Rapid Self-Assembly of Metal/Polymer Nanocomposite Particles as Nanoreactors and Their Kinetic Characterization


Abstract
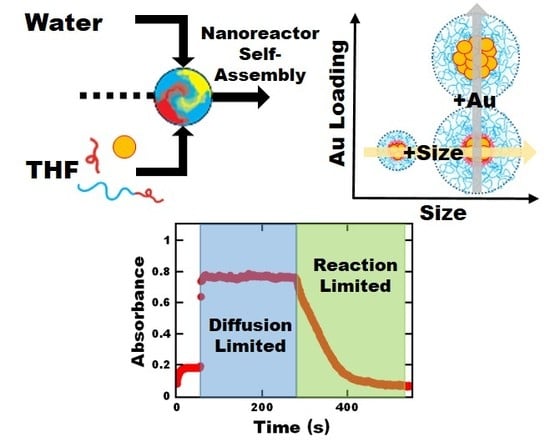
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoreactor Assembly
2.3. Nanoreactor Characterization
2.4. Kinetic Analysis
2.5. Langmuir-Hinshelwood Kinetics
2.6. NMR Measurements
3. Results and Discussion
3.1. Nanoreactor Self-Assembly
3.2. Initial Characterization of Nanoreactor Performance
3.3. Probing Potential Mass Transfer Limitations
3.3.1. Induction Time
3.3.2. Reaction Rate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Cardozo, A.F.; Chen, S.; Zhang, W.; Julcour, C.; Lansalot, M.; Blanco, J.F.; Gayet, F.; Delmas, H.; Charleux, B.; et al. Core-Shell Nanoreactors for Efficient Aqueous Biphasic Catalysis. Chem. A Eur. J. 2014, 20, 15505–15517. [Google Scholar] [CrossRef] [PubMed]
- Cotanda, P.; Petzetakis, N.; O’reilly, R.K. Catalytic Polymeric Nanoreactors: More than a Solid Supported Catalyst. MRS Commun. 2012, 2, 119–126. [Google Scholar] [CrossRef]
- Walther, A.; Muller, A.H.E. Janus Particles: Synthesis, Self-Assembly, Physical Properties, and Applications. Chem. Rev. 2013, 113, 5194–5261. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Ghorai, S. “Designer”-Surfactant-Enabled Cross-Couplings in Water at Room Temperature. Aldrichim. Acta 2012, 45, 3–16. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S. Transitioning Organic Synthesis from Organic Solvents to Water. What’s Your E-Factor? Green Chem. 2014, 16, 3660–3679. [Google Scholar] [CrossRef] [PubMed]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent Advances in Catalysis in Micellar Media. Green Chem. 2015, 17, 644–683. [Google Scholar] [CrossRef]
- Petrosko, S.H.; Johnson, R.; White, H.; Mirkin, C.A. Nanoreactors: Small Spaces, Big Implications in Chemistry. J. Am. Chem. Soc. 2016, 138, 7443–7445. [Google Scholar] [CrossRef] [PubMed]
- Vriezema, D.M.; Aragone, M.C.; Elemans, J.A.A.W.; Cornelissen, J.J.L.M.; Rowan, A.E.; Nolte, R.J.M. Self-Assembled Nanoreactors. Chem. Rev. 2005, 105, 1445–1489. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dimroth, J.; Weck, M. Compartmentalization of Incompatible Catalytic Transformations for Tandem Catalysis. J. Am. Chem. Soc. 2015, 137, 12984–12989. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Moatsou, D.; Hands-Portman, I.; Longbottom, D.A.; O’Reilly, R.K. Recyclable L-Proline Functional Nanoreactors with Temperature-Tuned Activity Based on Core-Shell Nanogels. ACS Macro Lett. 2014, 3, 1235–1239. [Google Scholar] [CrossRef]
- Cotanda, P.; Lu, A.; Patterson, J.P.; Petzetakis, N.; Reilly, R.K.O. Functionalized Organocatalytic Nanoreactors: Hydrophobic Pockets for Acylation Reactions in Water. Macromolecules 2012, 45, 2377–2384. [Google Scholar] [CrossRef]
- Gall, B.; Bortenschlager, M.; Nuyken, O.; Weberskirch, R. Cascade Reactions in Polymeric Nanoreactors: Mono (Rh)—And Bimetallic (Rh/Ir) Micellar Catalysis in the Hydroaminomethylation of 1-Octene. Macromol. Chem. Phys. 2008, 209, 1152–1159. [Google Scholar] [CrossRef]
- De Martino, M.T.; Abdelmohsen, L.K.E.A.; Rutjes, F.P.J.T.; Van Hest, J.C.M. Nanoreactors for Green Catalysis. Beilstein J. Org. Chem. 2018, 14, 716–733. [Google Scholar] [CrossRef] [PubMed]
- Wunder, S.; Lu, Y.; Albrecht, M.; Ballauff, M. Catalytic Activity of Faceted Gold Nanoparticles Studied by a Model Reaction: Evidence for Substrate-Induced Surface Restructuring. ACS Catal. 2011, 1, 908–916. [Google Scholar] [CrossRef]
- Cotanda, P.; O’Reilly, R.K. Molecular Recognition Driven Catalysis Using Polymeric Nanoreactors. Chem. Commun. 2012, 48, 10280–10282. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Yang, L.; Zhang, M.; Zhang, W.; Wang, S. Microreactor of Pd Nanoparticles Immobilized Hollow Microspheres for Catalytic Hydrodechlorination of Chlorophenols in Water. ACS Appl. Mater. Interfaces 2010, 2, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Angioletti-Uberti, S.; Lu, Y.; Ballauff, M.; Dzubiella, J. Theory of Solvation-Controlled Reactions in Stimuli-Responsive Nanoreactors. J. Phys. Chem. C 2015, 119, 15723–15730. [Google Scholar] [CrossRef]
- Peter, N.; Tan, B.; Lee, C.H.; Li, P. Green Synthesis of Smart Metal/Polymer Nanocomposite Particles and Their Tuneable Catalytic Activities. Polymers 2016, 8, 105. [Google Scholar] [CrossRef]
- Lempke, L.; Ernst, A.; Kahl, F.; Weberskirch, R.; Krause, N. Sustainable Micellar Gold Catalysis—Poly(2-Oxazolines) as Versatile Amphiphiles. Adv. Synth. Catal. 2016, 358, 1491–1499. [Google Scholar] [CrossRef]
- Choi, S.H.; Bates, F.S.; Lodge, T.P. Molecular Exchange in Ordered Diblock Copolymer Micelles. Macromolecules 2011, 44, 3594–3604. [Google Scholar] [CrossRef]
- Gindy, M.E.; Panagiotopoulos, A.Z.; Prud’Homme, R.K. Composite Block Copolymer Stabilized Nanoparticles: Simultaneous Encapsulation of Organic Actives and Inorganic Nanostructures. Langmuir 2008, 24, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, N.M.; Gindy, M.E.; Calero-Ddelc, V.L.; Wolfson, T.; Pagels, R.F.; Adler, D.; Gao, D.; Li, S.; Wang, R.; Zevon, M.; et al. Single-Step Assembly of Multimodal Imaging Nanocarriers: MRI and Long-Wavelength Fluorescence Imaging. Adv. Healthc. Mater. 2015, 4, 1376–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Amin, D.; Messersmith, P.B.; Anthony, J.E.; Prud’homme, R.K. Polymer Directed Self-Assembly of pH-Responsive Antioxidant Nanoparticles. Langmuir 2015, 31, 3612–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhu, Z.; Gian, H.; Wohl, A.R.; Beaman, C.J.; Hoye, T.R.; Macosko, C.W. A Simple Confined Impingement Jets Mixer for Flash Nanoprecipitation. J. Pharm. Sci. 2012, 101, 4018–4023. [Google Scholar] [CrossRef] [PubMed]
- Hervés, P.; Pérez-Lorenzo, M.; Liz-Marzán, L.M.; Dzubiella, J.; Lu, Y.; Ballauff, M. Catalysis by Metallic Nanoparticles in Aqueous Solution: Model Reactions. Chem. Soc. Rev. 2012, 41, 5577. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Wunder, S.; Lu, Y.; Ballauff, M.; Fenger, R.; Rademann, K.; Jaquet, B.; Zaccone, A. Kinetic Analysis of the Catalytic Reduction of 4-Nitrophenol by Metallic Nanoparticles. J. Phys. Chem. C 2014, 118, 18618–18625. [Google Scholar] [CrossRef]
- Momot, K.I.; Kuchel, P.W. Pulsed Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Drug Delivery Systems. Concepts Magn. Reson. 2003, 19A, 51–64. [Google Scholar] [CrossRef]
- Mun, E.A.; Hannell, C.; Rogers, S.E.; Hole, P.; Williams, A.C.; Khutoryanskiy, V.V. On the Role of Specific Interactions in the Diffusion of Nanoparticles in Aqueous Polymer Solutions. Langmuir 2014, 30, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H. Self-Diffusion Coefficients of Water. J. Phys. Chem. 1965, 69, 4412. [Google Scholar] [CrossRef]
- Johnson, B.K.; Prud, R.K. Chemical Processing and Micromixing in Confined Impinging Jets. AIChE J. 2003, 49, 2264–2282. [Google Scholar] [CrossRef]
- Liu, Y.; Kathan, K.; Saad, W.; Prud, R.K. Ostwald Ripening of β-Carotene Nanoparticles. Phys. Rev. Lett. 2007, 36102, 8–11. [Google Scholar] [CrossRef]
- Vaia, R.A.; Maguire, J.F. Polymer Nanocomposites with Prescribed Morphology: Going beyond Nanoparticle-Filled Polymers. Chem. Mater. 2007, 19, 2736–2751. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Nath, S.; Kundu, S.; Esumi, K.; Pal, T. Solvent and Ligand Effects on the Localized Surface Plasmon Resonance (LSPR) of Gold Colloids. J. Phys. Chem. B 2004, 108, 13963–13971. [Google Scholar] [CrossRef]
- Lange, H.; Juárez, B.H.; Carl, A.; Richter, M.; Bastús, N.G.; Weller, H.; Thomsen, C.; Von Klitzing, R.; Knorr, A. Tunable Plasmon Coupling in Distance-Controlled Gold Nanoparticles. Langmuir 2012, 28, 8862–8866. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Sosa, C.L.; Pagels, R.F.; Priestley, R.D.; Prud’homme, R.K. Efficient Preparation of Size Tunable PEGylated Gold Nanoparticles. J. Mater. Chem. B 2016, 4, 4813–4817. [Google Scholar] [CrossRef]
- Pagels, R.F.; Edelstein, J.; Tang, C.; Prud’homme, R.K. Controlling and Predicting Nanoparticle Formation by Block Copolymer Directed Rapid Precipitations. Nano Lett. 2018, 18, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, S.M.; Prud’homme, R.K. Controlling Drug Nanoparticle Formation by Rapid Precipitation. Adv. Drug Deliv. Rev. 2011, 63, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Prud’homme, R.K. Targeted Theragnostic Nanoparticles Via Flash Nanoprecipitation: Principles of Material Selection. In Polymer Nanoparticles for Nanomedicines; Springer: Cham, Switzerland, 2016; pp. 55–85. [Google Scholar]
- Zhao, P.; Feng, X.; Huang, D.; Yang, G.; Astruc, D. Basic Concepts and Recent Advances in Nitrophenol Reduction by Gold- and Other Transition Metal Nanoparticles. Coord. Chem. Rev. 2015, 287, 114–136. [Google Scholar] [CrossRef]
- Tadele, K.; Verma, S.; Nadagouda, M.N.; Gonzalez, M.A.; Varma, R.S. A Rapid Flow Strategy for the Oxidative Cyanation of Secondary and Tertiary Amines via C-H Activation. Sci. Rep. 2017, 7, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Kaiser, J.; Marzun, G.; Ott, A.; Lu, Y.; Ballauff, M.; Zaccone, A.; Barcikowski, S.; Wagener, P. Ligand-Free Gold Nanoparticles as a Reference Material for Kinetic Modelling of Catalytic Reduction of 4-Nitrophenol. Catal. Lett. 2015, 145, 1105–1112. [Google Scholar] [CrossRef]
- Fenger, R.; Fertitta, E.; Kirmse, H.; Thünemann, A.F.; Rademann, K. Size Dependent Catalysis with CTAB-Stabilized Gold Nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 9343–9349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, L.; Wu, C.; Fu, G.; Zhao, H.; He, B. Synthesis, Characterization and Application of Well-Defined Environmentally Responsive Polymer Brushes on the Surface of Colloid Particles. Polymer 2007, 48, 1989–1997. [Google Scholar] [CrossRef]
- Nigra, M.M.; Ha, J.M.; Katz, A. Identification of Site Requirements for Reduction of 4-Nitrophenol Using Gold Nanoparticle Catalysts. Catal. Sci. Technol. 2013, 3, 2976–2983. [Google Scholar] [CrossRef]
- Menumerov, E.; Hughes, R.A.; Neretina, S. Catalytic Reduction of 4-Nitrophenol: A Quantitative Assessment of the Role of Dissolved Oxygen in Determining the Induction Time. Nano Lett. 2016, 16, 7791–7797. [Google Scholar] [CrossRef] [PubMed]
- D’Addio, S.M.; Saad, W.; Ansell, S.M.; Squiers, J.J.; Adamson, D.H.; Herrera-Alonso, M.; Wohl, A.R.; Hoye, T.R.; MacOsko, C.W.; Mayer, L.D.; et al. Effects of Block Copolymer Properties on Nanocarrier Protection from in Vivo Clearance. J. Control. Release 2012, 162, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Budijono, S.J.; Russ, B.; Saad, W.; Adamson, D.H.; Prud’homme, R.K. Block Copolymer Surface Coverage on Nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2010, 360, 105–110. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chen, D.H. Catalytic Reduction of 4-Nitrophenol by Magnetically Recoverable Au Nanocatalyst. J. Hazard. Mater. 2009, 165, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Ncube, P.; Bingwa, N.; Baloyi, H.; Meijboom, R. Catalytic Activity of Palladium and Gold Dendrimer-Encapsulated Nanoparticles for Methylene Blue Reduction: A Kinetic Analysis. Appl. Catal. A Gen. 2015, 495, 63–71. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
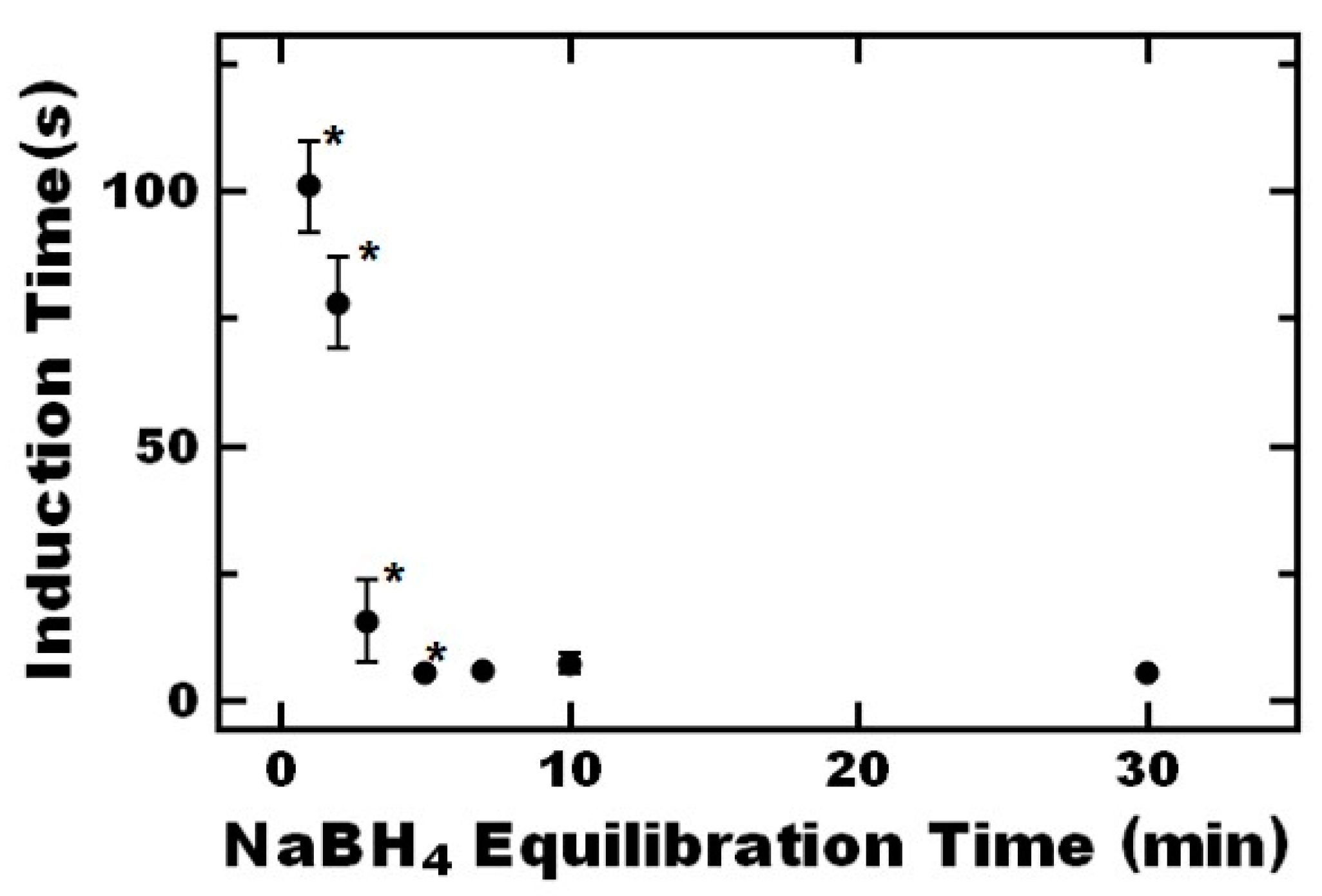{kind=link}
| Support | Diameter (nm) | k1 (L m−2 s−1) | Induction Time (s) | Reference |
|---|---|---|---|---|
| PS | 5 | 0.414 ± 0.095 | 229 ± 21 | This Paper |
| DDT | 5 | Undetected | N/A | This Paper |
| Citrate | 5 | 0.173 ± 0.026 | 5 ± 1 | This Paper |
| Ligand-Free | 7 | 0.17 | N/A | [41] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, A.; Vuong, T.T.; Zeevi, M.P.; Hittel, B.J.; Wi, S.; Tang, C. Rapid Self-Assembly of Metal/Polymer Nanocomposite Particles as Nanoreactors and Their Kinetic Characterization. Nanomaterials 2019, 9, 318. https://doi.org/10.3390/nano9030318
Harrison A, Vuong TT, Zeevi MP, Hittel BJ, Wi S, Tang C. Rapid Self-Assembly of Metal/Polymer Nanocomposite Particles as Nanoreactors and Their Kinetic Characterization. Nanomaterials. 2019; 9(3):318. https://doi.org/10.3390/nano9030318
Chicago/Turabian StyleHarrison, Andrew, Tien T. Vuong, Michael P. Zeevi, Benjamin J. Hittel, Sungsool Wi, and Christina Tang. 2019. "Rapid Self-Assembly of Metal/Polymer Nanocomposite Particles as Nanoreactors and Their Kinetic Characterization" Nanomaterials 9, no. 3: 318. https://doi.org/10.3390/nano9030318