Drug Resistance Mechanisms in Mycobacterium tuberculosis

Abstract
:1. Introduction
2. First-Line Anti-TB Drugs
2.1. Rifampicin
2.2. Isoniazid
2.3. Ethambutol
2.4. Pyrazinamide
2.5. Streptomycin
3. Second-Line Anti-TB Drugs
3.1. Fluoroquinolones
3.2. Kanamycin, Capreomycin, Amikacin, Viomycin
3.3. Ethionamide
3.4. Para-Amino Salicylic Acid
3.5. Cycloserine
3.6. Thioacetazone
3.7. Macrolides
3.8. Clofazimine
3.9. Linezolid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Gene | Mechanism Involved | Reference |
|---|---|---|---|
| Isoniazid | katG, inhA | Catalase/peroxidase; enoyl reductase | [26] |
| Rifampicin | rpoB | RNA polymerase | [11,12,13] |
| Pyrazinamide | pncA, rpsA | Pyrazinamidase; ribosomal protein 1 | |
| Ethambutol | embB | Arabinosyl transferase | [40,41] |
| Streptomycin | rpsL, rrs, gidB | S12 ribosomal protein, 16A rRNA, 7-methylguanosine methyltransferase | [59,60,61] |
| Quinolones | gyrA, gyrB | DNA gyrase | [67,68] |
| Capreomycin | rrs, tlyA | 16S rRNA, rRNA methyltransferase | [80,81] |
| Kanamycin/Amikacin | rrs | 16S rRNA | [83] |
| Ethionamide | ethA | Enoyl-ACP reductase | [85,86] |
| Para-aminosalicylic acid | thyA, folC | Thymidylate synthase A | [87,88,89] |
4. New Anti-TB Drugs
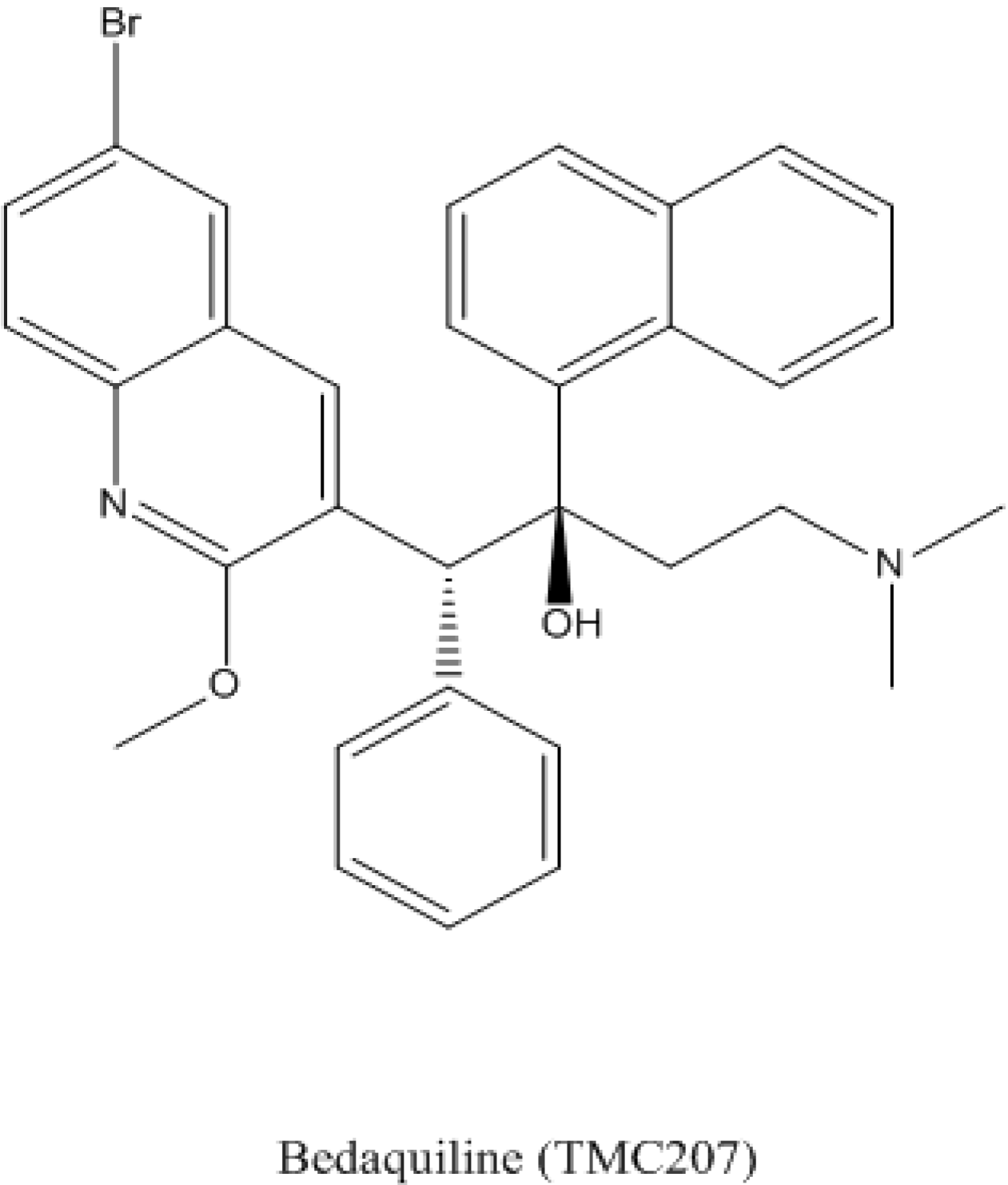
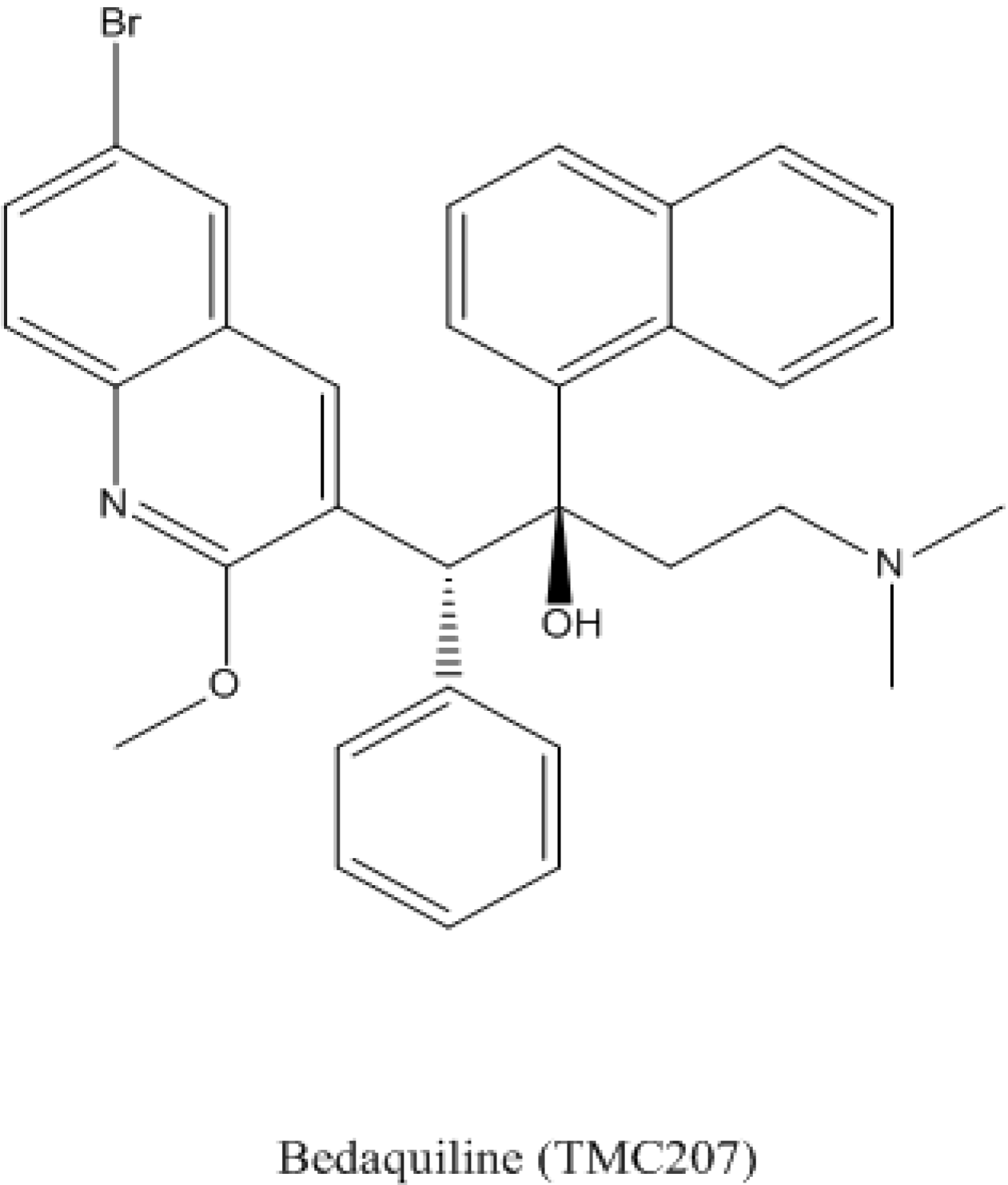
4.1. Bedaquiline
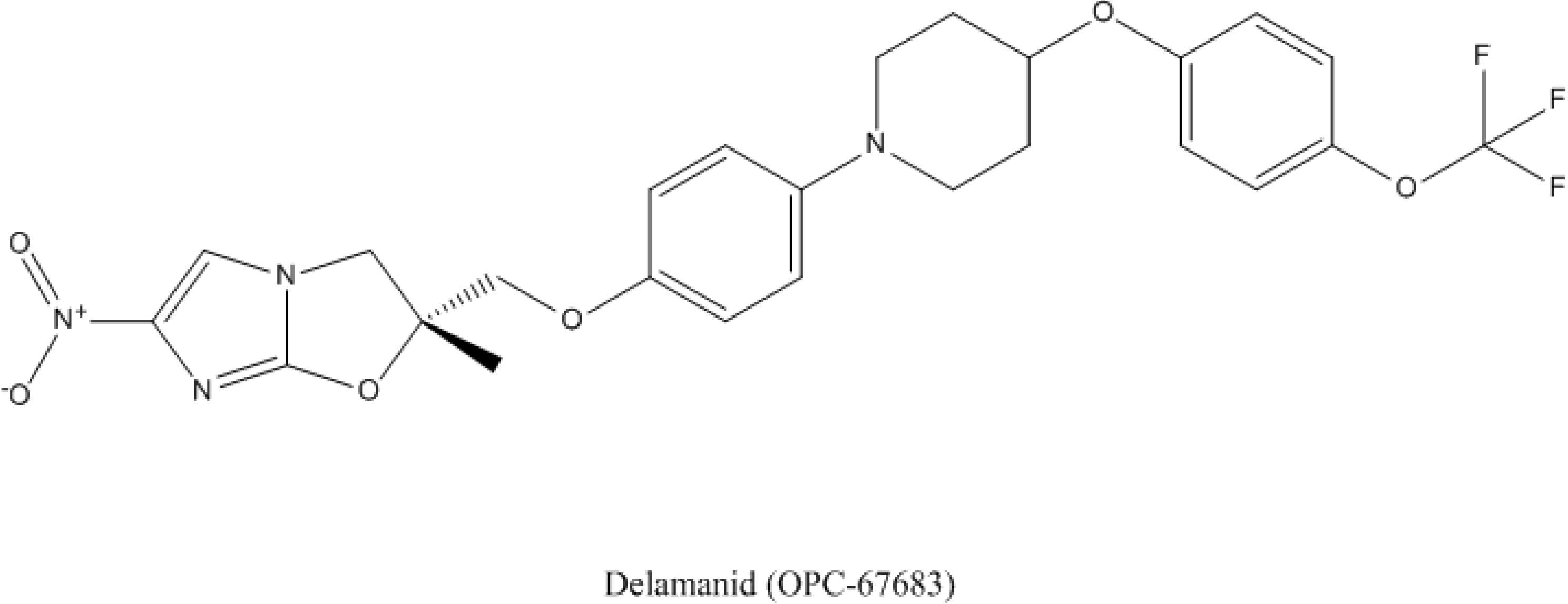
4.2. Delamanid
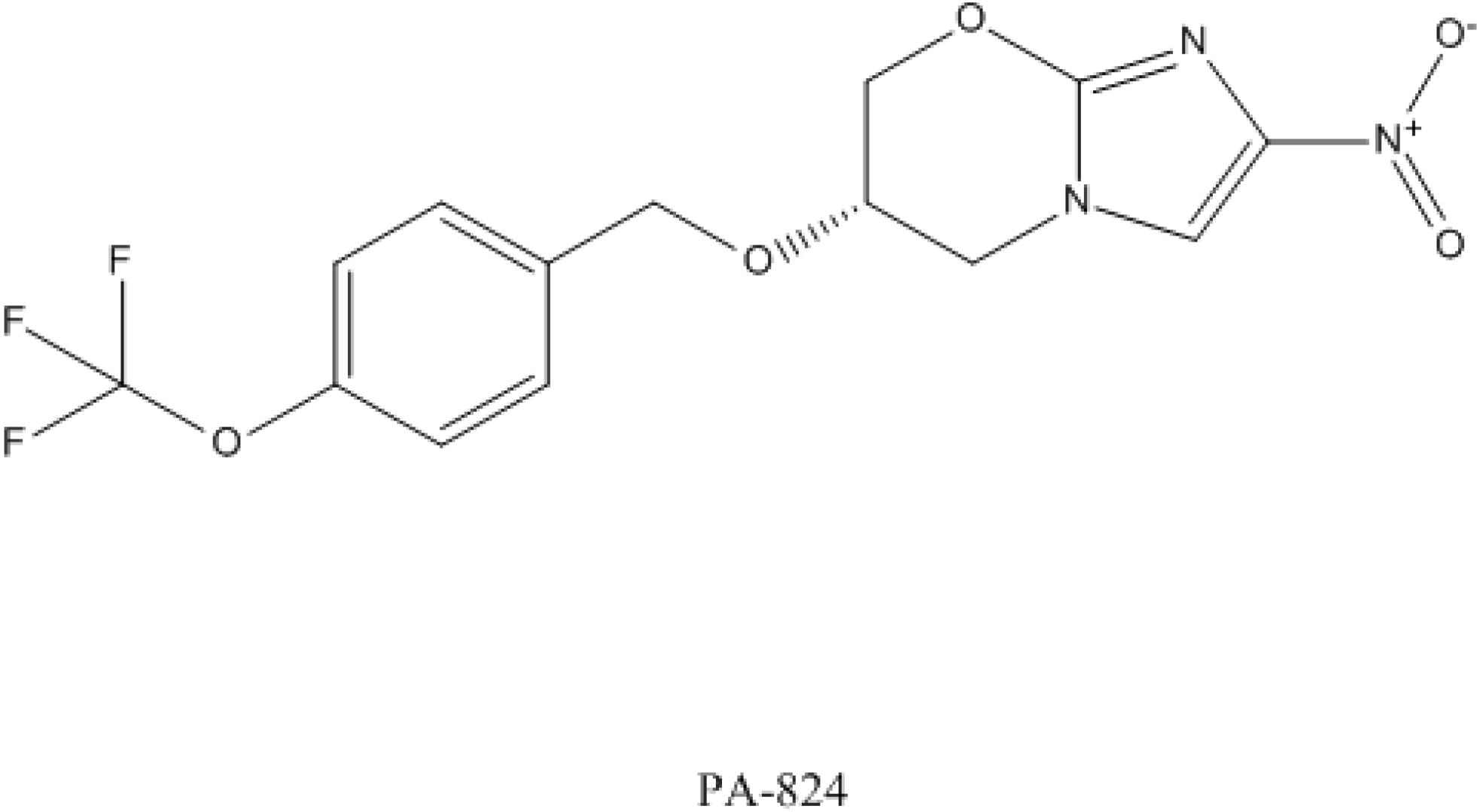
4.3. PA-824
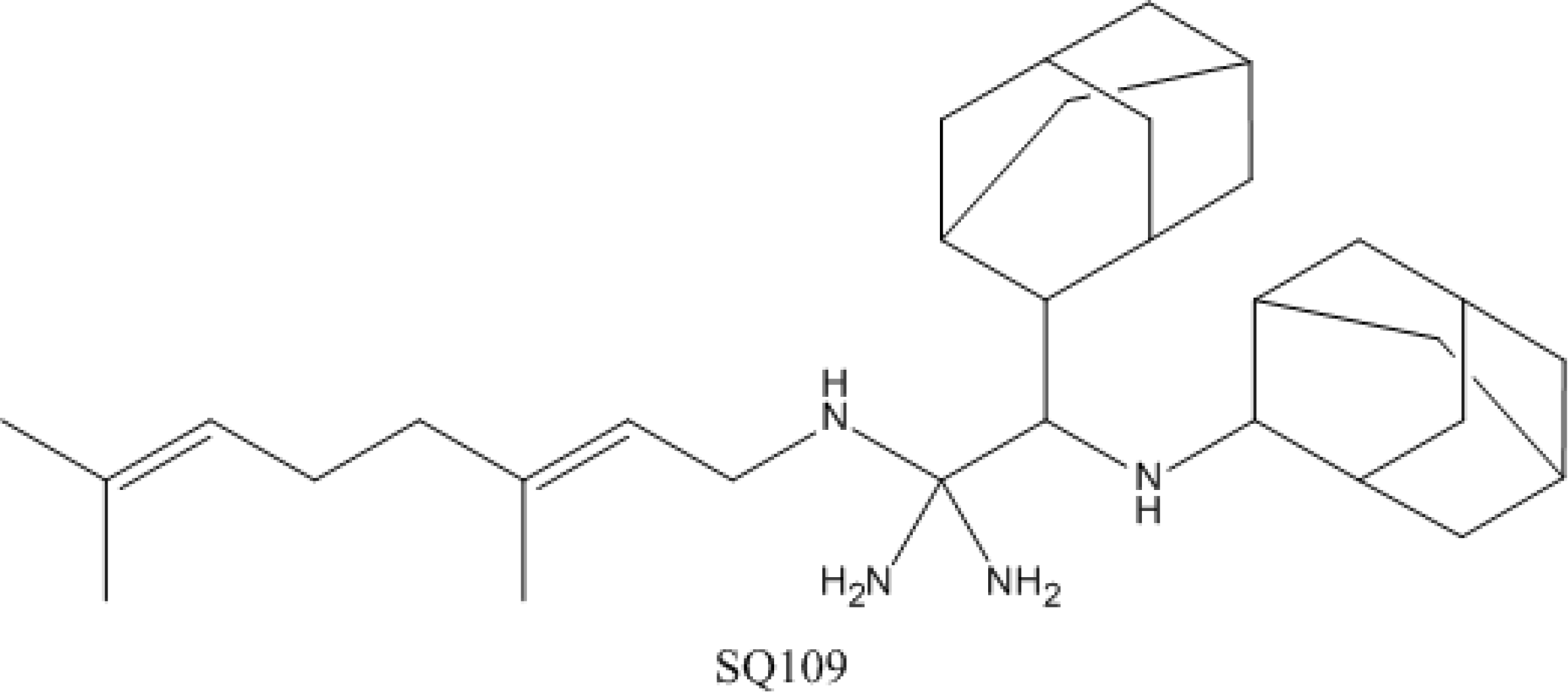
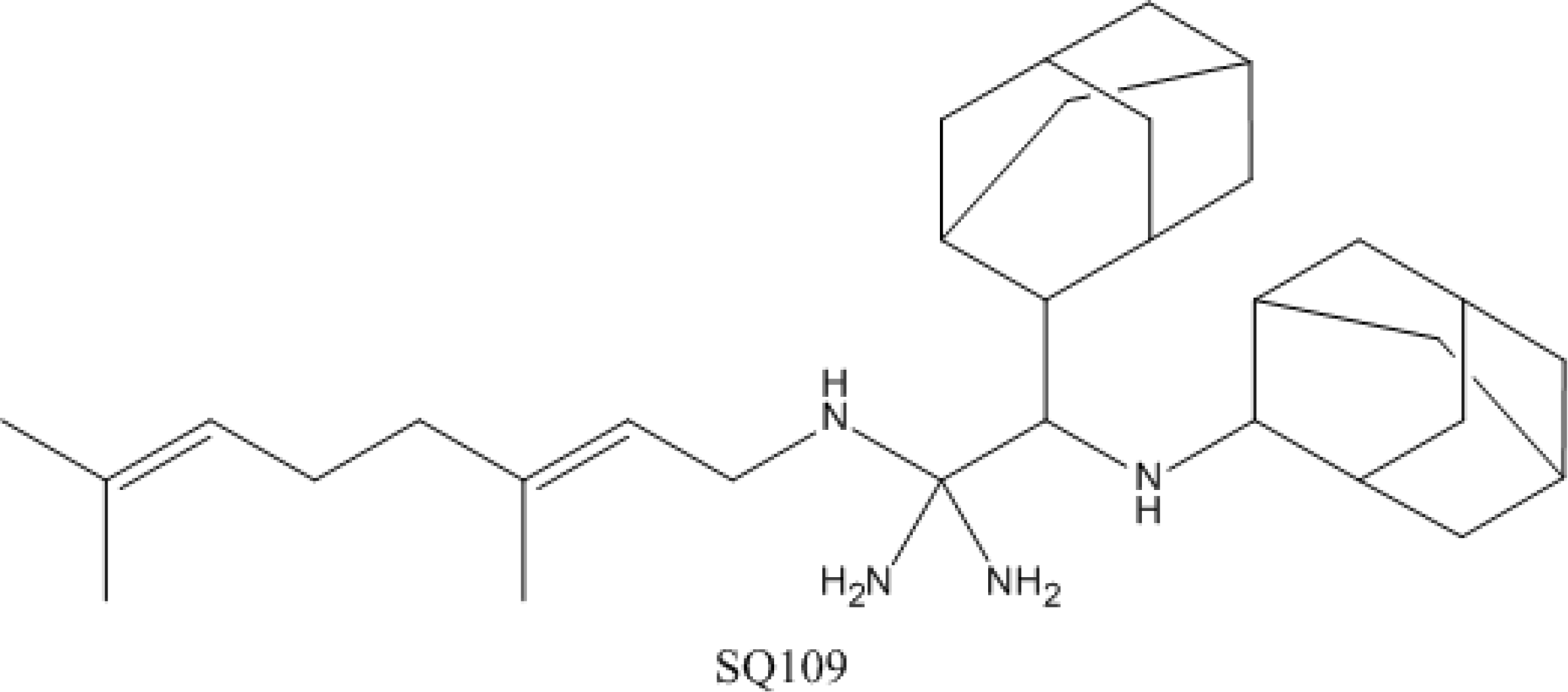
4.4. SQ-109
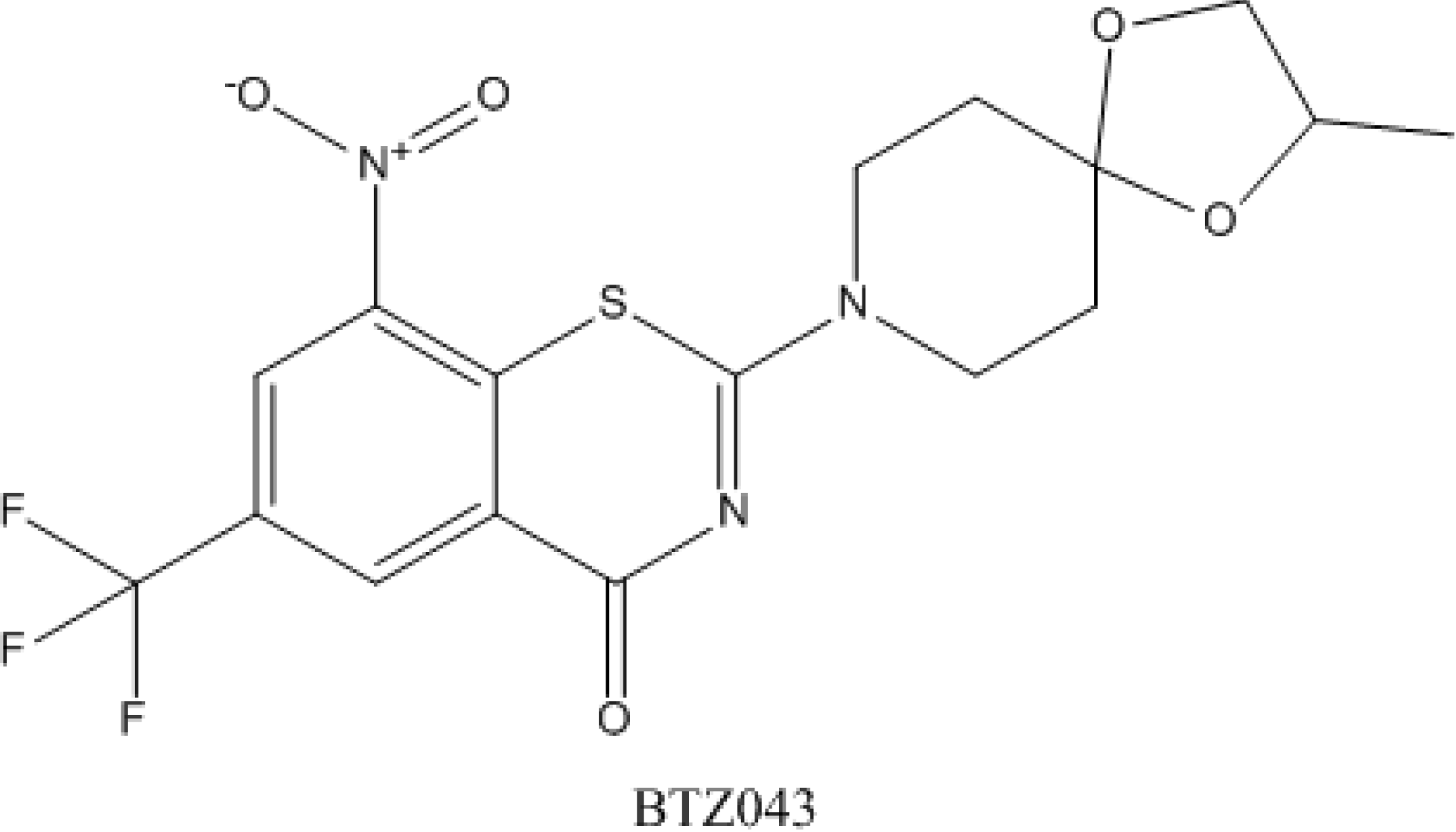
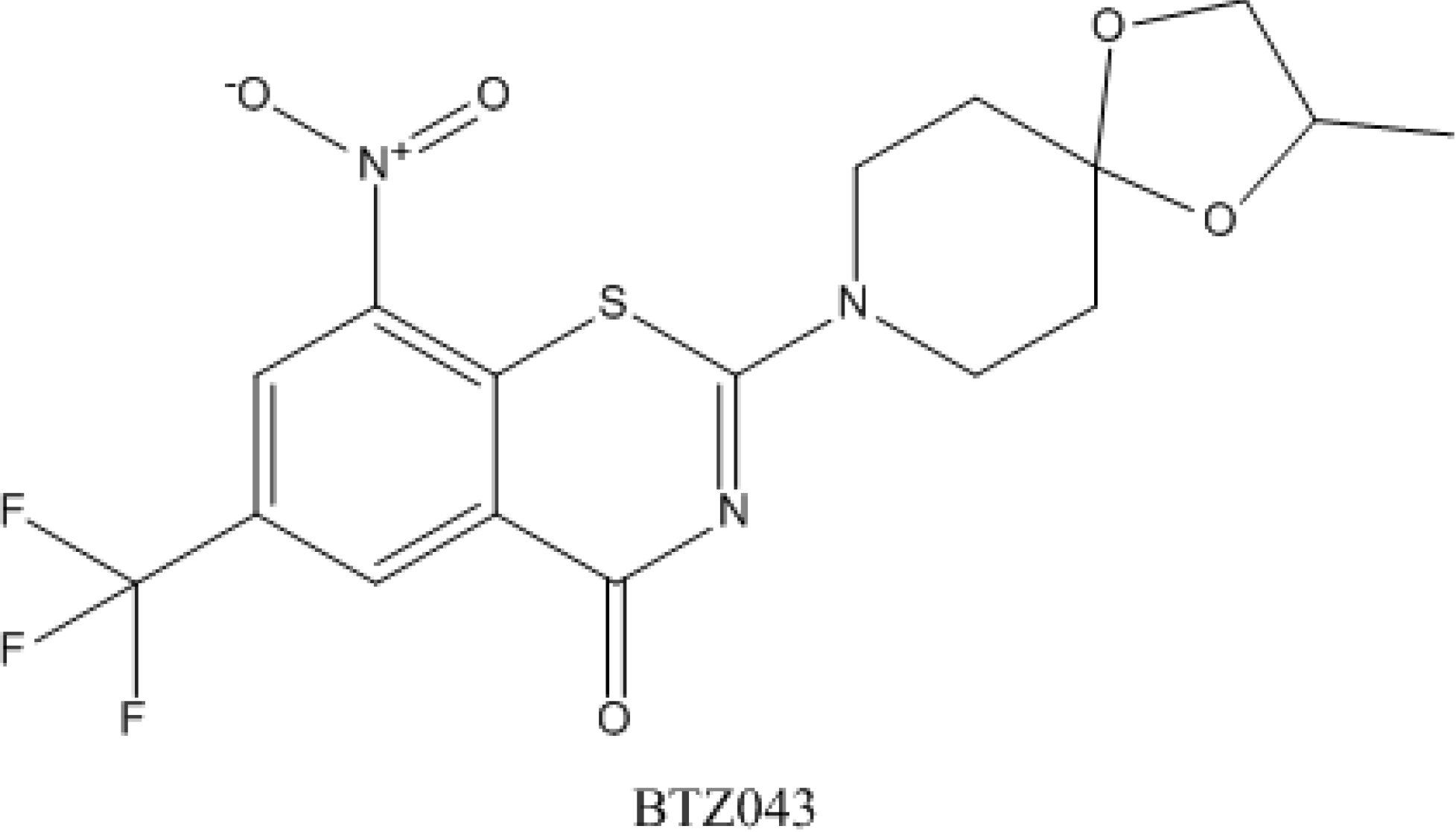
4.5. Benzothiazinones
5. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2013; WHO/HTM/TB/2013.11: Geneva, Switzerland, 2013. [Google Scholar]
- Centers for Disease Control and Prevention. Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs—worldwide, 2000–2004. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 301–305. [Google Scholar]
- World Health Organization. Multidrug and Extensively Drug-resistant TB (M/XDR-TB); 2010 Global Report on Surveillance and Response; WHO/HTM/TB/2010.3: Geneva, Switzerland, 2010. [Google Scholar]
- Gandhi, N.R.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govender, T.; Lalloo, U.; Zeller, K.; Andrews, J.; Friedland, G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006, 368, 1575–1580. [Google Scholar] [CrossRef]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; Ziazarifi, A.H.; Hoffner, S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: Super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Amale, R.A.; Ajbani, K.K.; Rodrigues, C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2012, 54, 579–581. [Google Scholar] [CrossRef]
- Migliori, G.B.; Centis, R.; D’Ambrosio, L.; Spanevello, A.; Borroni, E.; Cirillo, D.M.; Sotgiu, G. Totally drug-resistant and extremely drug-resistant tuberculosis: The same disease? Clin. Infect. Dis. 2012, 54, 1379–1380. [Google Scholar]
- Mitchison, D.A. Basic mechanisms of chemotherapy. Chest 1979, 76, 771–781. [Google Scholar] [CrossRef]
- Blanchard, J.S. Molecular mechanisms of drug resistance in Mycobacterium tuberculosis. Annu. Rev. Biochem. 1996, 65, 215–239. [Google Scholar] [CrossRef]
- Telenti, A.; Imboden, P.; Marchesi, F.; Lowrie, D.; Cole, S.; Colston, M.J.; Matter, L.; Schopfer, K.; Bodmer, T. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 1993, 341, 647–550. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Musser, J.M. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 1998, 79, 3–29. [Google Scholar] [CrossRef]
- Somoskovi, A.; Parsons, L.M.; Salfinger, M. The molecular basis of resistance to isoniazid, rifampin, and pyrazinamide in Mycobacterium tuberculosis. Respir. Res. 2001, 2, 164–168. [Google Scholar] [CrossRef]
- Caws, M.; Duy, P.M.; Tho, D.Q.; Lan, N.T.; Hoa, D.V.; Farrar, J. Mutations prevalent among rifampin and isoniazid-resistant Mycobacterium tuberculosis isolates from a hospital in Vietnam. J. Clin. Microbiol. 2006, 44, 2333–2337. [Google Scholar] [CrossRef] [Green Version]
- Heep, M.; Rieger, U.; Beck, D.; Lehn, N. Mutations in the beginning of the rpoB gene can induce resistance to rifamycins in both Helicobacter pylori and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2000, 44, 1075–1077. [Google Scholar] [CrossRef]
- Siu, G.K.; Zhang, Y.; Lau, T.C.; Lau, R.W.; Ho, P.L.; Yew, W.W.; Tsui, S.K.; Cheng, V.C.; Yuen, K.Y.; Yam, W.C. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2011, 66, 730–733. [Google Scholar] [CrossRef]
- Yang, B.; Koga, H.; Ohno, H.; Ogawa, K.; Fukuda, M.; Hirakata, Y.; Maesaki, S.; Tomono, K.; Tashiro, T.; Kohno, S. Relationship between antimycobacterial activities of rifampicin, rifabutin and KRM-1648 and rpoB mutations of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 1998, 42, 621–628. [Google Scholar] [CrossRef]
- Cavusoglu, C.; Karaca-Derici, Y.; Bilgic, A. In-vitro activity of rifabutin against rifampicin-resistant Mycobacterium tuberculosis isolates with known rpoB mutations. Clin. Microbiol. Infect. 2004, 10, 662–665. [Google Scholar] [CrossRef]
- Burman, W.J.; Jones, B.E. Treatment of HIV-related tuberculosis in the era of effective antiretroviral therapy. Am. J. Respir. Crit. Care Med. 2001, 164, 7–12. [Google Scholar] [CrossRef]
- Traore, H.; Fissette, K.; Bastian, I.; Devleeschouwer, M.; Portaels, F. Detection of rifampicin resistance in Mycobacterium tuberculosis isolates from diverse countries by a commercial line probe assay as an initial indicator of multidrug resistance. Int. J. Tuberc. Lung Dis. 2000, 4, 481–484. [Google Scholar]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2011, 44, 106–110. [Google Scholar]
- Brandis, G.; Hughes, D. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J. Antimicrob. Chemother. 2013, 68, 2493–2497. [Google Scholar] [CrossRef]
- De Vos, M.; Müller, B.; Borrell, S.; Black, P.A.; van Helden, P.D.; Warren, R.M.; Gagneux, S.; Victor, T.C. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 2013, 57, 827–832. [Google Scholar] [CrossRef]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Rawat, R.; Whitty, A.; Tonge, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar] [CrossRef]
- Ramaswamy, S.V.; Reich, R.; Dou, S.J.; Jasperse, L.; Pan, X.; Wanger, A.; Quitugua, T.; Graviss, E.A. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 1241–1250. [Google Scholar] [CrossRef]
- Hazbón, M.H.; Brimacombe, M.; Bobadilla del Valle, M.; Cavatore, M.; Guerrero, M.I.; Varma-Basil, M.; Billman-Jacobe, H.; Lavender, C.; Fyfe, J.; García-García, L.; et al. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2006, 50, 2640–2649. [Google Scholar] [CrossRef]
- Vilchèze, C.; Jacobs, W.R., Jr. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Grant, G.A.; Barton, D.H.; Jacobs, W.R., Jr.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar] [CrossRef]
- Fenner, L.; Egger, M.; Bodmer, T.; Altpeter, E.; Zwahlen, M.; Jaton, K.; Pfyffer, G.E.; Borrell, S.; Dubuis, O.; Bruderer, T.; et al. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3047–3053. [Google Scholar] [CrossRef]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R., Jr. InhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar]
- Larsen, M.H.; Vilchèze, C.; Kremer, L.; Besra, G.S.; Parsons, L.; Salfinger, M.; Heifets, L.; Hazbon, M.H.; Alland, D.; Sacchettini, J.C.; et al. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol. Microbiol. 2002, 46, 453–466. [Google Scholar] [CrossRef]
- Machado, D.; Perdigão, J.; Ramos, J.; Couto, I.; Portugal, I.; Ritter, C.; Boettger, E.C.; Viveiros, M. High-level resistance to isoniazid and ethionamide in multidrug-resistant Mycobacterium tuberculosis of the Lisboa family is associated with inhA double mutations. J. Antimicrob. Chemother. 2013, 68, 1728–1732. [Google Scholar] [CrossRef]
- Argyrou, A.; Vetting, M.W.; Aladegbami, B.; Blanchard, J.S. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat. Struct. Mol. Biol. 2006, 13, 408–413. [Google Scholar] [CrossRef]
- Argyrou, A.; Jin, L.; Siconilfi-Baez, L.; Angeletti, R.H.; Blanchard, J.S. Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry 2006, 45, 13947–13953. [Google Scholar] [CrossRef]
- Ho, Y.M.; Sun, Y.J.; Wong, S.Y.; Lee, A.S. Contribution of dfrA and inhA mutations to the detection of isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2009, 53, 4010–4012. [Google Scholar] [CrossRef]
- Wang, F.; Jain, P.; Gulten, G.; Liu, Z.; Feng, Y.; Ganesula, K.; Motiwala, A.S.; Ioerger, T.R.; Alland, D.; Vilchèze, C.; et al. Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob. Agents Chemother. 2010, 54, 3776–3782. [Google Scholar] [CrossRef]
- Rinder, H.; Thomschke, A.; Rüsch-Gerdes, S.; Bretzel, G.; Feldmann, K.; Rifai, M.; Löscher, T. Significance of ahpC promoter mutations for the prediction of isoniazid resistance in Mycobacterium tuberculosis. Eur. J. Clin. Microbiol. Infect. Dis. 1998, 17, 508–511. [Google Scholar]
- Sherman, D.R.; Mdluli, K.; Hickey, M.J.; Arain, T.M.; Morris, S.L.; Barry, C.E., 3rd.; Stover, C.K. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 1996, 272, 1641–1643. [Google Scholar]
- Heym, B.; Stavropoulos, E.; Honoré, N.; Domenech, P.; Saint-Joanis, B.; Wilson, T.M.; Collins, D.M.; Colston, M.J.; Cole, S.T. Effects of overexpression of the alkyl hydroperoxide reductase AhpC on the virulence and isoniazid resistance of Mycobacterium tuberculosis. Infect. Immun. 1997, 65, 1395–1401. [Google Scholar]
- Cardoso, R.F.; Cardoso, M.A.; Leite, C.Q.; Sato, D.N.; Mamizuka, E.M.; Hirata, R.D.; de Mello, F.F.; Hirata, M.H. Characterization of ndh gene of isoniazid resistant and susceptible Mycobacterium tuberculosis isolates from Brazil. Mem. Inst. Oswaldo Cruz 2007, 102, 59–61. [Google Scholar] [CrossRef]
- Ando, H.; Kitao, T.; Miyoshi-Akiyama, T.; Kato, S.; Mori, T.; Kirikae, T. Downregulation of katG expression is associated with isoniazid resistance in Mycobacterium tuberculosis. Mol. Microbiol. 2011, 79, 1615–1628. [Google Scholar] [CrossRef]
- Miesel, L.; Weisbrod, T.R.; Marcinkeviciene, J.A.; Bittman, R.; Jacobs, W.R., Jr. NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J. Bacteriol. 1998, 180, 2459–2467. [Google Scholar]
- Vilcheze, C.; Weisbrod, T.R.; Chen, B.; Kremer, L.; Hazbón, M.H.; Wang, F.; Alland, D.; Sachettini, J.C.; Jacobs, W.R., Jr. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob. Agents Chemother. 2005, 49, 708–720. [Google Scholar]
- Ando, H.; Miyoshi-Akiyama, T.; Watanabe, S.; Kirikae, T. A silent mutation in mabA confers isoniazid resistance on Mycobacterium tuberculosis. Mol. Microbiol. 2014, 91, 538–547. [Google Scholar] [CrossRef]
- Takayama, K.; Kilburn, J.O. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. [Google Scholar] [CrossRef]
- Mikusová, K.; Slayden, R.A.; Besra, G.S.; Brennan, P.J. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 1995, 39, 2484–2489. [Google Scholar] [CrossRef]
- Telenti, A.; Philipp, W.J.; Sreevatsan, S.; Bernasconi, C.; Stockbauer, K.E.; Wieles, B.; Musser, J.M.; Jacobs, W.R., Jr. The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 1997, 3, 567–570. [Google Scholar] [CrossRef]
- Sreevatsan, S.; Stockbauer, K.E.; Pan, X.; Kreiswirth, B.N.; Moghazeh, S.L.; Jacobs, W.R., Jr.; Telenti, A.; Musser, J.M. Ethambutol resistance in Mycobacterium tuberculosis: Critical role of embB mutations. Antimicrob. Agents Chemother. 1997, 41, 1677–1681. [Google Scholar]
- Ahmad, S.; Jaber, A.A.; Mokaddas, E. Frequency of embB codon 306 mutations in ethambutol-susceptible and -resistant clinical Mycobacterium tuberculosis isolates in Kuwait. Tuberculosis (Edinb.). 2007, 87, 123–129. [Google Scholar] [CrossRef]
- Hazbón, M.H.; Bobadilla del Valle, M.; Guerrero, M.I.; Varma-Basil, M.; Filliol, I.; Cavatore, M.; Colangeli, R.; Safi, H.; Billman-Jacobe, H.; Lavender, C.; et al. Role of embB codon 306 mutations in Mycobacterium tuberculosis revisited: A novel association with broad drug resistance and IS6110 clustering rather than ethambutol resistance. Antimicrob. Agents Chemother. 2005, 49, 3794–3802. [Google Scholar] [CrossRef]
- Safi, H.; Sayers, B.; Hazbón, M.H.; Alland, D. Transfer of embB codon 306 mutations into clinical Mycobacterium tuberculosis strains alters susceptibility to ethambutol, isoniazid, and rifampin. Antimicrob. Agents Chemother. 2008, 52, 2027–2034. [Google Scholar] [CrossRef]
- Safi, H.; Lingaraju, S.; Amin, A.; Kim, S.; Jones, M.; Holmes, M.; McNeil, M.; Peterson, S.N.; Chatterjee, D.; Fleischmann, R.; et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-d-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 2013, 45, 1190–1197. [Google Scholar]
- Mitchison, D.A. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 1985, 66, 219–225. [Google Scholar] [CrossRef]
- Konno, K.; Feldmann, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Zimhony, O.; Vilchèze, C.; Arai, M.; Welch, J.T.; Jacobs, W.R., Jr. Pyrazinoic acid and its n-propylester inhibit fatty acid synthase type I in replicating tubercle bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef]
- Zimhony, O.; Cox, J.S.; Welch, J.T.; Vilchèze, C.; Jacobs, W.R., Jr. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FASI) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E., 3rd.; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef]
- Scorpio, A.; Lindholm-Levy, P.; Heifets, L.; Gilman, R.; Siddiqi, S.; Cynamon, M.; Zhang, Y. Characterization of pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1997, 41, 540–543. [Google Scholar]
- Juréen, P.; Werngren, J.; Toro, J.C.; Hoffner, S. Pyrazinamide resistance and pncA gene mutations in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 1852–1854. [Google Scholar] [CrossRef]
- Cheng, S.J.; Thibert, L.; Sanchez, T.; Heifets, L.; Zhang, Y. pncA mutations as a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis: Spread of a monoresistant strain in Quebec, QC, Canada. Antimicrob. Agents Chemother. 2000, 44, 528–532. [Google Scholar]
- Alexander, D.C.; Ma, J.H.; Guthrie, J.L.; Blair, J.; Chedore, P.; Jamieson, F.B. Gene sequencing for routine verification of pyrazinamide resistance in Mycobacterium tuberculosis: A role for pncA but not rpsA. J. Clin. Microbiol. 2012, 50, 3726–3728. [Google Scholar] [CrossRef]
- Simons, S.O.; Mulder, A.; van Ingen, J.; Boeree, M.J.; van Soolingen, D. Role of rpsA gene sequencing in diagnosis of pyrazinamide resistance. J. Clin. Microbiol. 2013, 51, 382. [Google Scholar]
- Tan, Y.; Hu, Z.; Zhang, T.; Cai, X.; Kuang, H.; Liu, Y.; Chen, J.; Yang, F.; Zhang, K.; Tan, S.; et al. Role of pncA and rpsA gene sequencing in detection of pyrazinamide resistance in Mycobacterium tuberculosis isolates from southern China. J. Clin. Microbiol. 2014, 52, 291–297. [Google Scholar] [CrossRef]
- Crofton, J.; Mitchison, D.A. Streptomycin resistance in pulmonary tuberculosis. Br. Med. J. 1948, 2, 1009–1015. [Google Scholar] [CrossRef]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- Finken, M.; Kirschner, P.; Meier, A.; Wrede, A.; Böttger, E.C. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: Alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol. Microbiol. 1993, 9, 1239–1246. [Google Scholar]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef]
- Okamoto, S.; Tamaru, A.; Nakajima, C.; Nishimura, K.; Tanaka, Y.; Tokuyama, S.; Suzuki, Y.; Ochi, K. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol. Microbiol. 2007, 63, 1096–1106. [Google Scholar] [CrossRef]
- Spies, F.S.; da Silva, P.E.; Ribeiro, M.O.; Rossetti, M.L.; Zaha, A. Identification of mutations related to streptomycin resistance in clinical isolates of Mycobacterium tuberculosis and possible involvement of efflux mechanism. Antimicrob. Agents Chemother. 2008, 52, 2947–2949. [Google Scholar]
- Goss, W.A.; Deitz, W.H.; Cook, T.M. Mechanism of action of nalidixic acid on Escherichia coli. II. Inhibition of deoxyribonucleic acid synthesis. J. Bacteriol. 1965, 89, 1068–1074. [Google Scholar]
- Rustomjee, R.; Lienhardt, C.; Kanyok, T.; Davies, G.R.; Levin, J.; Mthiyane, T.; Reddy, C.; Sturm, A.W.; Sirgel, F.A.; Allen, J.; et al. A Phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 2008, 12, 128–138. [Google Scholar]
- Palomino, J.C.; Martin, A. Tuberculosis clinical trial update and the current anti-tuberculosis drug portfolio. Curr. Med. Chem. 2013, 20, 3785–3796. [Google Scholar]
- Fàbrega, A.; Madurga, S.; Giralt, E.; Vila, J. Mechanism of action of and resistance to quinolones. Microb. Biotechnol. 2009, 2, 40–61. [Google Scholar] [CrossRef]
- Aubry, A.; Pan, X.S.; Fisher, L.M.; Jarlier, V.; Cambau, E. Mycobacterium tuberculosis DNA gyrase: Interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 2004, 48, 1281–1288. [Google Scholar]
- Takiff, H.E.; Salazar, L.; Guerrero, C.; Philipp, W.; Huang, W.M.; Kreiswirth, B.; Cole, S.T.; Jacobs, W.R., Jr.; Telenti, A. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob. Agents Chemother. 1994, 38, 773–780. [Google Scholar]
- Cheng, A.F.; Yew, W.W.; Chan, E.W.; Chin, M.L.; Hui, M.M.; Chan, R.C. Multiplex PCR amplimer conformation analysis for rapid detection of gyrA mutations in fluoroquinolone-resistant Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 2004, 48, 596–601. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, J.; Zhang, X.; Wang, S.; Zhang, Y.; Li, C. Comparison of gyrA gene mutations between laboratory-selected ofloxacin-resistant Mycobacterium tuberculosis strains and clinical isolates. Int. J. Antimicrob. Agents 2008, 31, 115–121. [Google Scholar] [CrossRef]
- Maruri, F.; Sterling, T.R.; Kaiga, A.W.; Blackman, A.; van der Heijden, Y.F.; Mayer, C.; Cambau, E.; Aubry, A. A systematic review of gyrase mutations associated with fluoroquinolone-resistant Mycobacterium tuberculosis and a proposed gyrase numbering system. J. Antimicrob. Chemother. 2012, 67, 819–831. [Google Scholar] [CrossRef]
- Musser, J.M. Antimicrobial agent resistance in mycobacteria: Molecular genetic insights. Clin. Microbiol. Rev. 1995, 8, 496–514. [Google Scholar]
- Aubry, A.; Veziris, N.; Cambau, E.; Truffot-Pernot, C.; Jarlier, V.; Fisher, L.M. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: Functional analysis of mutant enzymes. Antimicrob. Agents Chemother. 2006, 50, 104–112. [Google Scholar]
- Von Groll, A.; Martin, A.; Jureen, P.; Hoffner, S.; Vandamme, P.; Portaels, F.; Palomino, J.C.; da Silva, P.A. Fluoroquinolone resistance in Mycobacterium tuberculosis and mutations in gyrA and gyrB. Antimicrob. Agents Chemother. 2009, 53, 4498–4500. [Google Scholar] [CrossRef]
- Escribano, I.; Rodríguez, J.C.; Llorca, B.; García-Pachon, E.; Ruiz, M.; Royo, G. Importance of the efflux pump systems in the resistance of Mycobacterium tuberculosis to fluoroquinolones and linezolid. Chemotherapy 2007, 53, 397–401. [Google Scholar] [CrossRef]
- Alangaden, G.J.; Kreiswirth, B.N.; Aouad, A.; Khetarpal, M.; Igno, F.R.; Moghazeh, S.L.; Manavathu, E.K.; Lerner, S.A. Mechanism of resistance to amikacin and kanamycin in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 1295–1297. [Google Scholar]
- Suzuki, Y.; Katsukawa, C.; Tamaru, A.; Abe, C.; Makino, M.; Mizuguchi, Y.; Taniguchi, H. Detection of kanamycin-resistant Mycobacterium tuberculosis by identifying mutations in the 16S rRNA gene. J. Clin. Microbiol. 1998, 36, 1220–1225. [Google Scholar]
- Krüüner, A.; Jureen, P.; Levina, K.; Ghebremichael, S.; Hoffner, S. Discordant resistance to kanamycin and amikacin in drug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 2971–2973. [Google Scholar] [CrossRef]
- Zaunbrecher, M.A.; Sikes, R.D., Jr.; Metchock, B.; Shinnick, T.M.; Posey, J.E. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 20004–20009. [Google Scholar]
- Campbell, P.J.; Morlock, G.P.; Sikes, R.D.; Dalton, T.L.; Metchock, B.; Starks, A.M.; Hooks, D.P.; Cowan, L.S.; Plikaytis, B.B.; Posey, J.E. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 2032–2041. [Google Scholar] [CrossRef]
- Stanley, R.E.; Blaha, G.; Grodzicki, R.L.; Strickler, M.D.; Steitz, T.A. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat. Struct. Mol. Biol. 2010, 17, 289–293. [Google Scholar] [CrossRef]
- McClatchy, J.K.; Kanes, W.; Davidson, P.T.; Moulding, T.S. Cross-resistance in M. tuberculosis to kanamycin, capreomycin and viomycin. Tubercle 1977, 58, 29–34. [Google Scholar]
- Johansen, S.K.; Maus, C.E.; Plikaytis, B.B.; Douthwaite, S. Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2'-O-methylations in 16S and 23S rRNAs. Mol. Cell 2006, 23, 173–182. [Google Scholar] [CrossRef]
- Georghiou, S.B.; Magana, M.; Garfein, R.S.; Catanzaro, D.G.; Catanzaro, A.; Rodwell, T.C. Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: A systematic review. PLoS One 2012, 7, e33275. [Google Scholar]
- Carette, X.; Blondiaux, N.; Willery, E.; Hoos, S.; Lecat-Guillet, N.; Lens, Z.; Wohlkönig, A.; Wintjens, R.; Soror, S.H.; Frénois, F. Structural activation of the transcriptional repressor EthR from Mycobacterium tuberculosis by single amino acid change mimicking natural and synthetic ligands. Nucleic Acids Res. 2012, 40, 3018–3030. [Google Scholar] [CrossRef]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.G.; Barry, C.E., 3rd. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9782. [Google Scholar] [CrossRef]
- Brossier, F.; Veziris, N.; Truffot-Pernot, C.; Jarlier, V.; Sougakoff, W. Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 355–560. [Google Scholar] [CrossRef]
- Vilchèze, C.; Av-Gay, Y.; Attarian, R.; Liu, Z.; Hazbón, M.H.; Colangeli, R.; Chen, B.; Liu, W.; Alland, D.; Sacchettini, J.C.; et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2008, 69, 1316–1329. [Google Scholar] [CrossRef]
- Rengarajan, J.; Sassetti, C.M.; Naroditskaya, V.; Sloutsky, A.; Bloom, B.R.; Rubin, E.J. The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol. Microbiol. 2004, 53, 275–282. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, X.D.; Erber, L.N.; Luo, M.; Guo, A.Z.; Yang, S.S.; Gu, J.; Turman, B.J.; Gao, Y.R.; Li, D.F.; et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 1479–1487. [Google Scholar] [CrossRef]
- Mathys, V.; Wintjens, R.; Lefevre, P.; Bertout, J.; Singhal, A.; Kiass, M.; Kurepina, N.; Wang, X.M.; Mathema, B.; Baulard, A.; et al. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 2100–2109. [Google Scholar] [CrossRef]
- Zhang, Y. The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 529–564. [Google Scholar] [CrossRef]
- Cáceres, N.E.; Harris, N.B.; Wellehan, J.F.; Feng, Z.; Kapur, V.; Barletta, R.G. Overexpression of the d-alanine racemase gene confers resistance to d-cycloserine in Mycobacterium smegmatis. J. Bacteriol. 1997, 179, 5046–5055. [Google Scholar]
- Chen, J.M.; Uplekar, S.; Gordon, S.V.; Cole, S.T. A point mutation in cycA partially contributes to the d-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS One 2012, 7, e43467. [Google Scholar]
- Grzegorzewicz, A.E.; Korduláková, J.; Jones, V.; Born, S.E.; Belardinelli, J.M.; Vaquié, A.; Gundi, V.A.; Madacki, J.; Slama, N.; Laval, F.; et al. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J. Biol. Chem. 2012, 287, 38434–38441. [Google Scholar] [CrossRef]
- Andini, N.; Nash, K.A. Intrinsic macrolide resistance of the Mycobacterium tuberculosis complex is inducible. Antimicrob. Agents Chemother. 2006, 50, 2560–2562. [Google Scholar]
- Bosne-David, S.; Barros, V.; Verde, S.C.; Portugal, C.; David, H.L. Intrinsic resistance of Mycobacterium tuberculosis to clarithromycin is effectively reversed by subinhibitory concentrations of cell wall inhibitors. J. Antimicrob. Chemother. 2000, 46, 391–395. [Google Scholar] [CrossRef]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O’Sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef]
- Browne, S.G.; Hogerzeil, L.M. “B 663” in the treatment of leprosy. Preliminary report of a pilot trial. Lepr. Rev. 1962, 33, 6–10. [Google Scholar]
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. [Google Scholar] [CrossRef]
- Yano, T.; Kassovska-Bratinova, S.; The, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 NADH: Quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar]
- Leach, K.L.; Brickner, S.J.; Noe, M.C.; Miller, P.F. Linezolid, the first oxazolidinone antibacterial agent. Ann. NY Acad. Sci. 2011, 1222, 49–54. [Google Scholar] [CrossRef]
- Richter, E.; Rüsch-Gerdes, S.; Hillemann, D. First linezolid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1534–1536. [Google Scholar] [CrossRef]
- Hillemann, D.; Rüsch-Gerdes, S.; Richter, E. In vitro-selected linezolid-resistant Mycobacterium tuberculosis mutants. Antimicrob. Agents Chemother. 2008, 52, 800–801. [Google Scholar] [CrossRef]
- Beckert, P.; Hillemann, D.; Kohl, T.A.; Kalinowski, J.; Richter, E.; Niemann, S.; Feuerriegel, S. rplC T460C identified as a dominant mutation in linezolid-resistant Mycobacterium tuberculosis strains. Antimicrob. Agents Chemother. 2012, 56, 2743–2745. [Google Scholar] [CrossRef]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.M.; Winkler, H.; van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Rustomjee, R.; Diacon, A.H.; Allen, J.; Venter, A.; Reddy, C.; Patientia, R.F.; Mthiyane, T.C.P.; de Marez, T.; van Heeswijk, R.; Kerstens, R.; et al. Early bactericidal activity and pharmacokinetics of the diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 2831–2835. [Google Scholar] [CrossRef]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G.; et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef]
- Chahine, E.B.; Karaoui, L.R.; Mansour, H. Bedaquiline: A novel diarylquinoline for multidrug-resistant tuberculosis. Ann. Pharmacother. 2014, 48, 107–115. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A. TMC207 becomes bedaquiline, a new anti-TB drug. Future Microbiol. 2013, 8, 1071–1080. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; von Groote-Bidlingmaier, F.; Symons, G.; Venter, A.; Donald, P.R.; van Niekerk, C.; Everitt, D.; Winter, H.; Becker, P.; et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: A randomised trial. Lancet 2012, 380, 986–993. [Google Scholar] [CrossRef]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Abdillahi-Ibrahim, R.; Wagner, M.J.; Krab, K.; Vergauwen, K.; Guillemont, J.; Andries, K.; Lill, H.; Koul, A.; Bald, D. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. [Google Scholar] [CrossRef]
- Petrella, S.; Cambau, E.; Chauffour, A.; Andries, K.; Jarlier, V.; Sougakoff, W. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob. Agents Chemother. 2006, 50, 2853–2856. [Google Scholar] [CrossRef]
- Segala, E.; Sougakoff, W.; Nevejans-Chauffour, A.; Jarlier, V.; Petrella, S. New mutations in the mycobacterial ATP synthase: New insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-ring structure. Antimicrob. Agents Chemother. 2012, 56, 2326–2634. [Google Scholar] [CrossRef]
- Huitric, E.; Verhasselt, P.; Koul, A.; Andries, K.; Hoffner, S.; Andersson, D.I. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2010, 54, 1022–1028. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; Hanekom, M.; Narunsky, K.; Venter, A.; Hittel, N.L.; Geiter, J.; Wells, C.D.; Paccaly, A.J.; Donald, P.R. Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int. J. Tuberc. Lung Dis. 2011, 15, 949–954. [Google Scholar] [CrossRef]
- Gler, M.T.; Skripconoka, V.; Sanchez-Garavito, E.; Xiao, H.; Cabrera-Rivero, J.L.; Vargas-Vasquez, D.E.; Gao, M.; Awad, M.; Park, S.K.; Shim, T.S.; et al. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Lenaerts, A.J.; Gruppo, V.; Marietta, K.S.; Johnson, C.M.; Driscoll, D.K.; Tompkins, N.M.; Rose, J.D.; Reynolds, R.C.; Orme, I.M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. [Google Scholar] [CrossRef]
- Ginsberg, A.M.; Laurenzi, M.W.; Rouse, D.J.; Whitney, K.D.; Spigelman, M.K. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob. Agents Chemother. 2009, 53, 3720–3725. [Google Scholar] [CrossRef]
- Diacon, A.H.; Dawson, R.; du Bois, J.; Narunsky, K.; Venter, A.; Donald, P.R.; van Niekerk, C.; Erondu, N.; Ginsberg, A.M.; Becker, P.; et al. Phase II dose-ranging trial of the early bactericidal activity of PA-824. Antimicrob. Agents Chemother. 2012, 56, 3027–3231. [Google Scholar] [CrossRef]
- Manjunatha, U.H.; Boshoff, H.; Dowd, C.S.; Zhang, L.; Albert, T.J.; Norton, J.E.; Daniels, L.; Dick, T.; Pang, S.S.; Barry, C.E., 3rd. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 431–436. [Google Scholar] [CrossRef]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef]
- Reddy, V.M.; Einck, L.; Andries, K.; Nacy, C.A. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 2010, 54, 2840–2846. [Google Scholar] [CrossRef]
- Reddy, V.M.; Dubuisson, T.; Einck, L.; Wallis, R.S.; Jakubiec, W.; Ladukto, L.; Campbell, S.; Nacy, C.A. SQ109 and PNU-100480 interact to kill Mycobacterium tuberculosis in vitro. J. Antimicrob. Chemother. 2012, 67, 1163–1166. [Google Scholar] [CrossRef]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E., 3rd; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Myers, T.G.; Copp, B.R.; McNeil, M.R.; Wilson, M.A.; Barry, C.E., 3rd. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: Novel insights into drug mechanisms of action. J. Biol. Chem. 2004, 279, 40174–40184. [Google Scholar]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., 3rd; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Pasca, M.R.; Degiacomi, G.; Ribeiro, A.L.; Zara, F.; De Mori, P.; Heym, B.; Mirrione, M.; Brerra, R.; Pagani, L.; Pucillo, L.; et al. Clinical isolates of Mycobacterium tuberculosis in four European hospitals are uniformly susceptible to benzothiazinones. Antimicrob. Agents Chemother. 2010, 54, 1616–1618. [Google Scholar] [CrossRef]
- Mikusová, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl arabinofuranose, the donor of the d-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs that covalently modify the decaprenylphosphoryl-β-d-ribose 2'-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; Ribeiro, A.L.; Buroni, S.; Skovierová, H.; Dianišková, P.; Mikušová, K.; et al. Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiazinone resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Zhang, H.; Li, D.; Zhao, L.; Fleming, J.; Lin, N.; Wang, T.; Liu, Z.; Li, C.; Galwey, N.; Deng, J.; et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 2013, 45, 1255–1260. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Palomino, J.C.; Martin, A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317-340. https://doi.org/10.3390/antibiotics3030317
Palomino JC, Martin A. Drug Resistance Mechanisms in Mycobacterium tuberculosis. Antibiotics. 2014; 3(3):317-340. https://doi.org/10.3390/antibiotics3030317
Chicago/Turabian StylePalomino, Juan Carlos, and Anandi Martin. 2014. "Drug Resistance Mechanisms in Mycobacterium tuberculosis" Antibiotics 3, no. 3: 317-340. https://doi.org/10.3390/antibiotics3030317