Rifampicin Resistance: Fitness Costs and the Significance of Compensatory Evolution

{kind=link}
{kind=link}
Abstract
:1. Introduction
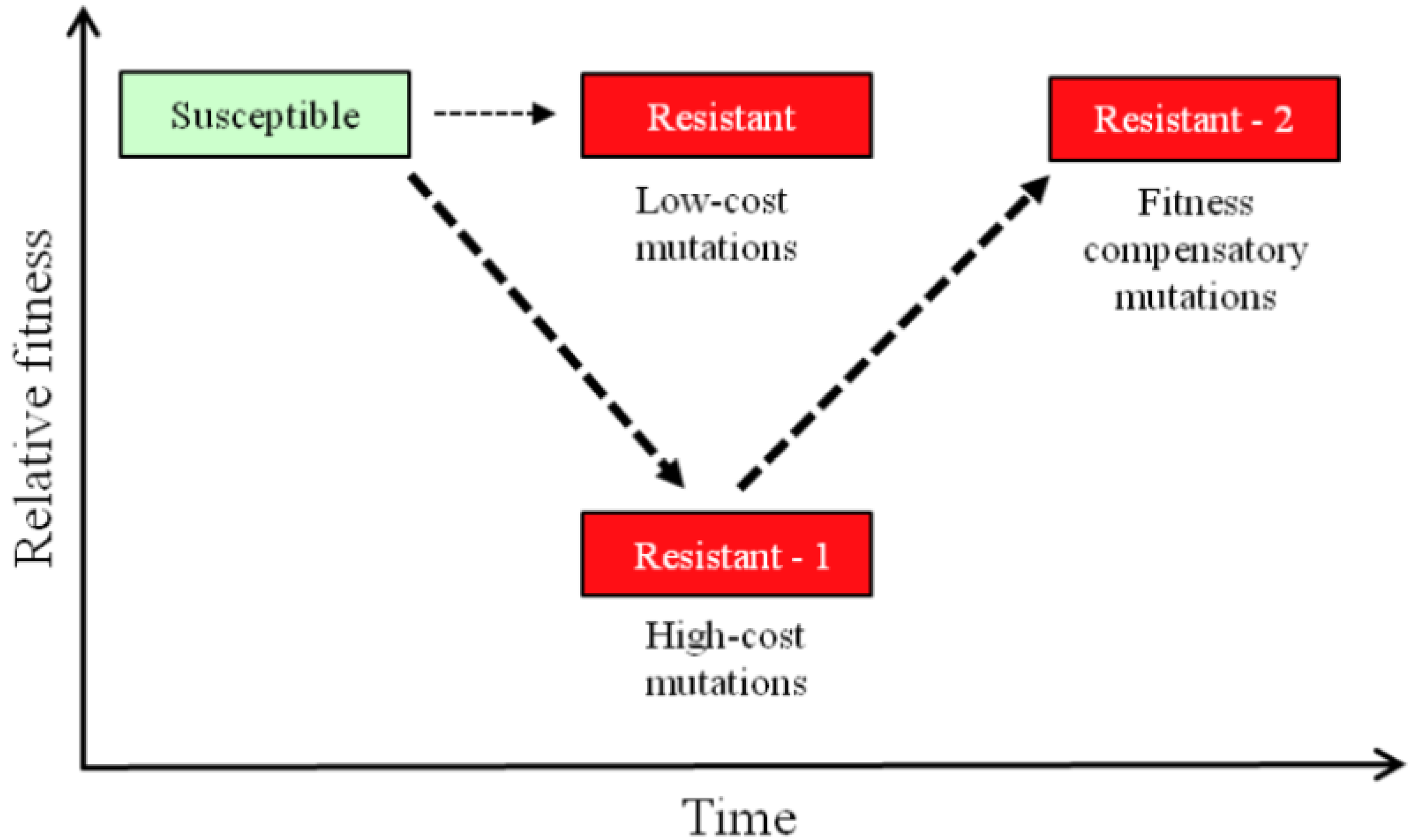
2. Resistance and Fitness
3. Measuring Fitness Costs

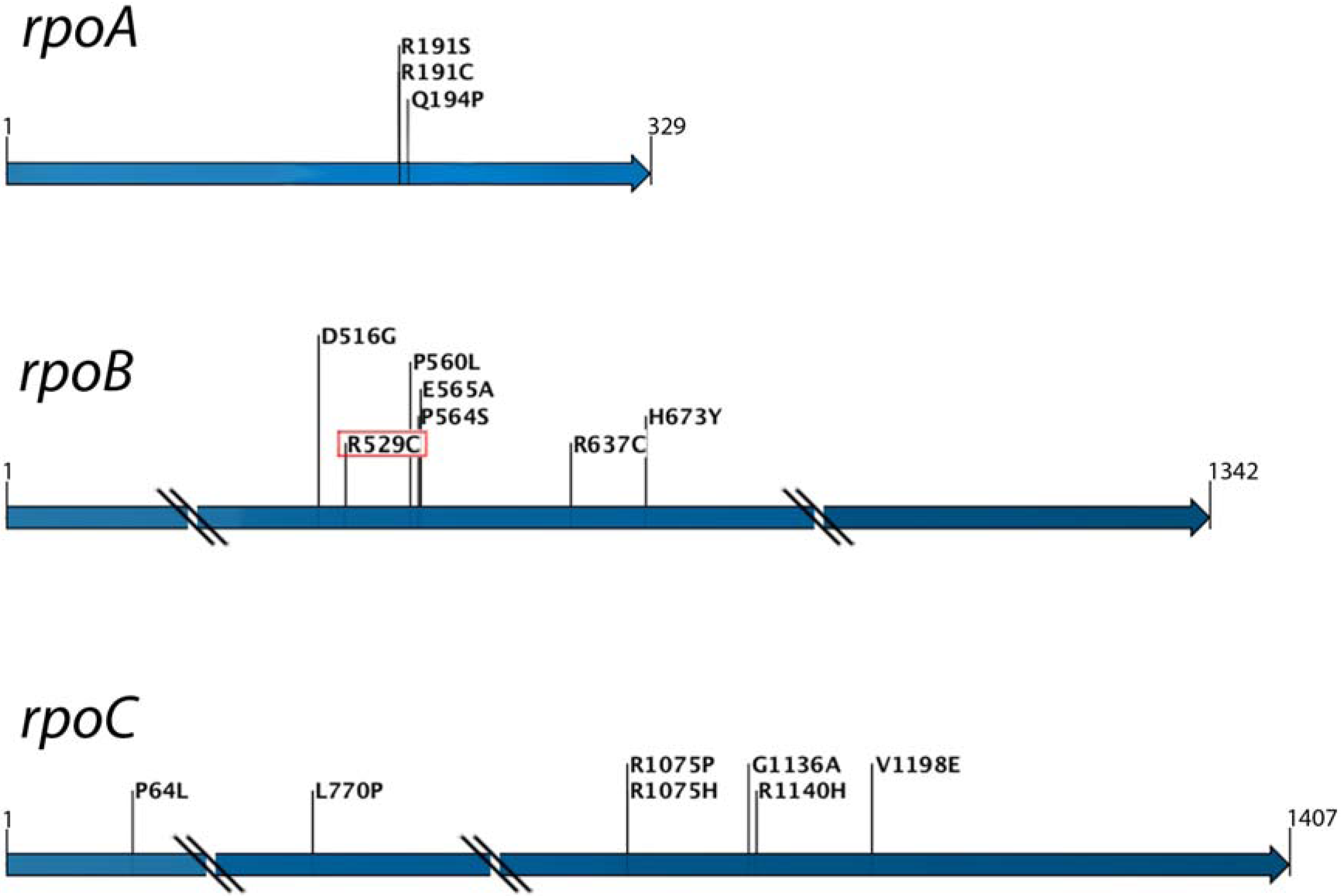
4. Resistance and Genomics Analysis

5. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Kimbrough, W.; Saliba, V.; Dahab, M.; Haskew, C.; Checchi, F. The burden of tuberculosis in crisis-affected populations: A systematic review. Lancet Infect. Dis. 2012, 12, 950–965. [Google Scholar] [CrossRef]
- Chang, K.C.; Nuermberger, E.L. 2011: The year in review. Part I: Tuberculosis. Int. J. Tuberc. Lung Dis. 2012, 16, 740–748. [Google Scholar] [CrossRef]
- Diacon, A.H.; von Groote-Bidlingmaier, F.; Donald, P.R. From magic mountain to table mountain. Swiss Med. Wkly 2012, 142, w13665. [Google Scholar]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; Gonzalez, R.; et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef]
- Almeida Da Silva, P.E.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef]
- Chang, K.C.; Yew, W.W. Management of difficult multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis: Update 2012. Respirology 2013, 18, 8–21. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Selection of resistance at lethal and non-lethal antibiotic concentrations. Curr. Opin. Microbiol. 2012, 15, 555–560. [Google Scholar] [CrossRef]
- Maggi, N.; Pasqualucci, C.R.; Ballotta, R.; Sensi, P. Rifampicin: A new orally active rifamycin. Chemotherapy 1966, 11, 285–292. [Google Scholar] [CrossRef]
- Grumbach, F.; Canetti, G.; Le Lirzin, M. Rifampicin in daily and intermittent treatment of experimental murine tuberculosis, with emphasis on late results. Tubercle 1969, 50, 280–293. [Google Scholar] [CrossRef]
- World Health Organization, Treatment of tuberculosis: Guidelines, 4th ed2010.
- Jindani, A.; Dore, C.J.; Mitchison, D.A. Bactericidal and sterilizing activities of antituberculosis drugs during the first 14 days. Am. J. Respir. Crit. Care Med. 2003, 167, 1348–1354. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Jin, D.J.; Gross, C.A. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J. Mol. Biol. 1988, 202, 45–58. [Google Scholar] [CrossRef]
- Telenti, A.; Imboden, P.; Marchesi, F.; Lowrie, D.; Cole, S.; Colston, M.J.; Matter, L.; Schopfer, K.; Bodmer, T. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 1993, 341, 647–650. [Google Scholar]
- Wrande, M.; Roth, J.R.; Hughes, D. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11863–11868. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Huovinen, T.; Fishwick, C.W.; Chopra, I. Molecular genetic and structural modeling studies of Staphylococcus aureus RNA polymerase and the fitness of rifampin resistance genotypes in relation to clinical prevalence. Antimicrob. Agents Chemother. 2006, 50, 298–309. [Google Scholar] [CrossRef]
- Heep, M.; Brandstatter, B.; Rieger, U.; Lehn, N.; Richter, E.; Rusch-Gerdes, S.; Niemann, S. Frequency of rpoB mutations inside and outside the cluster I region in rifampin-resistant clinical Mycobacterium tuberculosis isolates. J. Clin. Microbiol. 2001, 39, 107–110. [Google Scholar]
- Ramaswamy, S.; Musser, J.M. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber. Lung Dis. 1998, 79, 3–29. [Google Scholar] [CrossRef]
- Williamson, D.A.; Roberts, S.A.; Bower, J.E.; Vaughan, R.; Newton, S.; Lowe, O.; Lewis, C.A.; Freeman, J.T. Clinical failures associated with rpob mutations in phenotypically occult multidrug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2012, 16, 216–220. [Google Scholar] [CrossRef]
- Gumbo, T. New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob. Agents Chemother. 2010, 54, 1484–1491. [Google Scholar] [CrossRef]
- Brandis, G.; Wrande, M.; Liljas, L.; Hughes, D. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 2012, 85, 142–151. [Google Scholar] [CrossRef]
- Namouchi, A.; Didelot, X.; Schock, U.; Gicquel, B.; Rocha, E.P. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination, and natural selection. Genome Res. 2012, 22, 721–734. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. 2010, 8, 260–271. [Google Scholar]
- Bjorkman, J.; Hughes, D.; Andersson, D.I. Virulence of antibiotic-resistant Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 1998, 95, 3949–3953. [Google Scholar]
- Andersson, D.I.; Hughes, D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 2011, 35, 901–911. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D. Effects of Antibiotic Resistance on Bacterial Fitness, Virulence, and Transmission. In Evolutionary Biology of Bacterial and Fungal Pathogens; Baquero, F., Nombela, C., Cassell, G.H., Gutiérrez-Fuentes, J.A., Eds.; ASM Press: Washington, DC, USA, 2007; pp. 307–318. [Google Scholar]
- Rozen, D.E.; McGee, L.; Levin, B.R.; Klugman, K.P. Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2007, 51, 412–416. [Google Scholar] [CrossRef]
- Marcusson, L.L.; Frimodt-Moller, N.; Hughes, D. Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog. 2009, 5, e1000541. [Google Scholar] [CrossRef]
- Hall, A.R.; MacLean, R.C. Epistasis buffers the fitness effects of rifampicin-resistance mutations in Pseudomonas aeruginosa. Evolution 2011, 65, 2370–2379. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilback, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef]
- Sander, P.; Springer, B.; Prammananan, T.; Sturmfels, A.; Kappler, M.; Pletschette, M.; Bottger, E.C. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob. Agents Chemother. 2002, 46, 1204–1211. [Google Scholar] [CrossRef]
- Bottger, E.C.; Springer, B. Tuberculosis: Drug resistance, fitness, and strategies for global control. Eur. J. Pediatr. 2008, 167, 141–148. [Google Scholar] [CrossRef]
- Billington, O.J.; McHugh, T.D.; Gillespie, S.H. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1999, 43, 1866–1869. [Google Scholar]
- Ward, H.; Perron, G.G.; Maclean, R.C. The cost of multiple drug resistance in Pseudomonas aeruginosa. J. Evol. Biol. 2009, 22, 997–1003. [Google Scholar]
- Fenner, L.; Egger, M.; Bodmer, T.; Altpeter, E.; Zwahlen, M.; Jaton, K.; Pfyffer, G.E.; Borrell, S.; Dubuis, O.; Bruderer, T.; et al. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3047–3053. [Google Scholar]
- Shcherbakov, D.; Akbergenov, R.; Matt, T.; Sander, P.; Andersson, D.I.; Bottger, E.C. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol. Microbiol. 2010, 77, 830–840. [Google Scholar]
- Williams, D.L.; Spring, L.; Collins, L.; Miller, L.P.; Heifets, L.B.; Gangadharam, P.R.; Gillis, T.P. Contribution of rpoB mutations to development of rifamycin cross-resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 1853–1857. [Google Scholar]
- Pang, Y.; Lu, J.; Wang, Y.; Song, Y.; Wang, S.; Zhao, Y. Study of the rifampin mono-resistance mechanism in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 893–900. [Google Scholar] [CrossRef]
- Trautinger, B.W.; Lloyd, R.G. Modulation of DNA repair by mutations flanking the DNA channel through rna polymerase. EMBO J. 2002, 21, 6944–6953. [Google Scholar]
- Gagneux, S.; Long, C.D.; Small, P.M.; Van, T.; Schoolnik, G.K.; Bohannan, B.J. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 2006, 312, 1944–1946. [Google Scholar]
- Mariam, D.H.; Mengistu, Y.; Hoffner, S.E.; Andersson, D.I. Effect of rpob mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2004, 48, 1289–1294. [Google Scholar] [CrossRef]
- Reynolds, M.G. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 2000, 156, 1471–1481. [Google Scholar]
- Bartlett, M.S.; Gaal, T.; Ross, W.; Gourse, R.L. RNA polymerase mutants that destabilize RNA polymerase-promoter complexes alter NTP-sensing by rrn P1 promoters. J. Mol. Biol. 1998, 279, 331–345. [Google Scholar] [CrossRef]
- Zhou, Y.N.; Jin, D.J. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc. Natl. Acad. Sci. USA 1998, 95, 2908–2913. [Google Scholar] [CrossRef]
- Comas, I.; Borrell, S.; Roetzer, A.; Rose, G.; Malla, B.; Kato-Maeda, M.; Galagan, J.; Niemann, S.; Gagneux, S. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 2012, 44, 106–110. [Google Scholar]
- Casali, N.; Nikolayevskyy, V.; Balabanova, Y.; Ignatyeva, O.; Kontsevaya, I.; Harris, S.R.; Bentley, S.D.; Parkhill, J.; Nejentsev, S.; Hoffner, S.E.; et al. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 2012, 22, 735–745. [Google Scholar] [CrossRef]
- De Vos, M.; Muller, B.; Borrell, S.; Black, P.; van Helden, P.; Warren, R.; Gagneux, S.; Victor, T. Putative compensatory mutations in the rpoC gene of rifampicin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 2012, 57, 827–832. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hughes, D.; Brandis, G. Rifampicin Resistance: Fitness Costs and the Significance of Compensatory Evolution. Antibiotics 2013, 2, 206-216. https://doi.org/10.3390/antibiotics2020206
Hughes D, Brandis G. Rifampicin Resistance: Fitness Costs and the Significance of Compensatory Evolution. Antibiotics. 2013; 2(2):206-216. https://doi.org/10.3390/antibiotics2020206
Chicago/Turabian StyleHughes, Diarmaid, and Gerrit Brandis. 2013. "Rifampicin Resistance: Fitness Costs and the Significance of Compensatory Evolution" Antibiotics 2, no. 2: 206-216. https://doi.org/10.3390/antibiotics2020206