. Electrochemically modified glassy carbon for capacitor electrodes characterization of thick anodic layers by cyclic voltammetry, differential electrochemical mass spectrometry, spectroscopic ellipsometry, X-ray photoelectron spectroscopy, FTIR, and AFM. J. Electrochem. Soc. 2000, 147, 2636–2643. [Google Scholar] [CrossRef]
- Wenzel, R.N. Resistance of Solid Surfaces to Wetting by Water. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Marmur, A. Wetting on hydrophobic rough surfaces: To be heterogeneous or not to be? Langmuir 2003, 19, 8343–8348. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bromberg, L.A.; Xia, J.; Rooney, R.; Dimitrov, N. Enhanced Adhesion of Continuous Nanoporous Au Layers by Thermochemical Oxidation of Glassy Carbon. Coatings 2014, 4, 416-432. https://doi.org/10.3390/coatings4030416
Bromberg LA, Xia J, Rooney R, Dimitrov N. Enhanced Adhesion of Continuous Nanoporous Au Layers by Thermochemical Oxidation of Glassy Carbon. Coatings. 2014; 4(3):416-432. https://doi.org/10.3390/coatings4030416
Chicago/Turabian StyleBromberg, Lori Ana, Jiaxin Xia, Ryan Rooney, and Nikolay Dimitrov. 2014. "Enhanced Adhesion of Continuous Nanoporous Au Layers by Thermochemical Oxidation of Glassy Carbon" Coatings 4, no. 3: 416-432. https://doi.org/10.3390/coatings4030416
APA StyleBromberg, L. A., Xia, J., Rooney, R., & Dimitrov, N. (2014). Enhanced Adhesion of Continuous Nanoporous Au Layers by Thermochemical Oxidation of Glassy Carbon. Coatings, 4(3), 416-432. https://doi.org/10.3390/coatings4030416