Peroxide-Dependent Analyte Conversion by the Heme Prosthetic Group, the Heme Peptide “Microperoxidase-11” and Cytochrome c on Chitosan Capped Gold Nanoparticles Modified Electrodes



Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents
2.2. Preparation of Electrodes
2.3. Apparatus and Electrochemical Measurements
3. Results and Discussion
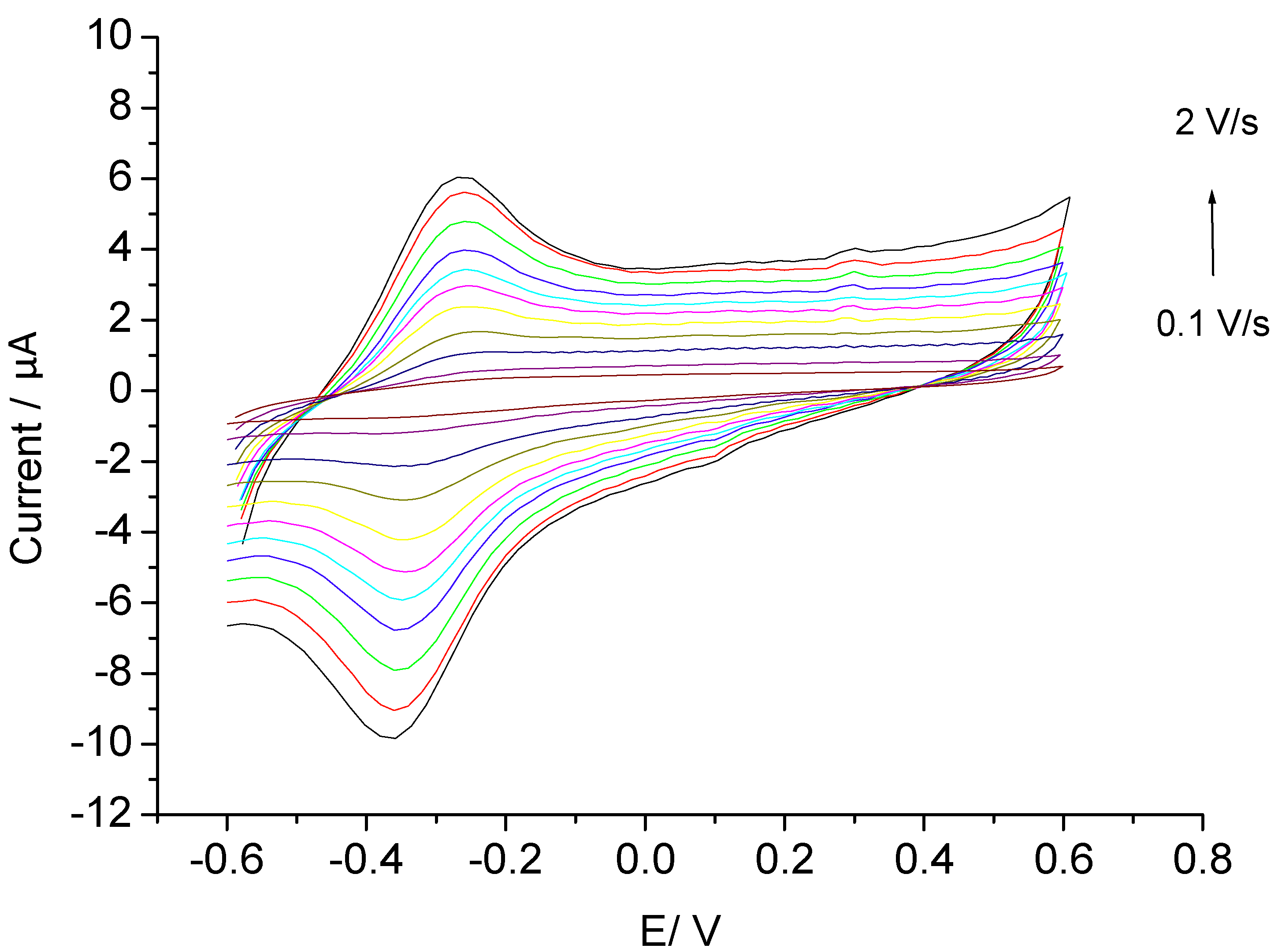
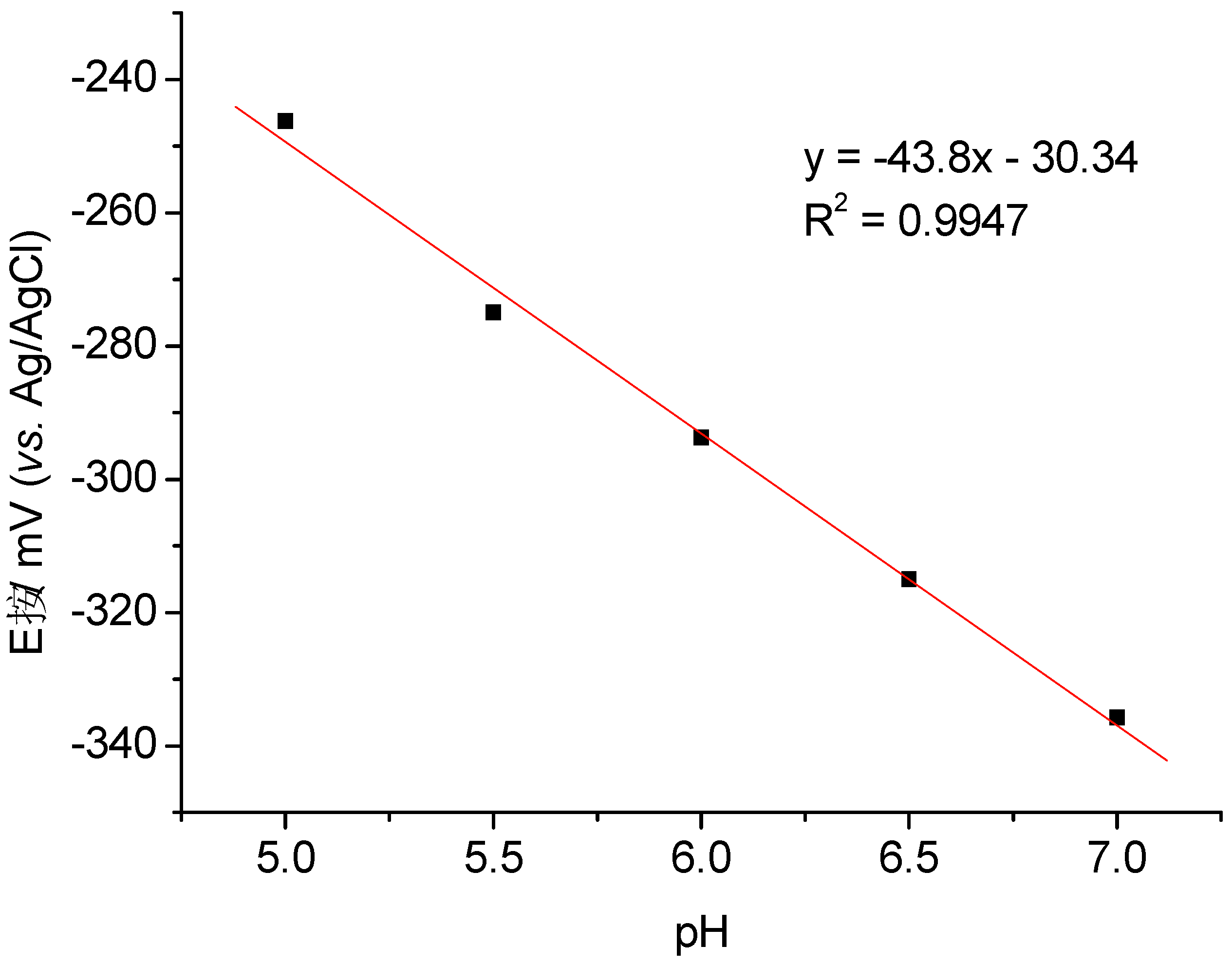
3.1. Direct Electron Transfer
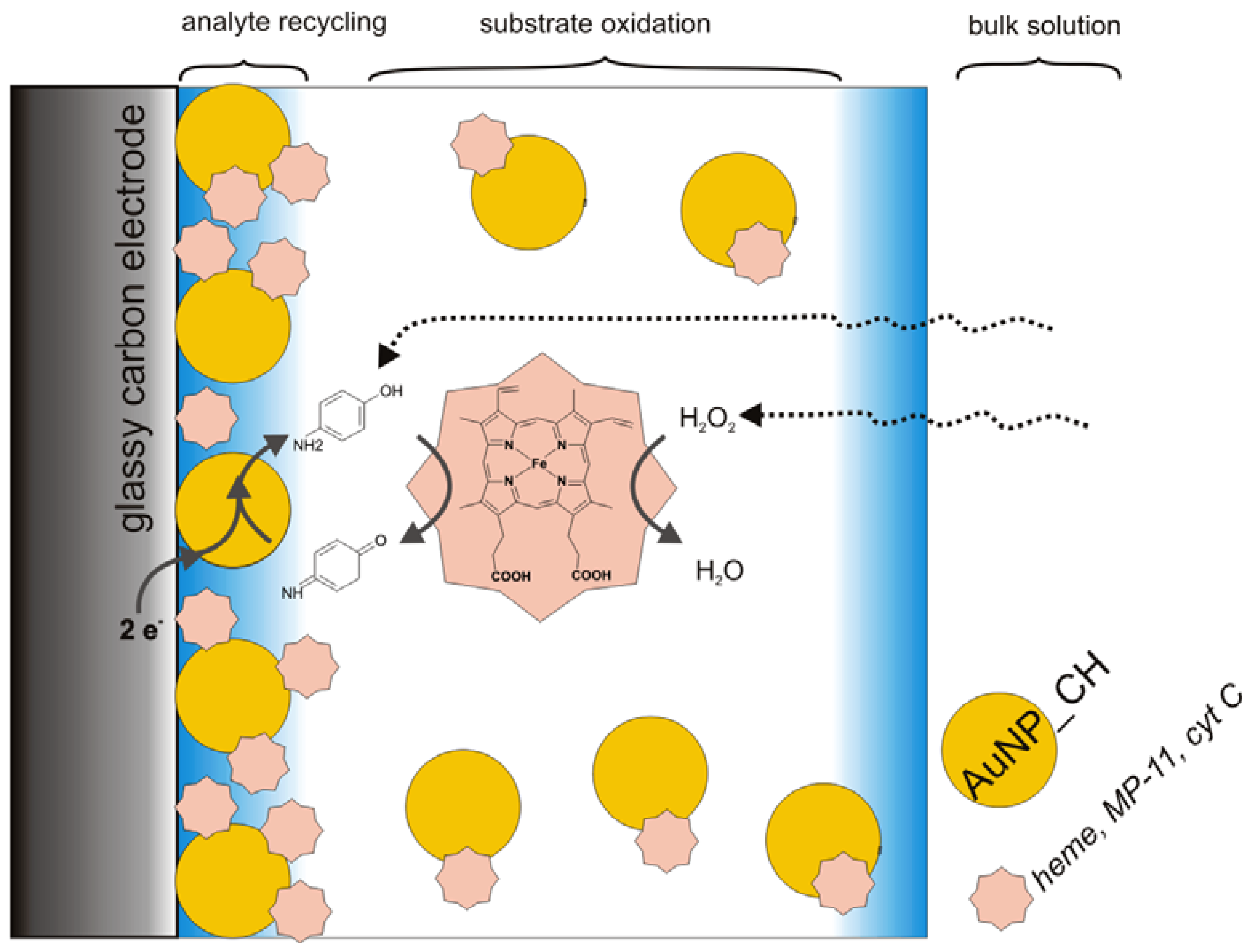
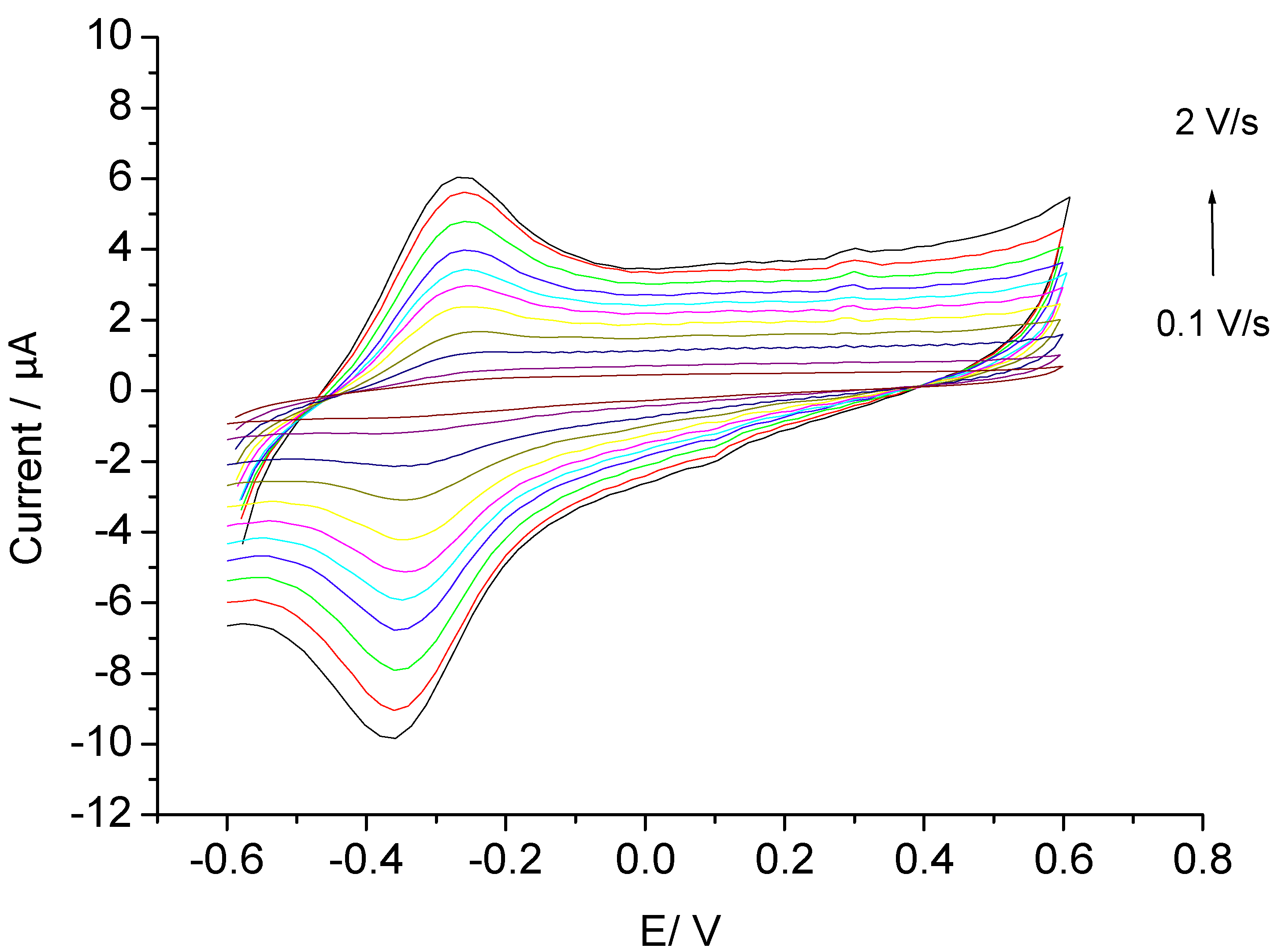

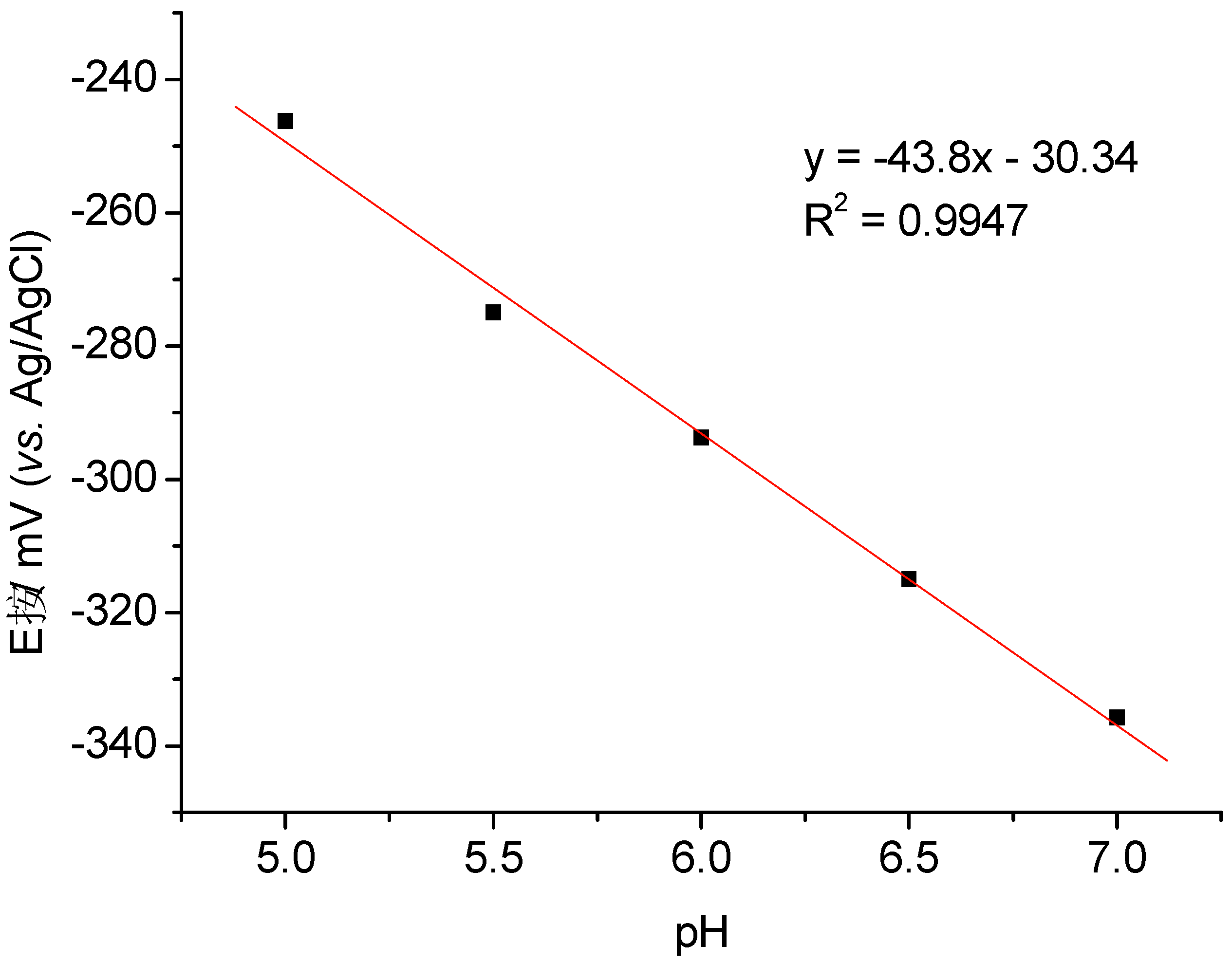
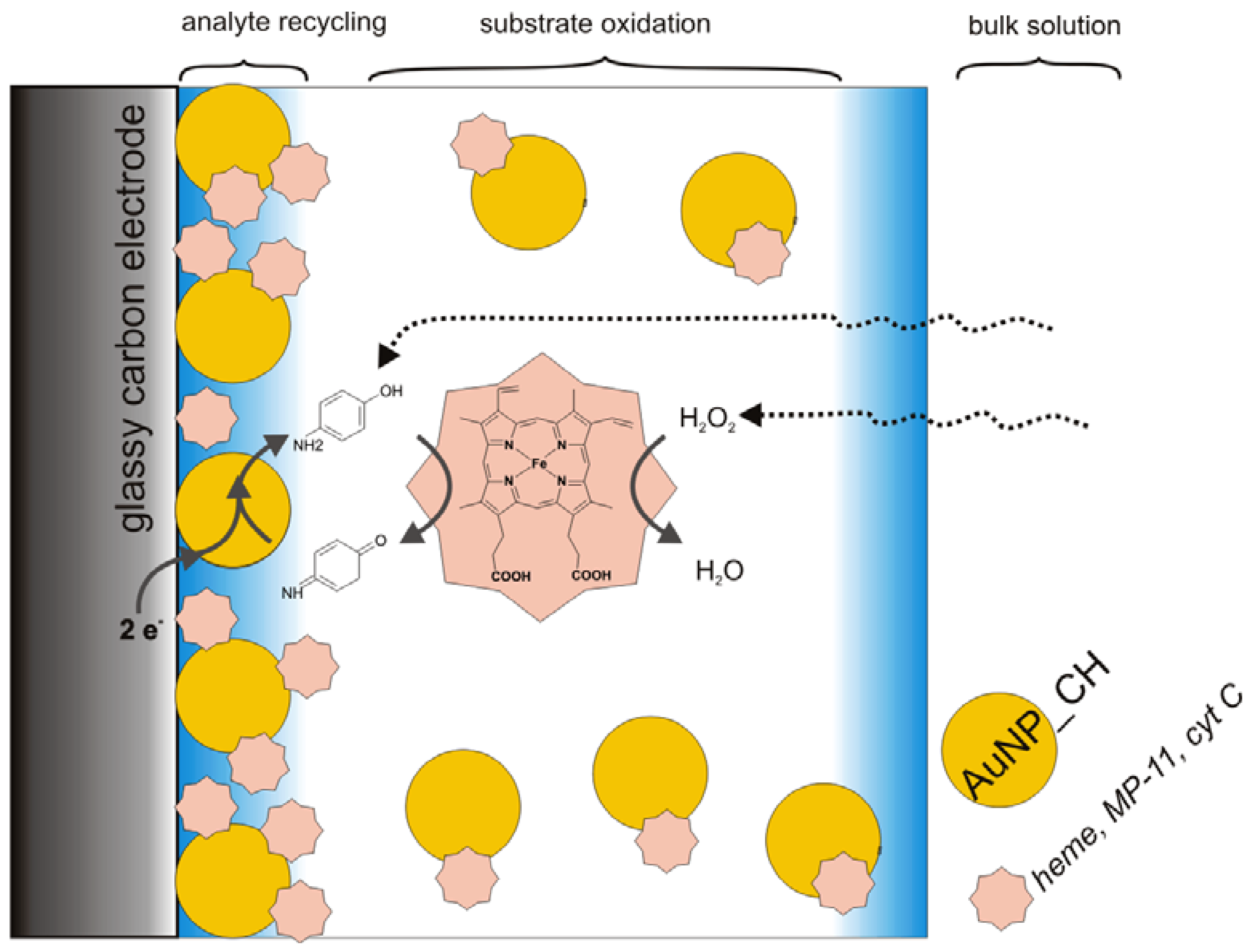
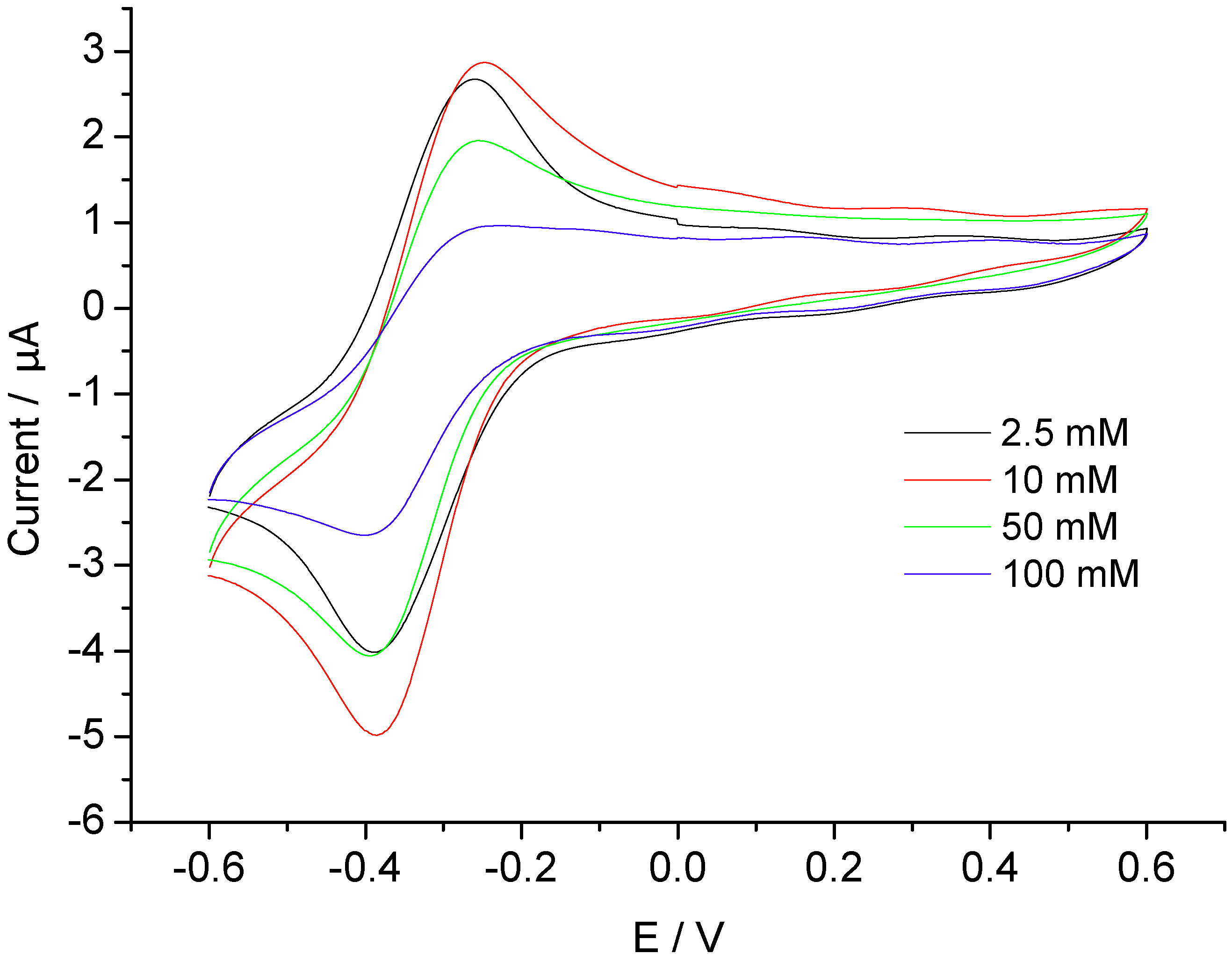
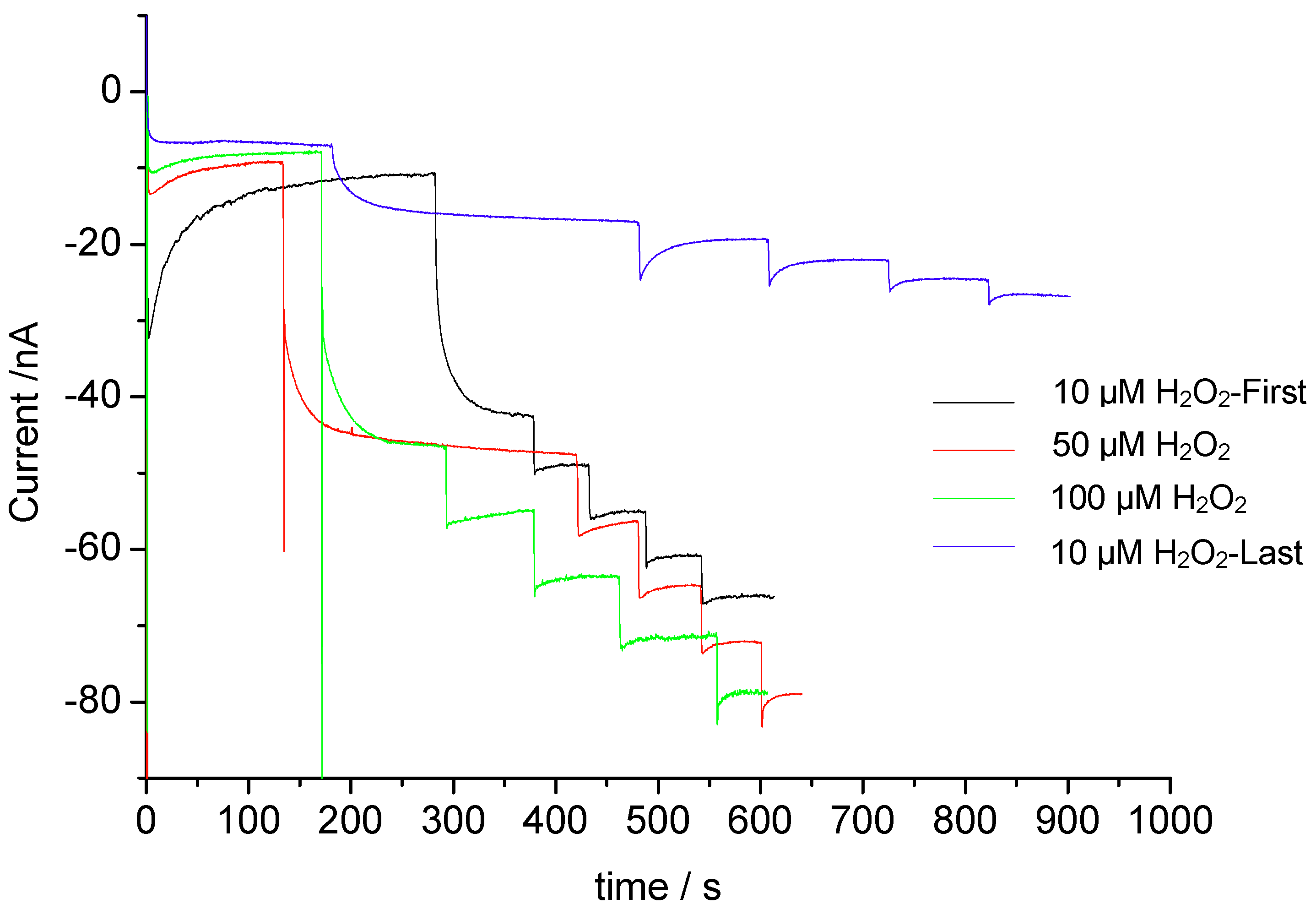
3.2. Catalysis of Cathodic Reduction of Peroxide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode preparation | Linear range (µM) | LOD | KM | Reference |
|---|---|---|---|---|
| MP-11-CPE | 55(±12) mM | [31] | ||
| MP-11-HCPE | - | - | 6.4(±1.1) mM | |
| MP-8-polypyrrole | 1–10 | 10 nM | [32] | |
| MP-11-chitosan–graphene-nanocomposite | 2.5–135 | 2 µM | 0.54 mM | [24] |
| MP-11-MWNT/GC | - | - | 2.4 mM | [33] |
| MP-11-GNP-MWNT/GC | 10–200 | 3 µM | 0.32 mM | [34] |
| MP/ZnO NPs/PG | 1–700 | 30 µM | - | [35] |
| MP-11-DDAB/GC | 2.4 mM and 20 µM | 0.8 µM | - | [36] |
| (MP11/PNTs/PAH)n = 4/ITO | - | 6 µM | - | [37] |
| MP-11/RW | 30 µM-4 mM | 5–10 mM | [27] | |
| MP-11-AuNP-CH/GC | 1–7 | 0.27 µM | 4.43 µM | [14] |
| MP-11-MWNT | 5–70 | 3.8 pM | [38] | |
| MP-11-Nanopolyurethane/GC | 0.02–1.3 | 10 pM | (1.87 ± 0.05) µM | [39] |
| MP-11-SnO2-PLL | 0.05–30 | 50 nM | [30] |
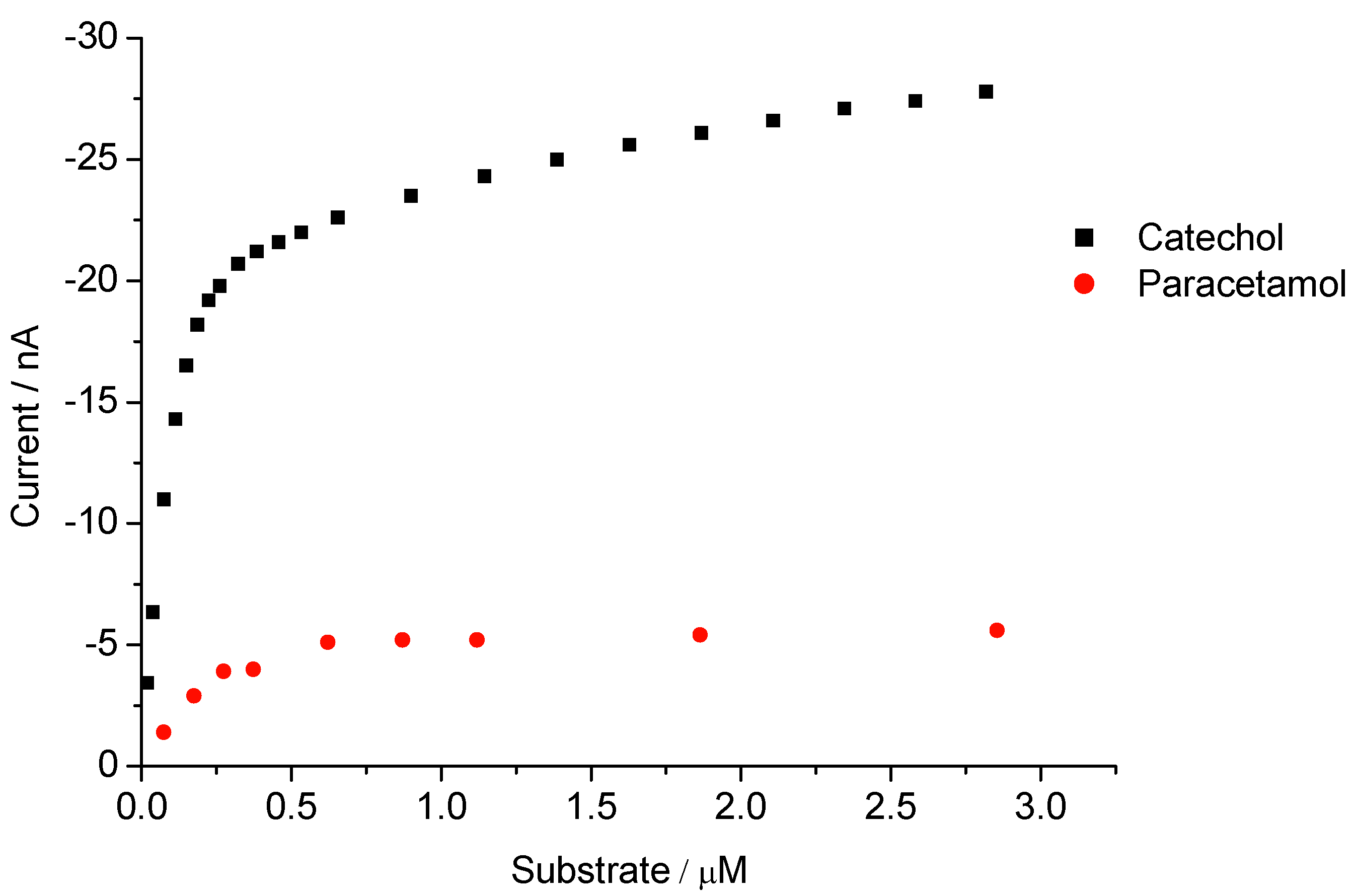
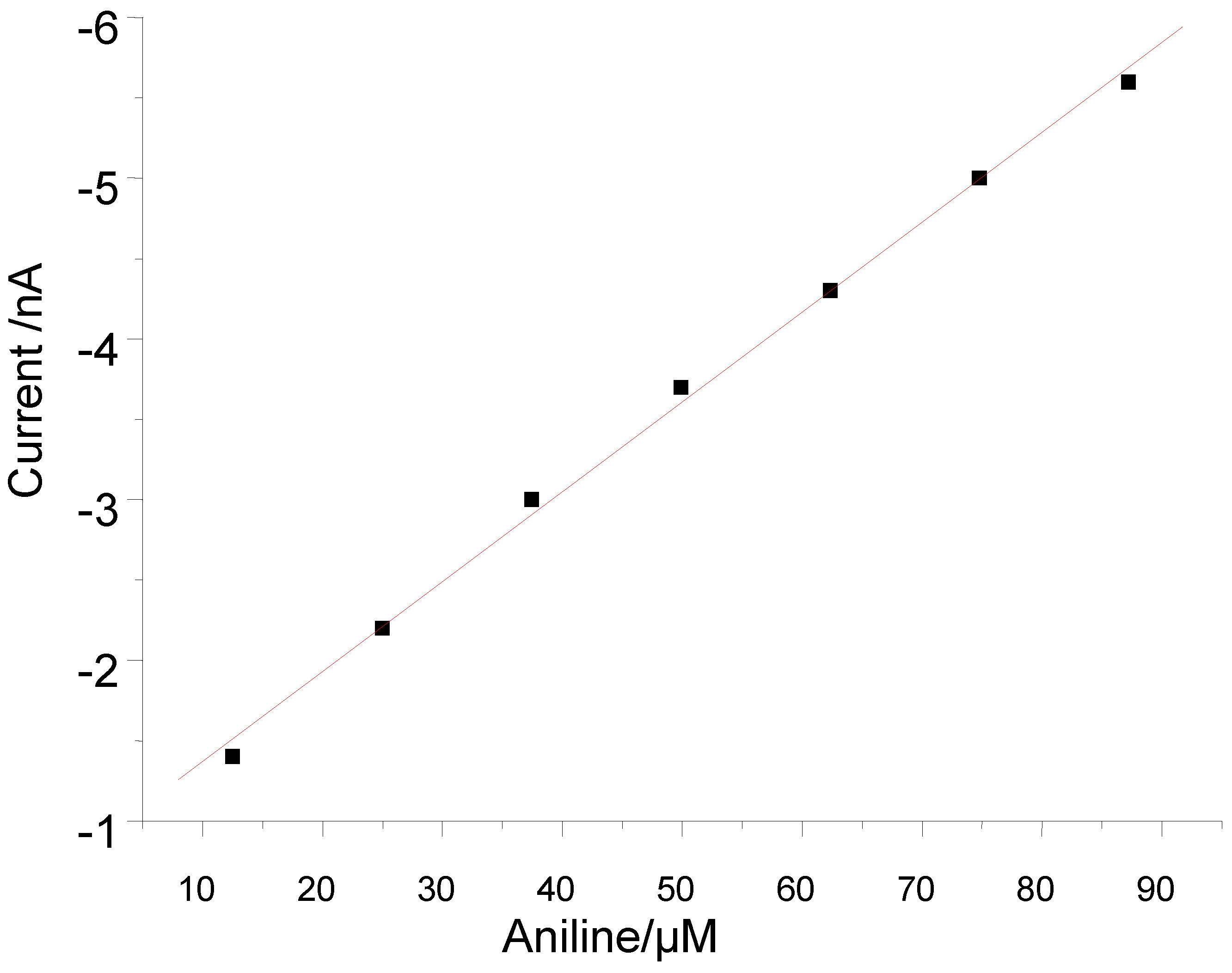
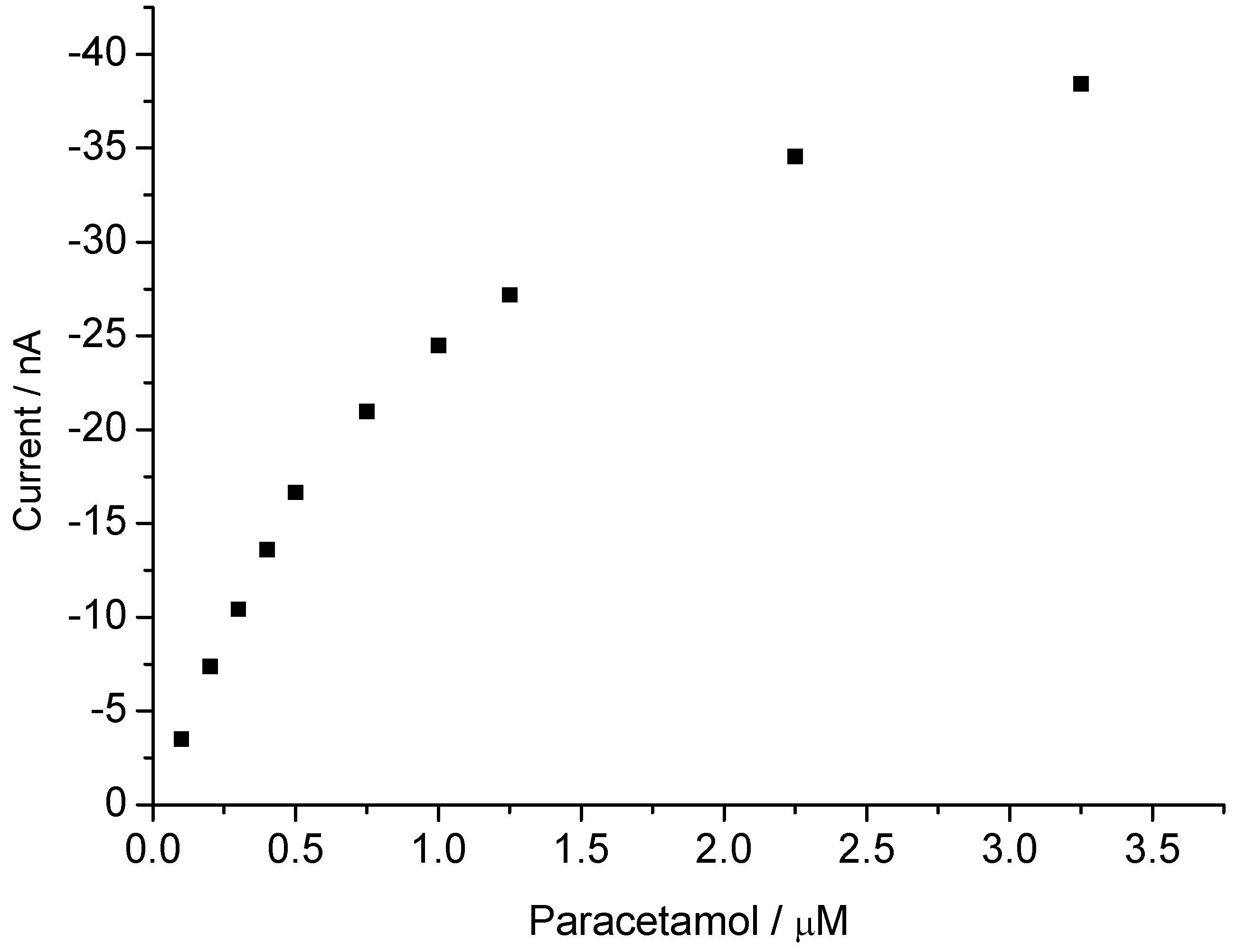
3.2. Peroxide-Dependent Substrate Oxidation by Hemin, Microperoxidase-11, and Cytochrome C

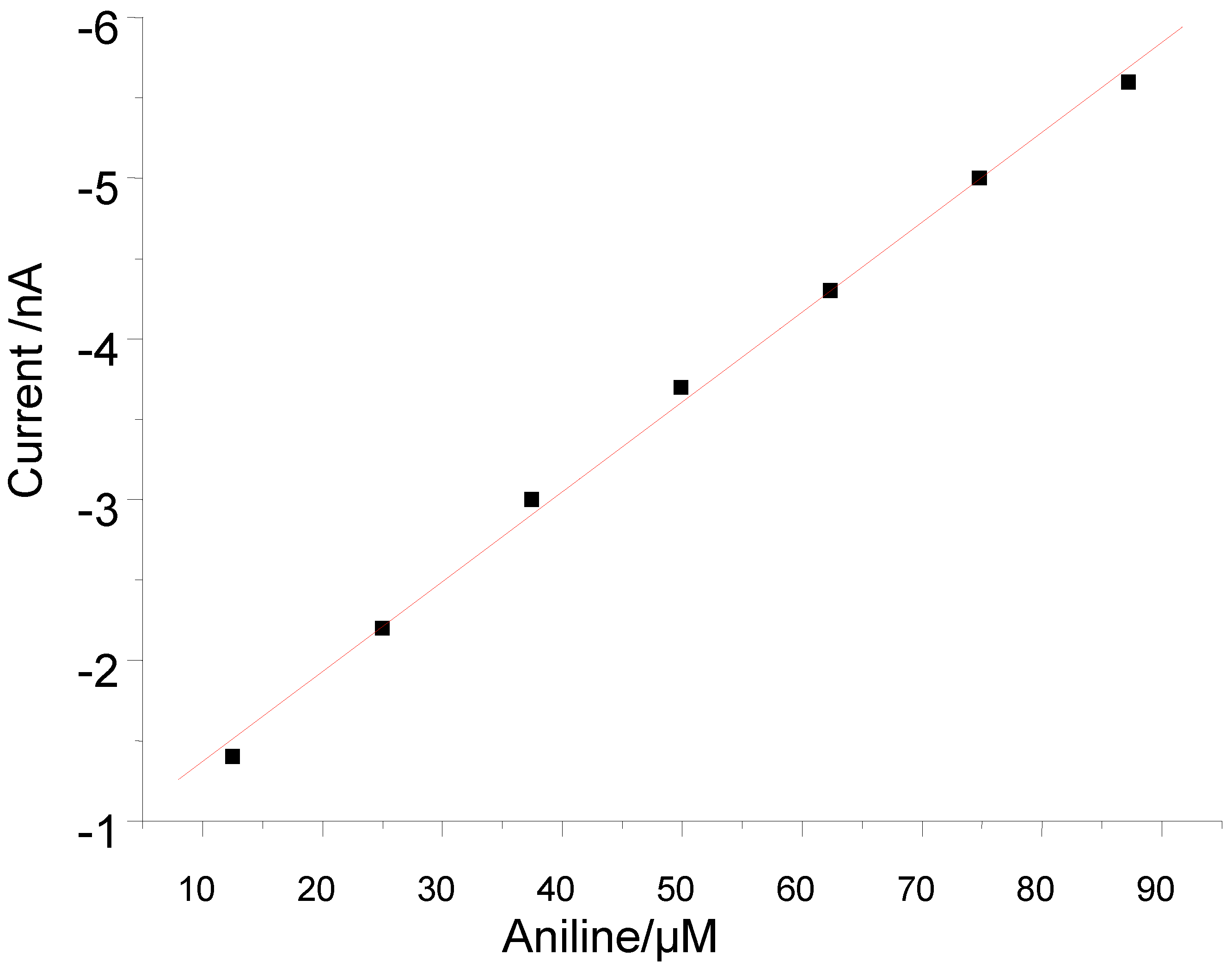
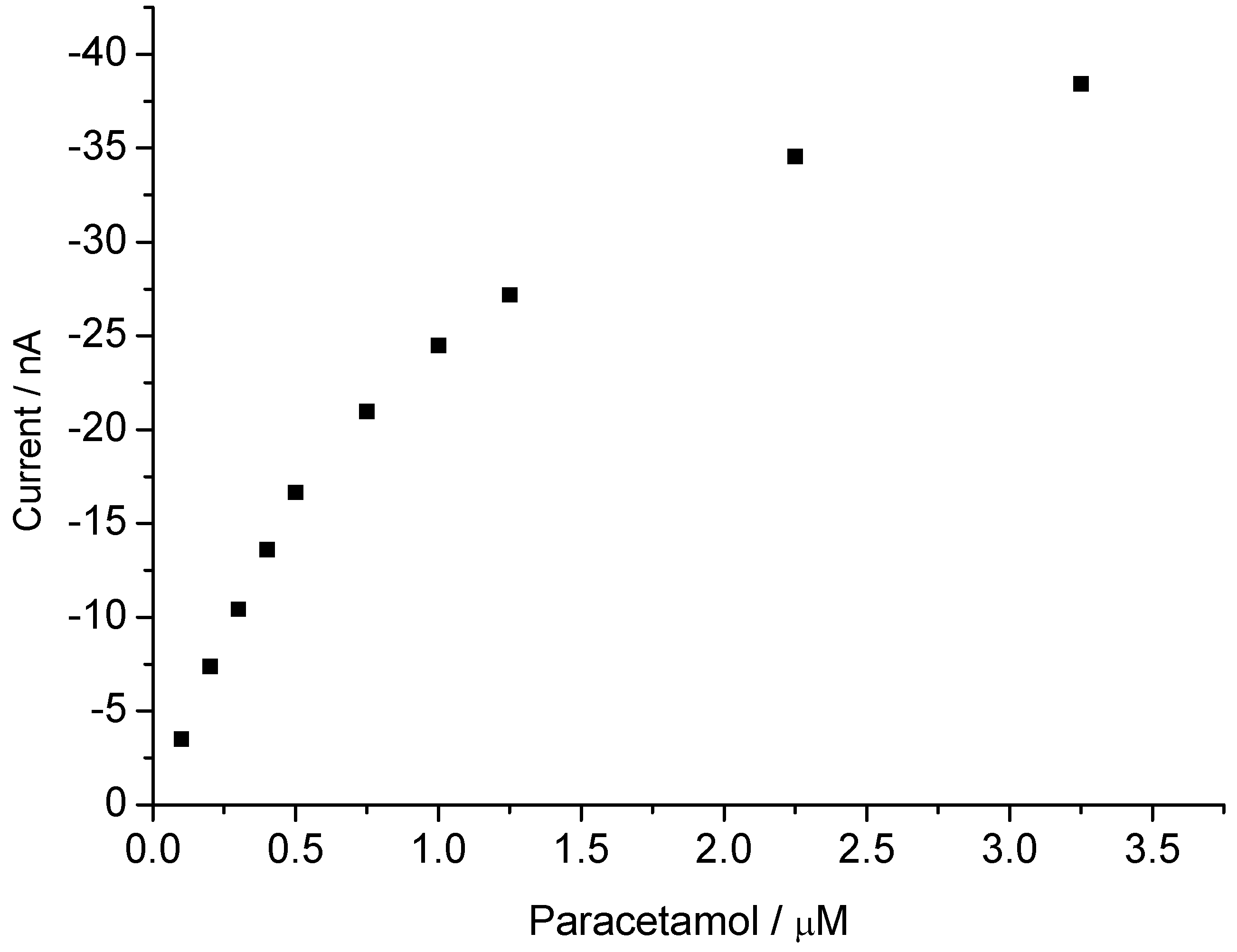

4. Conclusions
Acknowledgments
References
- Diederix, R.E.M.; Ubbink, M.; Canters, G.W. Effect of the protein matrix of cytochrome c in suppressing the inherent peroxidase activity of its heme prosthetic group. ChemBioChem 2002, 3, 110–112. [Google Scholar]
- Lohmann, W.; Karst, U. Biomimetic modelling of oxidative drug metabolism. Anal. Bioanal. Chem. 2011, 391, 79–96. [Google Scholar]
- Ahluwalia, U.; Nayeem, S.M.; Deep, S. The non-native conformations of cytochrome c in sodium dodecyl sulfate and their modulation by ATP. Eur. Biophys. J. 2011, 40, 259–271. [Google Scholar]
- Vazquez-Duhalt, R. Cytochrome c as a biocatalyst. J. Mol. Catal. B Enzym. 1999, 7, 241–249. [Google Scholar]
- Laszlo, J.A.; Compton, D.L. Comparison of peroxidase activities of hemin, cytochrome c, and microperoxidase-11 in molecular solvents and imidazolium-based ionic liquids. J. Mol. Catal. B Enzym. 2002, 18, 109–120. [Google Scholar]
- Everse, J.; Liu, C.-J.J.; Coates, P.W. Physical and catalytic properties of a peroxidase derived from cytochrome c. Biochim. Biophys. Acta 2011, 1812, 1138–1145. [Google Scholar]
- Kluck, R.M.; Ellerby, L.M.; Ellerby, H.M.; Naiem, S.; Yaffe, M.P.; Margoliash, E.; Bredesen, D.; Mauk, A.G.; Sherman, F.; Newmeyer, D.D. Determinants of cytochrome c pro-apoptotic activity. J. Biol. Chem. 2000, 275, 16127–16133. [Google Scholar]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of Apoptosis: The role of the cytochrome c. Biochim. Biophys. Acta 1998, 1366, 139–149. [Google Scholar]
- Yarman, A.; Badalyan, A.; Gajovic-Eichelmann, N.; Wollenberger, U.; Scheller, F.W. Enzyme electrode for aromatic compounds exploiting the catalytic activities of microperoxidase-11. Biosens. Bioelectron. 2011, 30, 320–323. [Google Scholar]
- Bonnard, C.; Papermaster, D.S.; Kiraehenbuhl, J.-P. The streptavidin-biotin bridge technique: Applications in light and electron microscope immunocytochemistry. In Immunolabeling for Electron Microscopy; Elsevier: New York, NY, USA, 1984. [Google Scholar]
- Peng, L.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. Bioelectrocatalytic properties of Agrocybe aegerita peroxygenase. Electrochim. Acta 2010, 55, 7809–7813. [Google Scholar]
- Marcus, R.A.; Sutin, N. Electron transfer in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar]
- Lötzbeyer, T.; Schuhmann, W.; Schmidt, H.-L. Minienzymes: A review for the development of reagentless amperometric biosensors based on direct electron-transfer process. Bioelectrochem. Bioenerg. 1997, 42, 1–6. [Google Scholar]
- Yarman, A.; Nagel, T.; Gajovic-Eichelmann, N.; Fischer, A.; Wollenberger, U.; Scheller, F.W. Bioelectrocatalysis by microperoxidase-11 in a multilayer architecture of chitosan embedded gold nanoparticles. Electroanalysis 2011, 23, 611–618. [Google Scholar]
- Liu, J.; Qiu, J.; Sun, K.; Chen, J.; Miao, Y. Electrochemistry of hemin self-assembled from aqueoushexadecyltrimethylammonium bromide (CTAB) solution on single-wall-carbon-nanotube-modified glassy carbon electrodes. Helv. Chim. Acta 2009, 92, 462–469. [Google Scholar]
- Feng, Z.Q.; Sagara, T.; Niki, K. Electroreflectance study of hemin adsorbed on pyrolytic graphite electrode surface and its coadsorption with with methylene blue. J. Electroanal. Chem. 1993, 349, 159–171. [Google Scholar]
- Chen, J.; Wollenberger, U.; Lisdat, F.; Ge, B.; Scheller, F.W. Superoxide sensor based on hemin modified electrode. Sens. Actuat. B 2000, 70, 115–120. [Google Scholar]
- Zheng, N.; Zeng, Y.; Osborne, P.G.; Li, Y.; Chang, W.; Wang, Z. Electrocatalytic reduction of dioxygen on hemin based carbon paste electrode. J. Appl. Electrochem. 2002, 32, 129–133. [Google Scholar]
- Feng, J.-J.; Li, Z.-H.; Li, Y.-F.; Wang, A.-J.; Zhang, P.-P. Electrochemical determination of dioxygen and hydrogen peroxide using Fe3O4@SiO2@hemin microparticles. Microchim. Acta 2002, 32, 129–133. [Google Scholar]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.D.; Ruzgas, T.; Gazaryan, I. Direct electron transfer between heme-containing enzymes and electrodes as basis for third generation biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar]
- Marques, M.H. Insights into porphyrin chemistry provided by the microperoxidases, the haempeptides derived from cytochrome c. Dalton Trans. 2007, 39, 4371–4385. [Google Scholar]
- Ruzgas, T.; Gaigalas, A.; Gorton, L. Diffusionless electron transfer of microperoxidase-11 on gold electrodes. J. Electroanal. Chem. 1999, 469, 123–131. [Google Scholar]
- Zhou, Y.; Liu, S.; Jiang, H.-J.; Yang, H.; Chen, H.Y. Direct electrochemistry and bioelectrocatalysis of microperoxidase-11 immobilized on chitosan-graphene nanocomposite. Electroanalysis 2010, 22, 1323–1328. [Google Scholar]
- Feng, J.-J.; Zhao, G.; Xu, J.-J.; Chen, H.-Y. Direct electrochemistry and electrocatalysis of heme proteins immobilized on gold nanoparticles stabilized by chitosan. Anal. Biochem. 2005, 342, 280–286. [Google Scholar] [CrossRef]
- Dallacosta, C.; Casella, L.; Monzani, E. Modified microperoxidases exhibit different reactivity towards phenolic substrates. ChemBioChem 2004, 5, 1692–1699. [Google Scholar]
- Csöregi, E.; Jönsson-Petterson, G.; Gorton, L. Mediatorless electrocatalytic reduction of hydrogen peroxide at graphite electrodes chemically modified with peroxidases. J. Biotechnol. 1993, 30, 315–337. [Google Scholar]
- Ranieri, A.; Battistuzzi, G.; Borsari, M.; Bortolotti, C.A.; Di Rocco, G.; Monari, S.; Sola, M. A bis-histidine-ligated unfolded cytochrome c immobilized on anionic SAM shows pseudo-peroxidase activity. Electrochem. Commun. 2012, 14, 29–31. [Google Scholar]
- Dunford, H.B. Oxidations of iron(II)/(III) by hydrogen peroxide: From aquo to enzyme. Coord. Chem. Rev. 2002, 311, 233–234. [Google Scholar]
- Astuti, Y.; Topoglidis, E.; Durrant, J.R. Use of microperoxidase-11 to functionalize tin dioxide electrodes for the optical and electrochemical sensing of hydrogen peroxide. Anal. Chim. Acta 2011, 686, 126–132. [Google Scholar]
- Razumas, V.; Kazlauskaitė, J.; Vidžiūnaitė, R. Electrocatalytic reduction of hydrogen peroxide on the microperoxidase-11 modified carbon paste and graphite electrodes. Bioelectrochem. Bioenerg. 1996, 39, 139–143. [Google Scholar]
- Youssoufi-Korri, H.; Desbenoit, N.; Ricoux, R.; Mahy, J.P.; Lecomte, S. Eleboration of a new hydrogen peroxide biosensor using microperoxidase 8 (MP8) immobilised on a polypyyrole coated electrode. Mater. Sci. Eng. C 2008, 28, 855–860. [Google Scholar]
- Wang, M.; Shen, Y.; Liu, Y.; Wang, T.; Zhao, F.; Liu, F.; Dong, S. Direct electrochemistry of microperoxidase-11 using carbon nanotube modified electrodes. J. Electroanal. Chem. 2005, 578, 121–127. [Google Scholar]
- Liu, Y.; Wang, M.; Zhao, F.; Guo, Z.; Chen, H.; Dong, S. Direct electron transfer and electrocatalysis of microperoxidase immobilised on nanohybrid film. J. Electroanal. Chem. 2005, 581, 1–10. [Google Scholar]
- Zhu, X.; Yuri, I.; Gan, X.; Suzuki, I.; Li, G. Electrochemical study of the effect of nano-zinc oxide on microperoxidase and its application to more sensitive hydrogen peroxide biosensor preparation. Biosens. Bioelectron. 2007, 22, 1600–1604. [Google Scholar]
- Huang, W.; Jia, J.; Zhang, Z.; Han, X.; Tang, J.; Wang, J.; Dong, S.; Wang, E. Hydrogen peroxide biosensor based on microperoxidase-11 entrapped in lipid membrane. Biosens. Bioelectron. 2003, 18, 1225–1230. [Google Scholar]
- Cipriano, T.C.; Takahashi, P.M.; de Lima, D.; Oliveira, V.X.; Souza, J.A.; Martinho, H.; Alves, W.A. Spatial organization of peptide nanotubes for electrochemical devices. J. Mater. Sci. 2010, 45, 5101–5108. [Google Scholar]
- Wan, J.; Bi, J.; Du, P.; Zhang, S. Biosensor based on the biocatalysis of microperoxidase-11 in nanocomposite material of multiwalled carbon nanotubes/room temperature ionic liquid for amperometric determination of hydrogen peroxide. Anal. Biochem. 2009, 386, 256–261. [Google Scholar]
- Chen, Z.; Sun, D.; Zhou, Y.; Zhao, J.; Lu, T.; Huang, X.; Cai, C.; Shen, J. Nano polyurethane-assisted ultrasensitive biodetection of hydrogen peroxide over immobilized Microperoxidase-11. Biosens.Bioelectron. 2011, 29, 53–59. [Google Scholar]
- Ju, H.; Liu, S.; Ge, B.; Lisdat, F.; Scheller, F.W. Electrochemistry of cytochrome c immobilized on colloidal gold modified carbon paste electrodes and its electrocatalytic activity. Electroanalysis 2002, 14, 141–147. [Google Scholar] [CrossRef]
- Wang, L.; Waldeck, D.H. Denaturation of cytochrome c and its peroxidase activity when immobilized on SAM films. J.Phys.Chem.C 2008, 112, 1351–1356. [Google Scholar]
- Jänchen, M.; Scheller, F.; Prümke, H.-J.; Mohr, P. Polarographische untersuchungen zur peroxidaseaktivität von ligandiertem deuterohämin. Acta Biol. Medica Ger. 1975, 34, 319–324. [Google Scholar]
- Yang, S.; Li, Y.; Jiang, X.; Chen, Z.; Lin, X. Horseradish peroxidase biosensor based on layer-by-layer technique for the determination of phenolic compounds. Sens. Actuat. B 2006, 114, 774–780. [Google Scholar]
- Veeger, C. Does P450-type catalysis proceed through a peroxo-iron intermediate? A review of studies with microperoxidase. J. Inorg. Biochem. 2002, 91, 35–45. [Google Scholar]
- Lin, M.S.; Jan, B.I.; Chen, P.Y.; Cheng, W.C.; Chen, C.H. New determination scheme of p-aminophenol by MnO2 modified electrode coupled with flow injection analysis. Electroanalysis 2010, 22, 1278–1281. [Google Scholar]
- Wollenberger, U.; Lisdat, F.; Rose, A.; Streffer, K. Bioelectrochemistry: Fundamentals, Experimental Techniques and Applications; Bartlett, P., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Wollenberger, U.; Neumann, B. Quinoprotein glucose dehydrogenase modified carbon paste electrode for the detection of phenolic compounds. Electroanalysis 1997, 9, 366–371. [Google Scholar]
- Mie, Y.; Kowata, K.; Hirano, Y.; Niwa, O.; Mizutani, F. Comparison of enzymatic recycling electrodes for measuring aminophenol: Development of a highly sensitive natriuretic peptide assay system. Anal. Sci. 2008, 24, 577–582. [Google Scholar]
- Solná, R.; Skládal, P. Amperometric flow-injection determination of phenolic compounds using a biosensor with immobilized laccase, peroxidase and tyrosinase. Electroanalysis 2005, 17, 2137–2146. [Google Scholar]
- Zhu, Y.; Li, J.; Dong, S. Dimerization of hydroxylated species of m-aminophenolby cytochrome c with hydrogen peroxide. J. Mol. Catal. B Enzym. 1998, 5, 475–482. [Google Scholar]
- Kawakami, M.; Akamatsu, N.; Koya, H.; Amada, K. Amperometric detection of phenol with cytochrome c-modified gold electrode using dual working electrode system. Anal. Lett. 2005, 38, 549–561. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yarman, A.; Neumann, B.; Bosserdt, M.; Gajovic-Eichelmann, N.; Scheller, F.W. Peroxide-Dependent Analyte Conversion by the Heme Prosthetic Group, the Heme Peptide “Microperoxidase-11” and Cytochrome c on Chitosan Capped Gold Nanoparticles Modified Electrodes. Biosensors 2012, 2, 189-204. https://doi.org/10.3390/bios2020189
Yarman A, Neumann B, Bosserdt M, Gajovic-Eichelmann N, Scheller FW. Peroxide-Dependent Analyte Conversion by the Heme Prosthetic Group, the Heme Peptide “Microperoxidase-11” and Cytochrome c on Chitosan Capped Gold Nanoparticles Modified Electrodes. Biosensors. 2012; 2(2):189-204. https://doi.org/10.3390/bios2020189
Chicago/Turabian StyleYarman, Aysu, Bettina Neumann, Maria Bosserdt, Nenad Gajovic-Eichelmann, and Frieder W. Scheller. 2012. "Peroxide-Dependent Analyte Conversion by the Heme Prosthetic Group, the Heme Peptide “Microperoxidase-11” and Cytochrome c on Chitosan Capped Gold Nanoparticles Modified Electrodes" Biosensors 2, no. 2: 189-204. https://doi.org/10.3390/bios2020189
APA StyleYarman, A., Neumann, B., Bosserdt, M., Gajovic-Eichelmann, N., & Scheller, F. W. (2012). Peroxide-Dependent Analyte Conversion by the Heme Prosthetic Group, the Heme Peptide “Microperoxidase-11” and Cytochrome c on Chitosan Capped Gold Nanoparticles Modified Electrodes. Biosensors, 2(2), 189-204. https://doi.org/10.3390/bios2020189