
Synthesis of Pt@TiO2@CNTs Hierarchical Structure Catalyst by Atomic Layer Deposition and Their Photocatalytic and Photoelectrochemical Activity

Abstract
:1. Introduction
2. Experimental
2.1. Acid Treatment of CNTs
2.2. Fabrication of TiO2@CNTs Structure by ALD
2.3. Fabrication of Pt@TiO2@CNTs Composites by PEALD
2.4. Photocatalytic Activity and Characterization
3. Results and Discussion
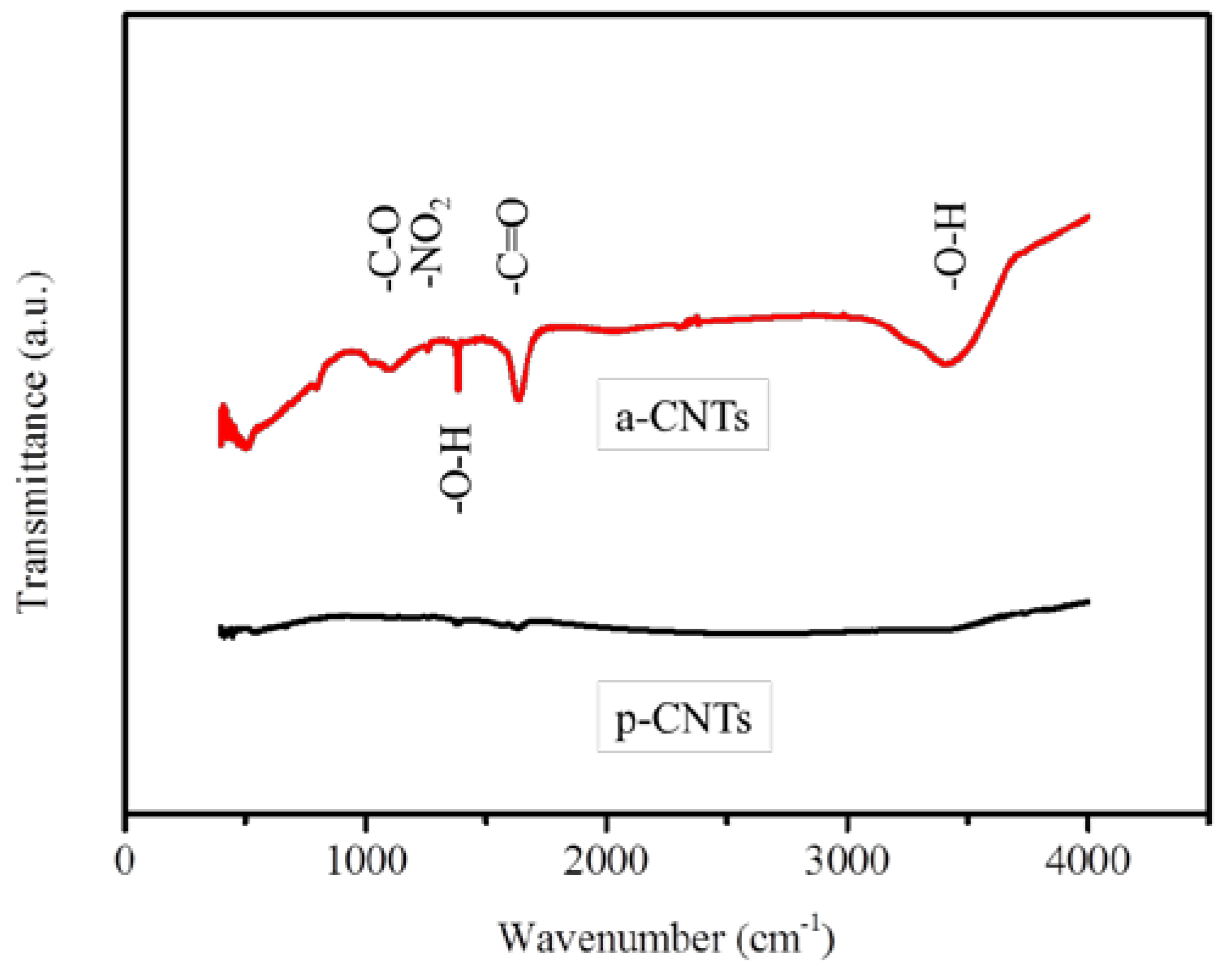
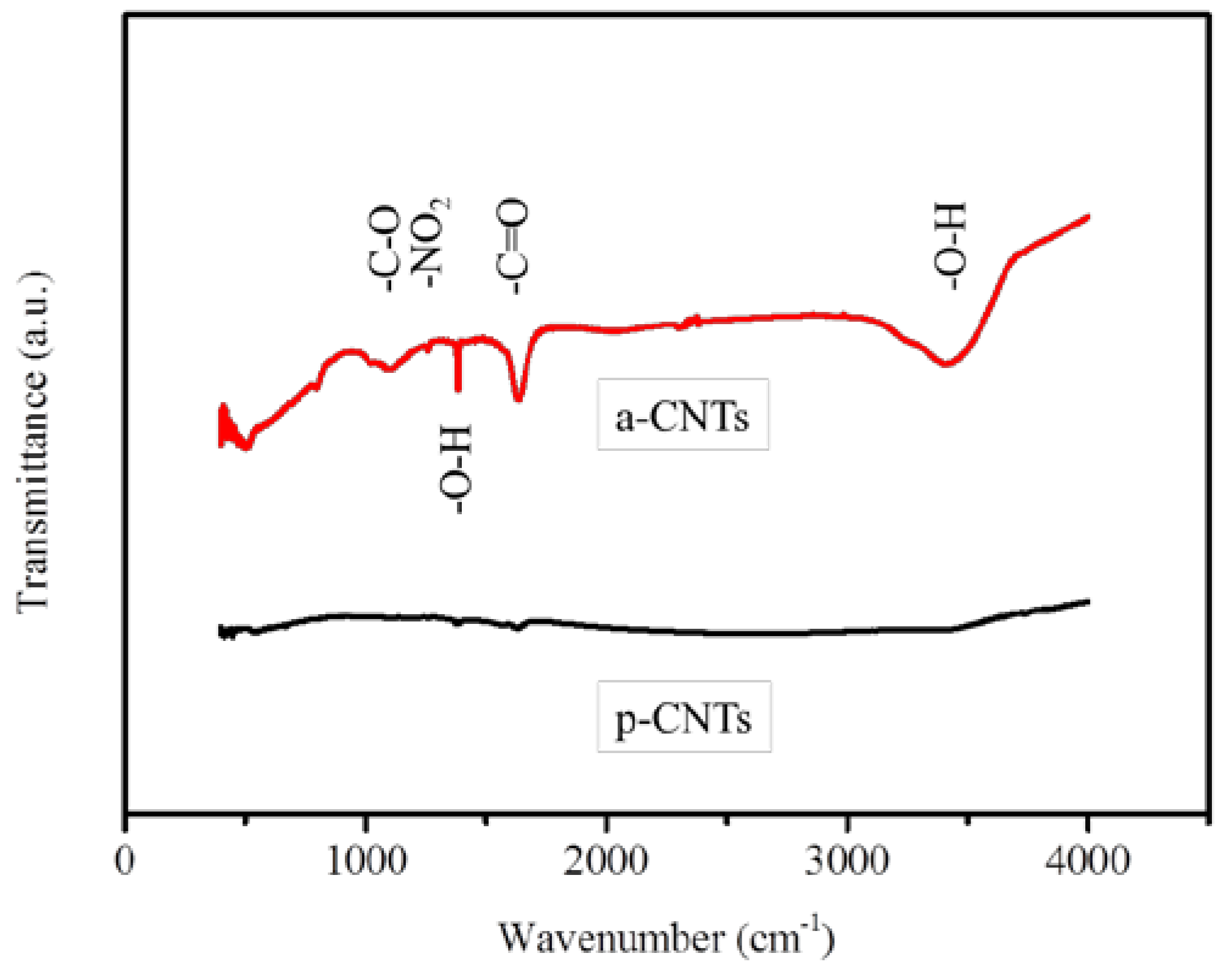
3.1. Modification of Multiwall CNTs
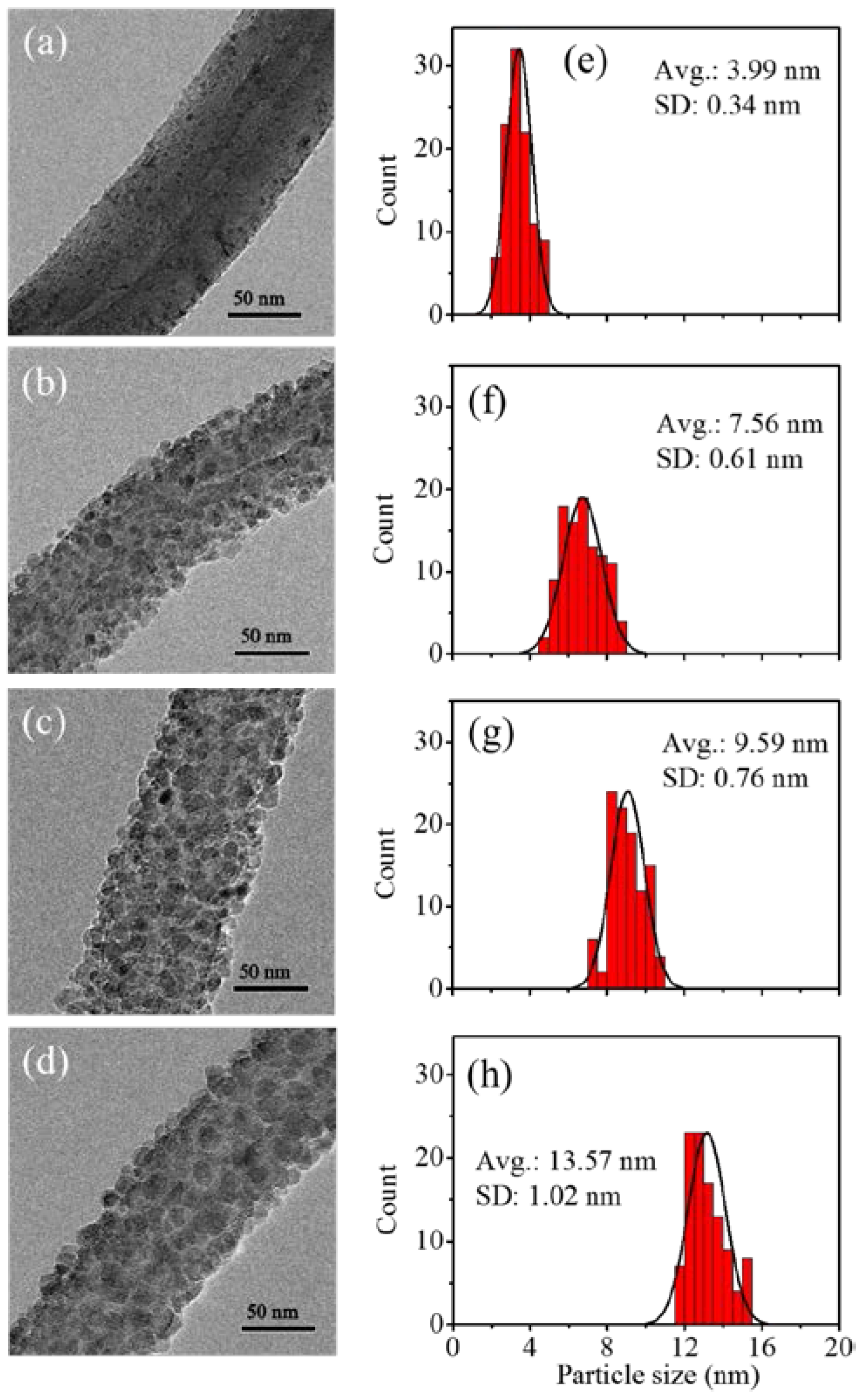
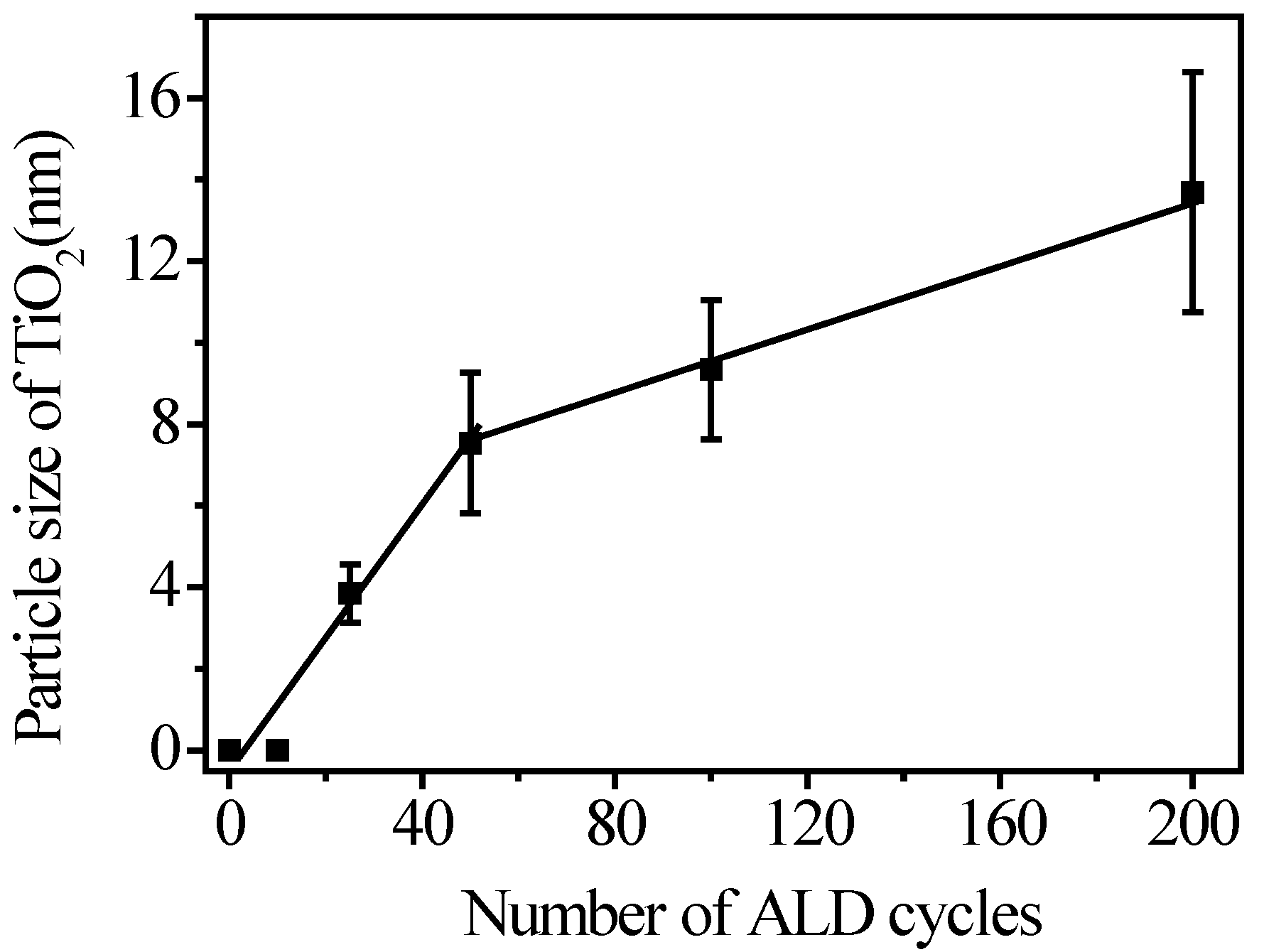
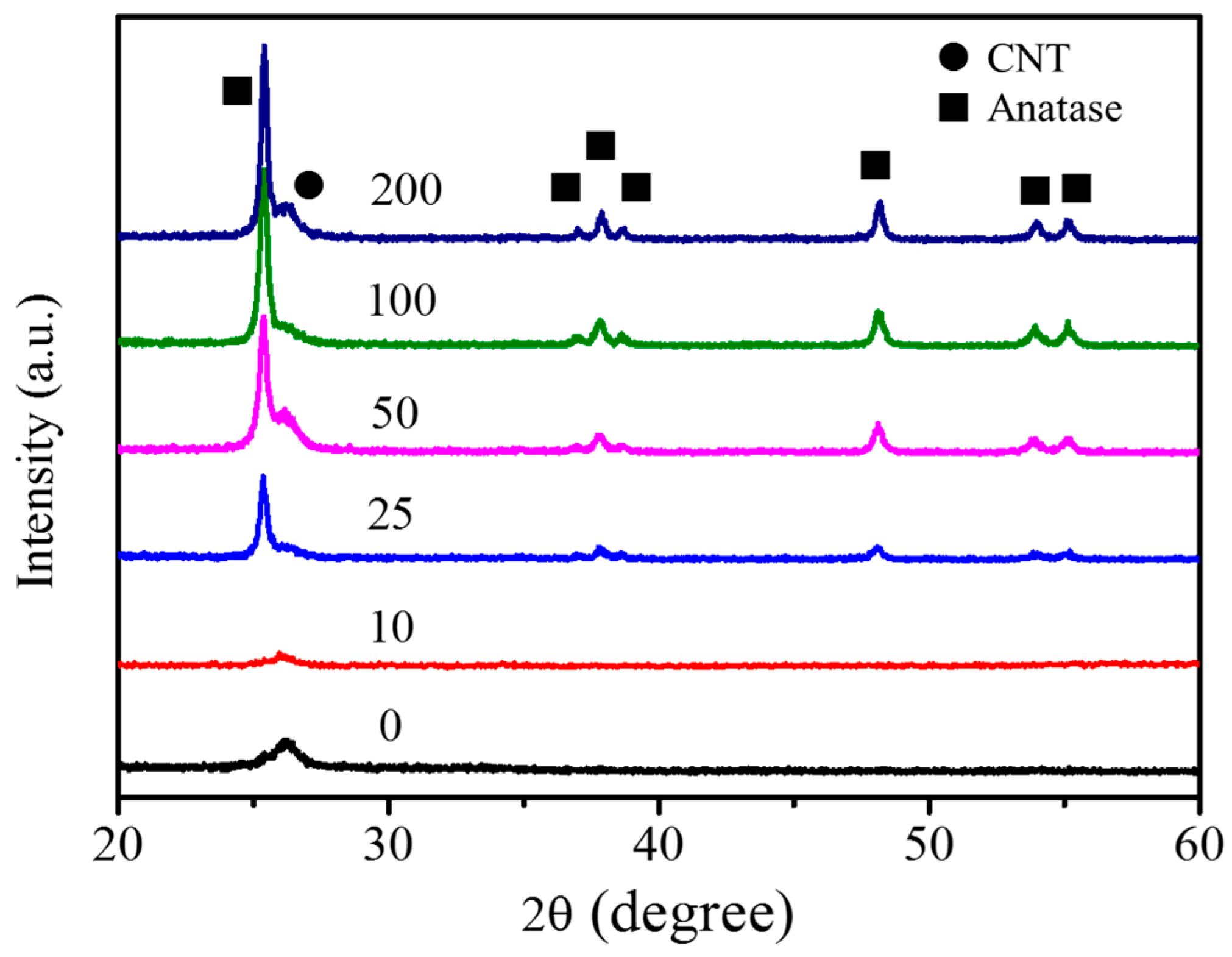
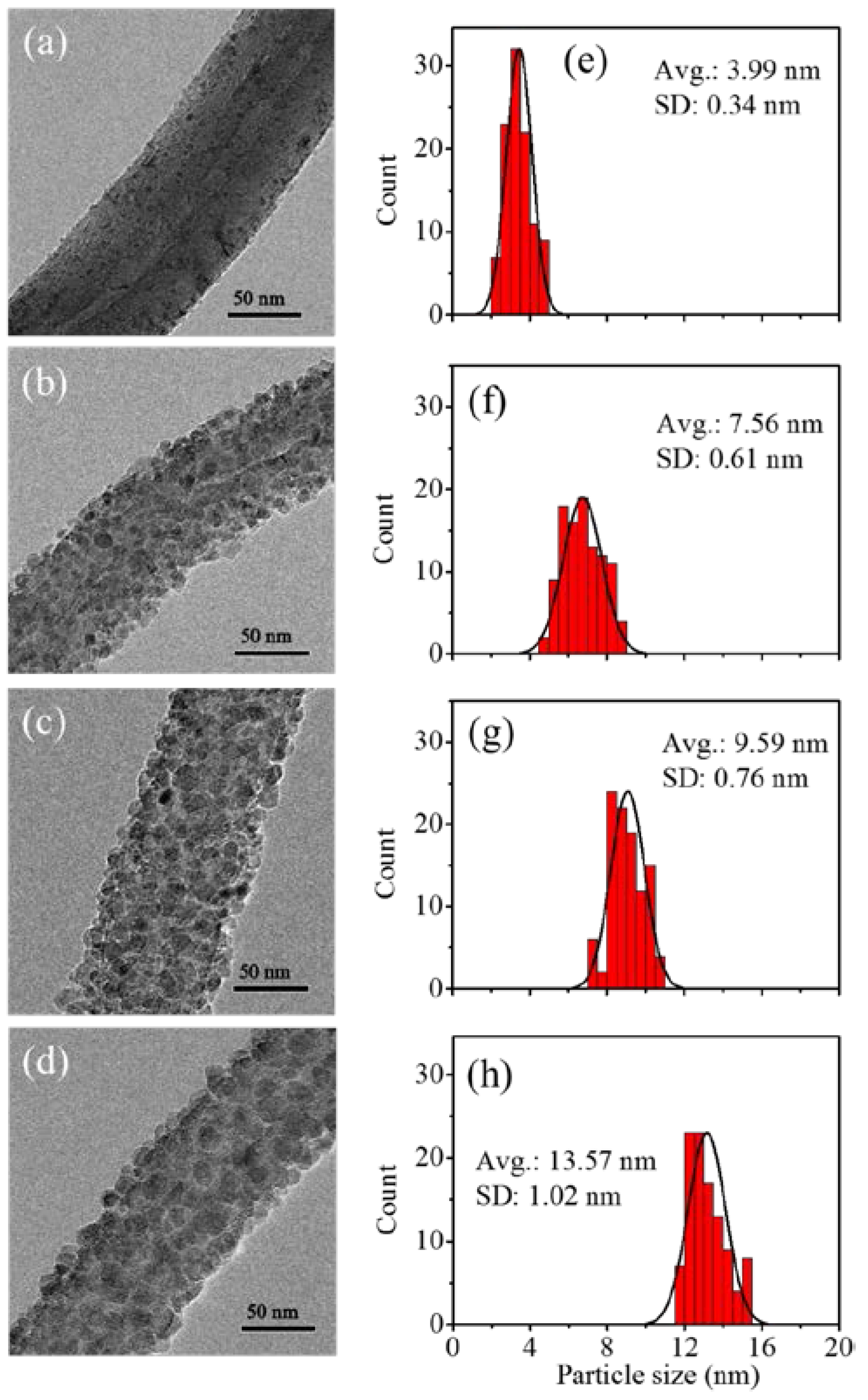
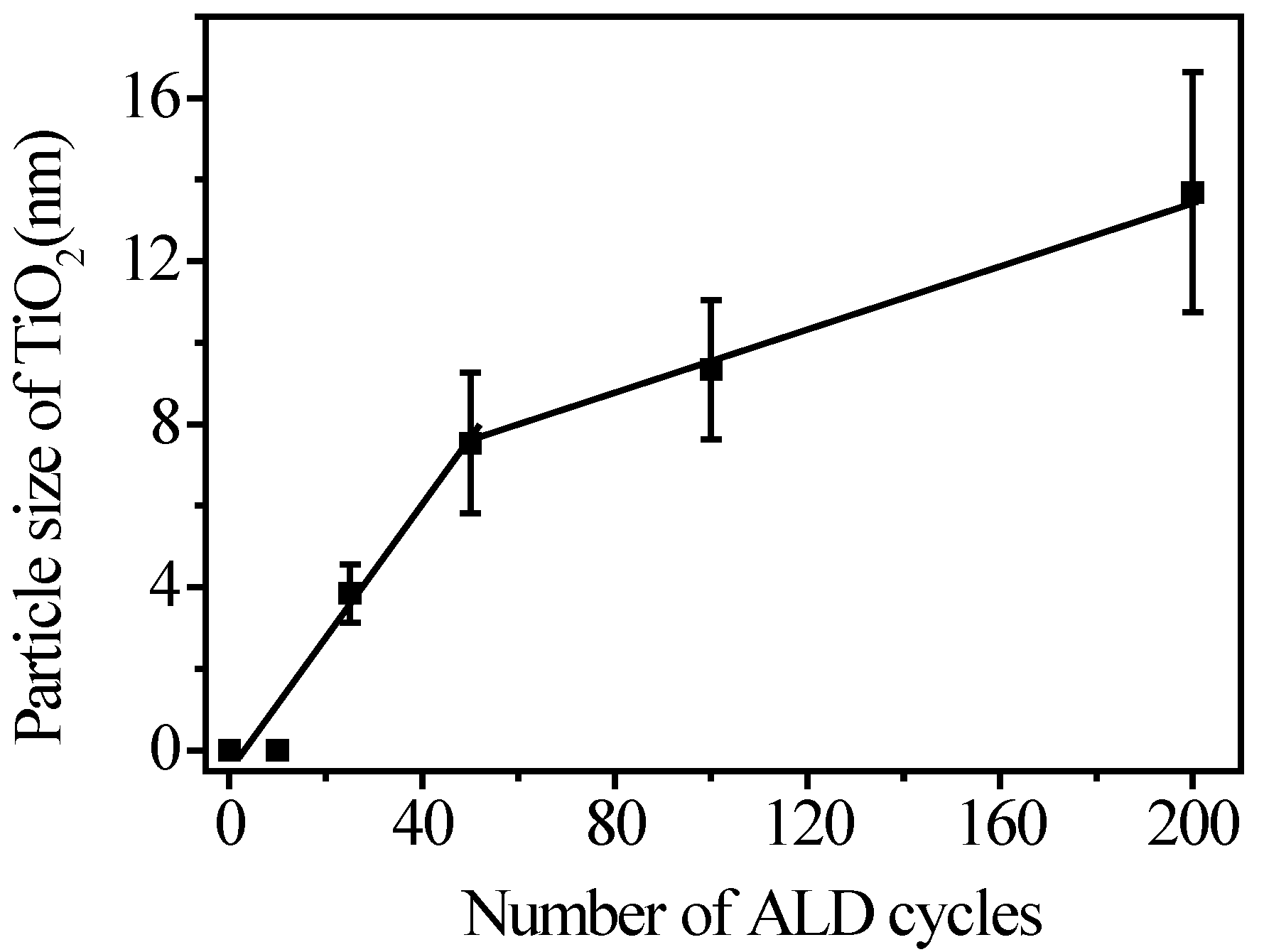
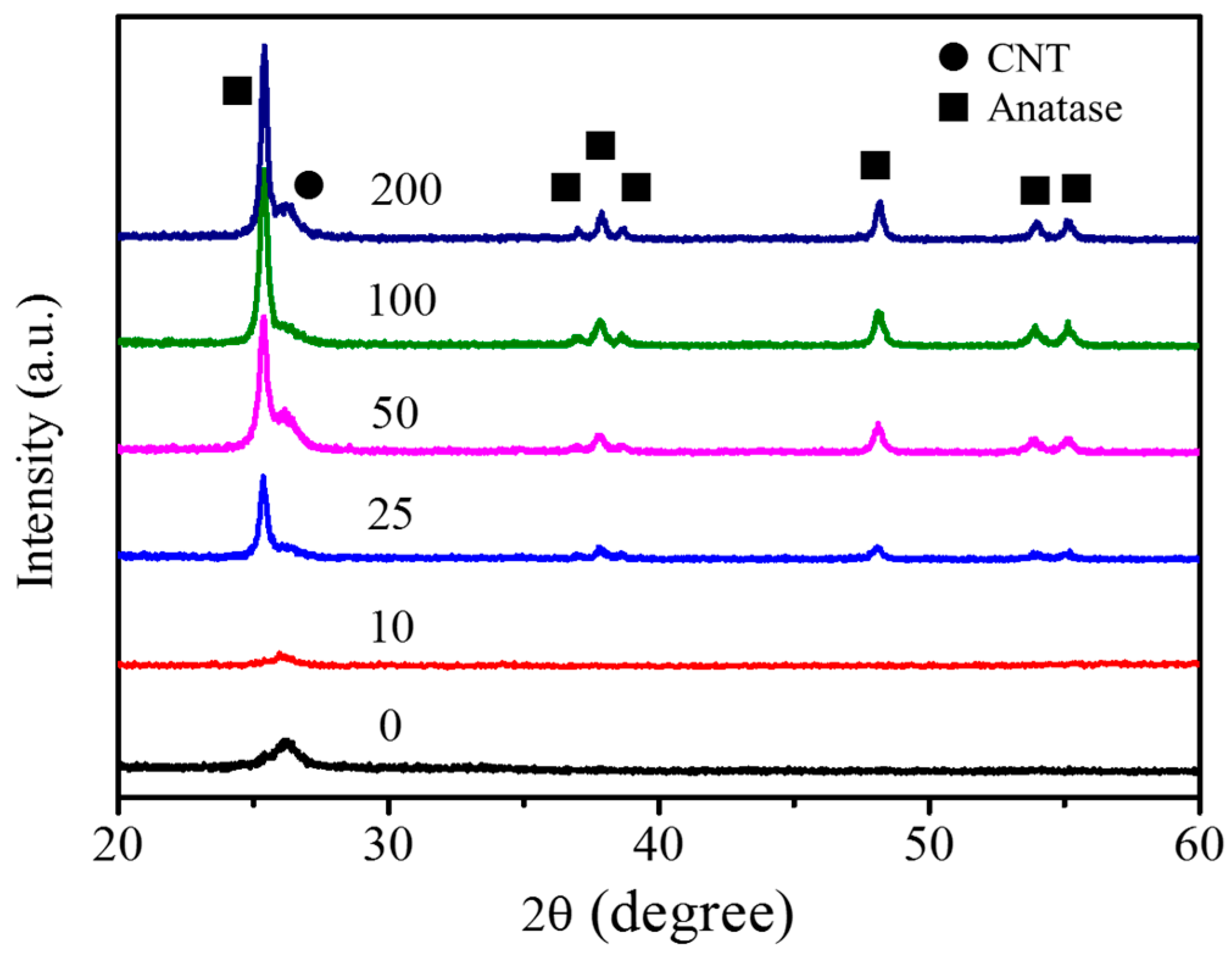
3.2. Fabrication and Characterization of TiO2@CNTs Structures
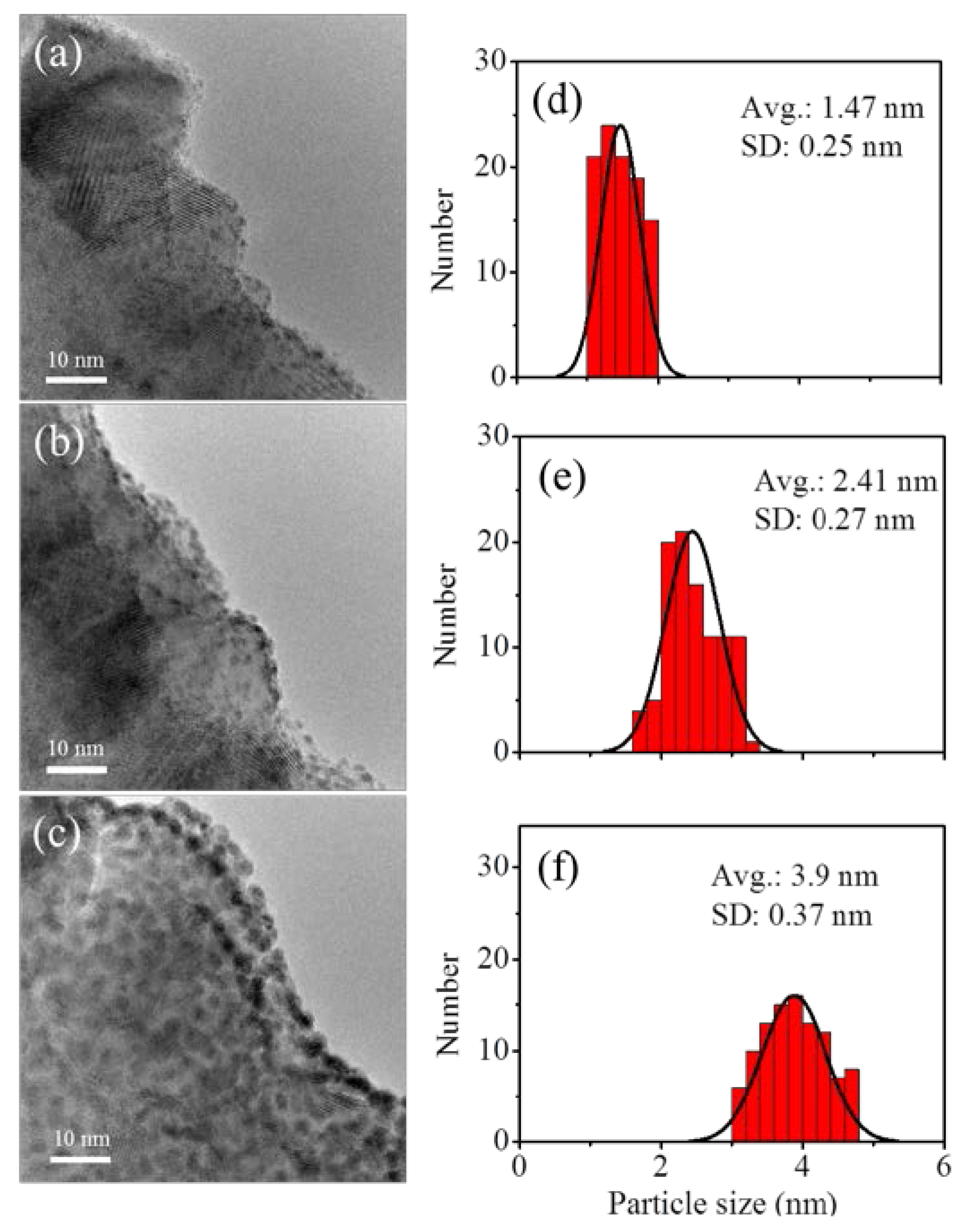
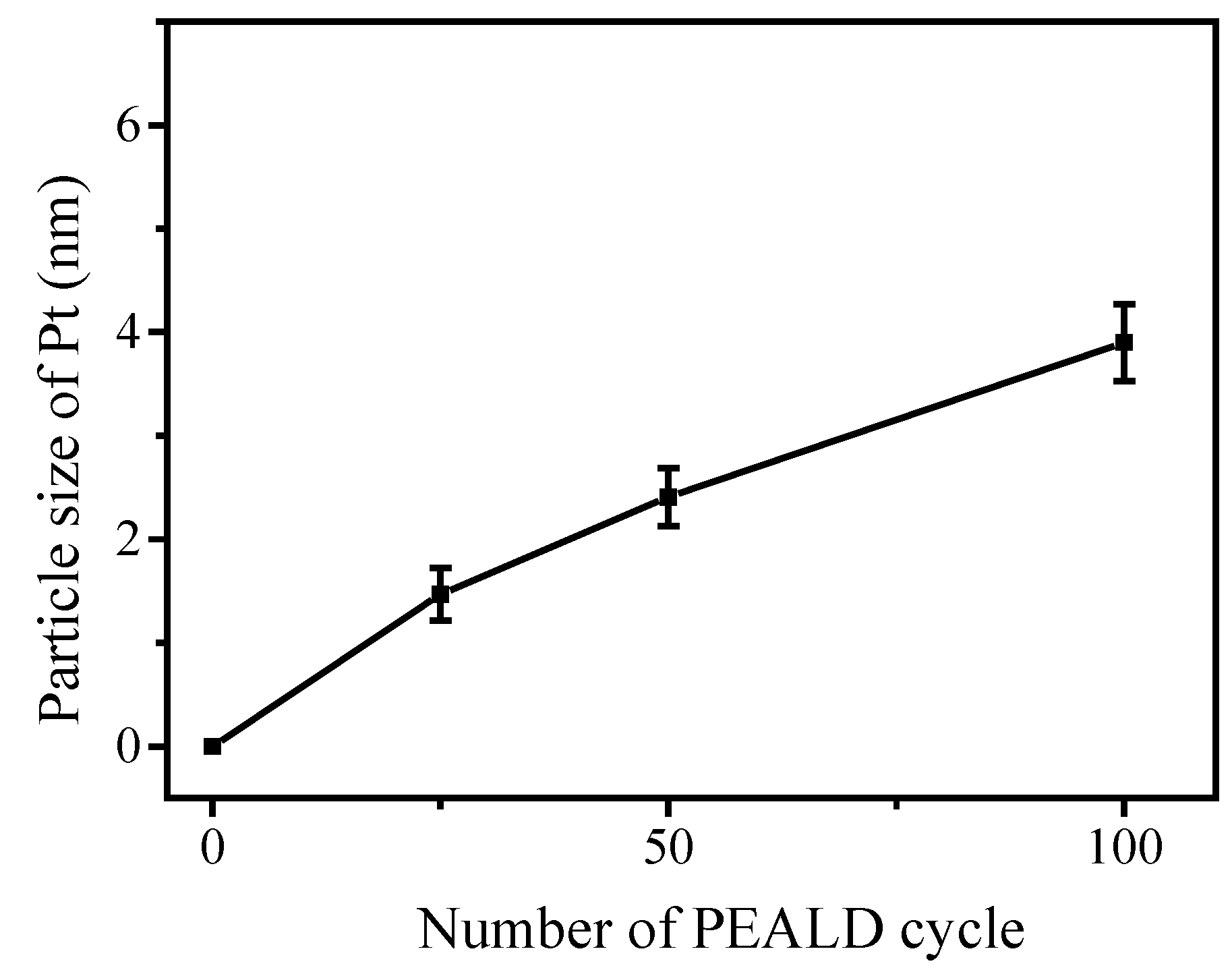
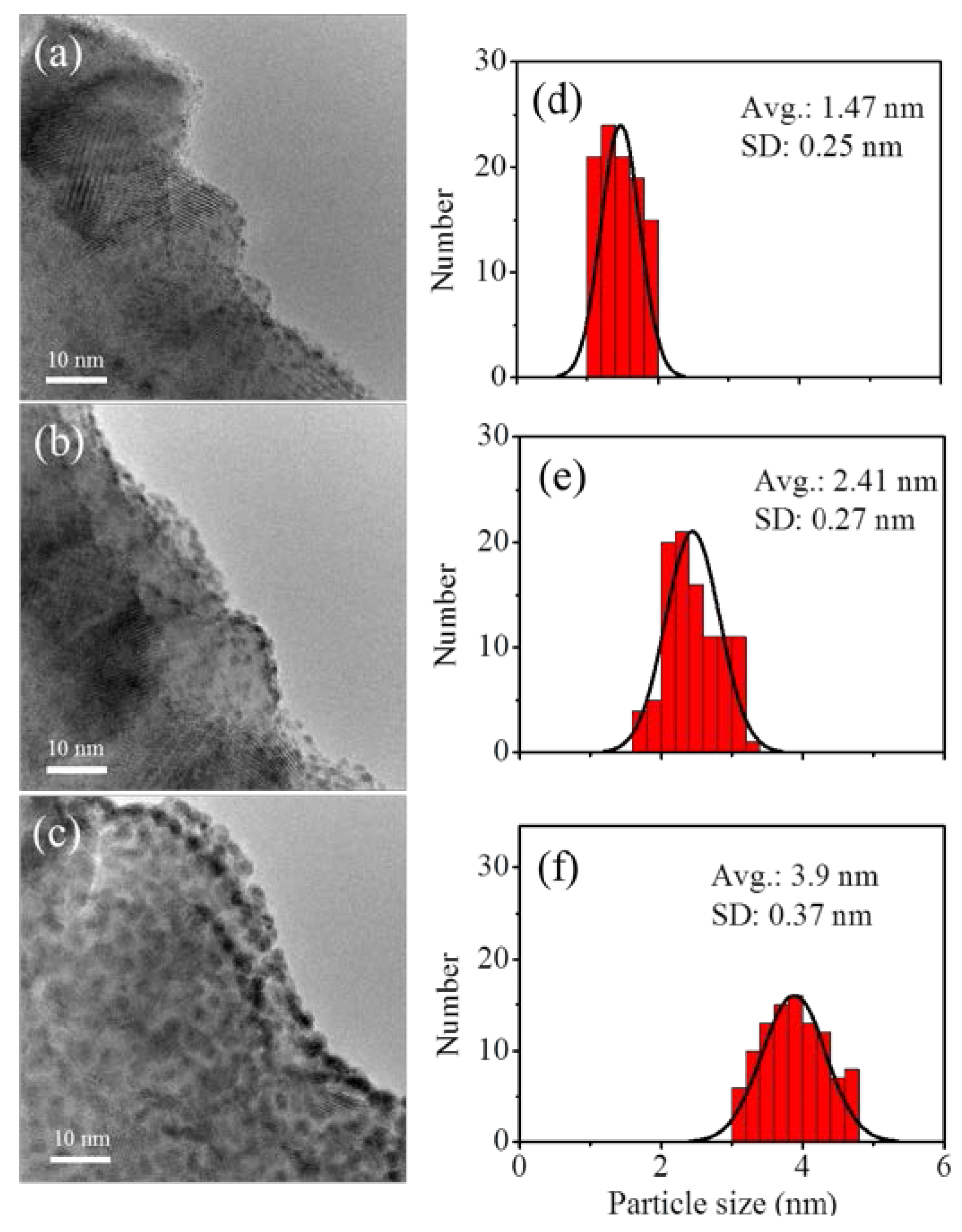
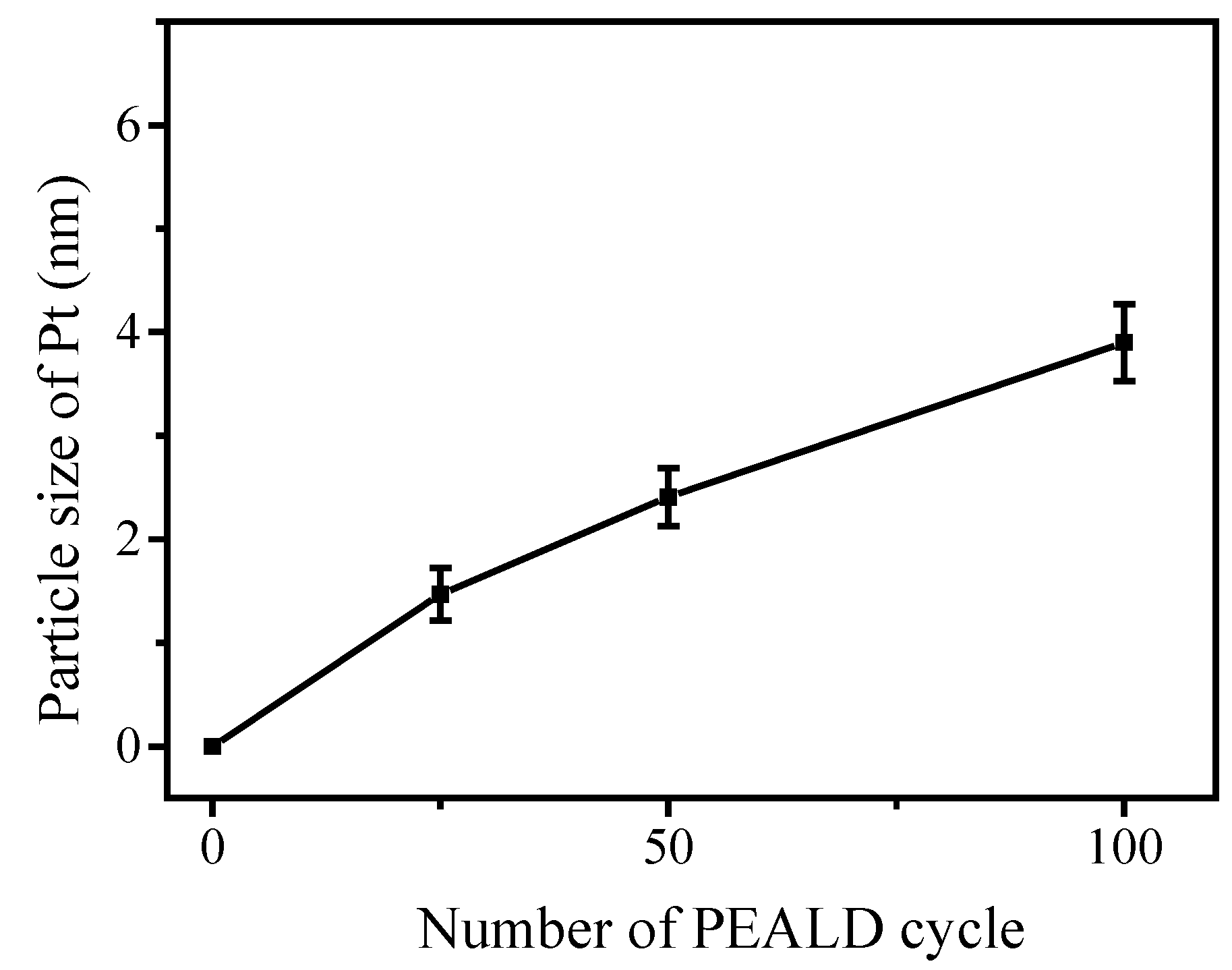
3.3. Fabrication and Characterization of Pt@TiO2@CNTs Structures
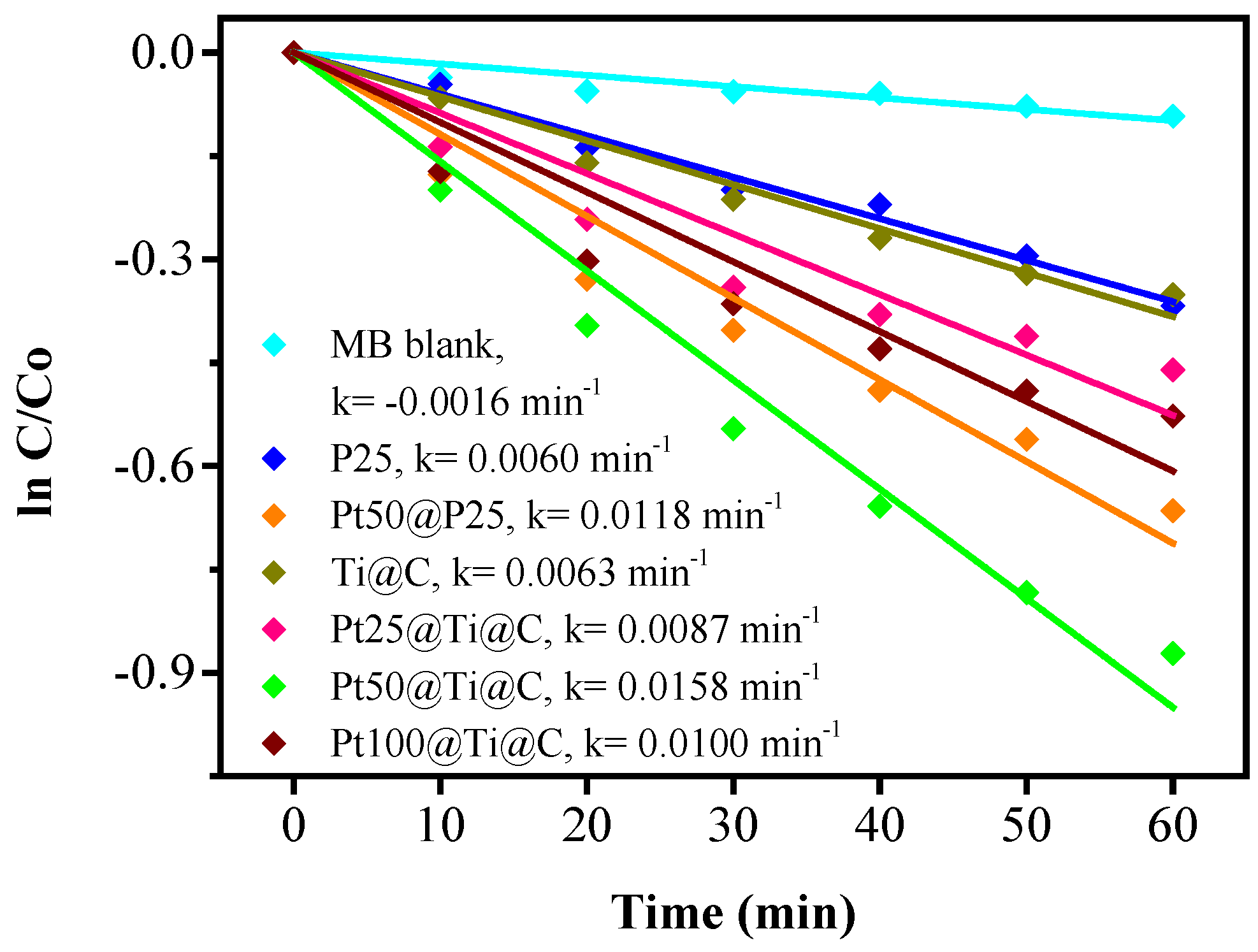
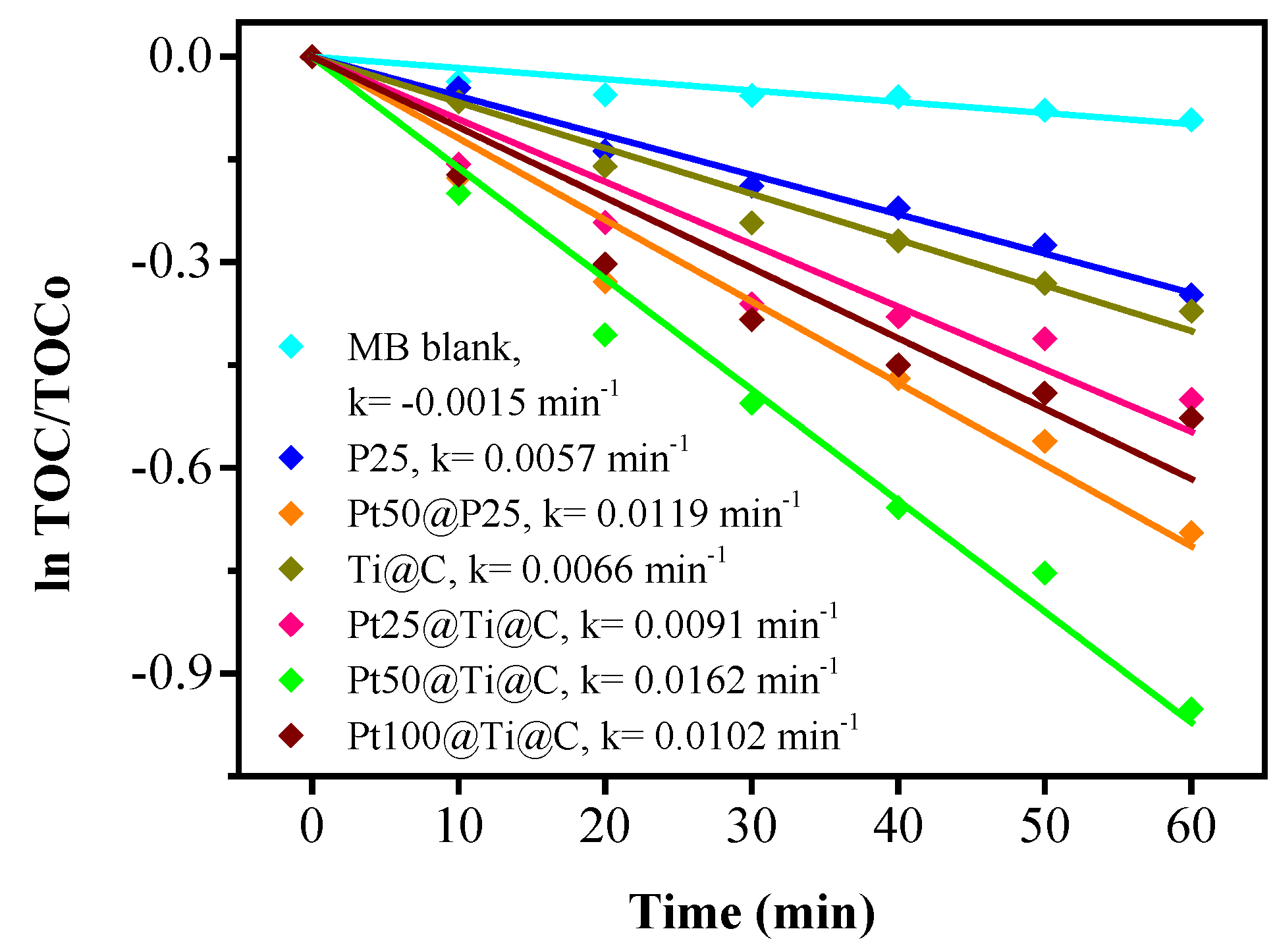
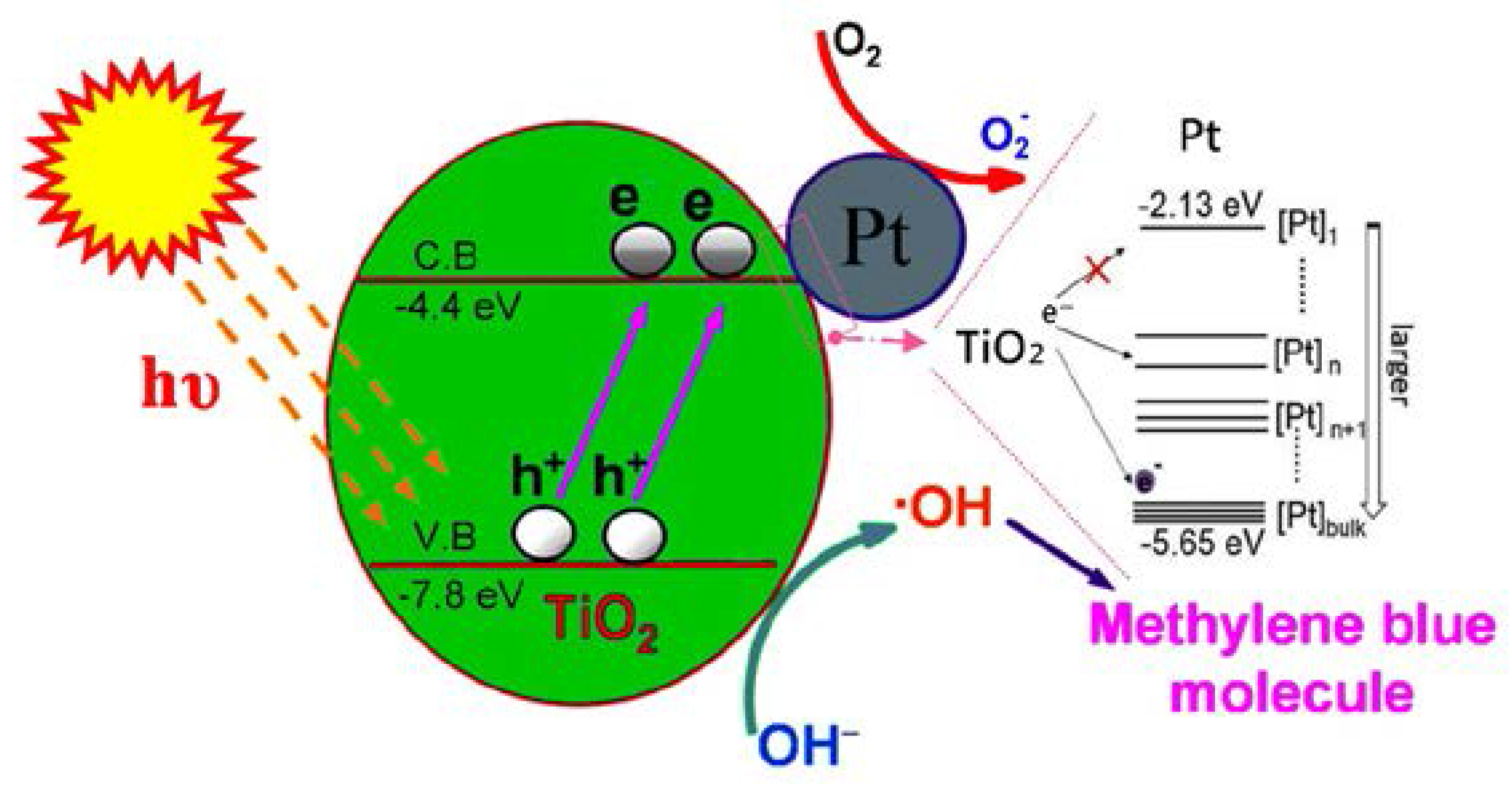
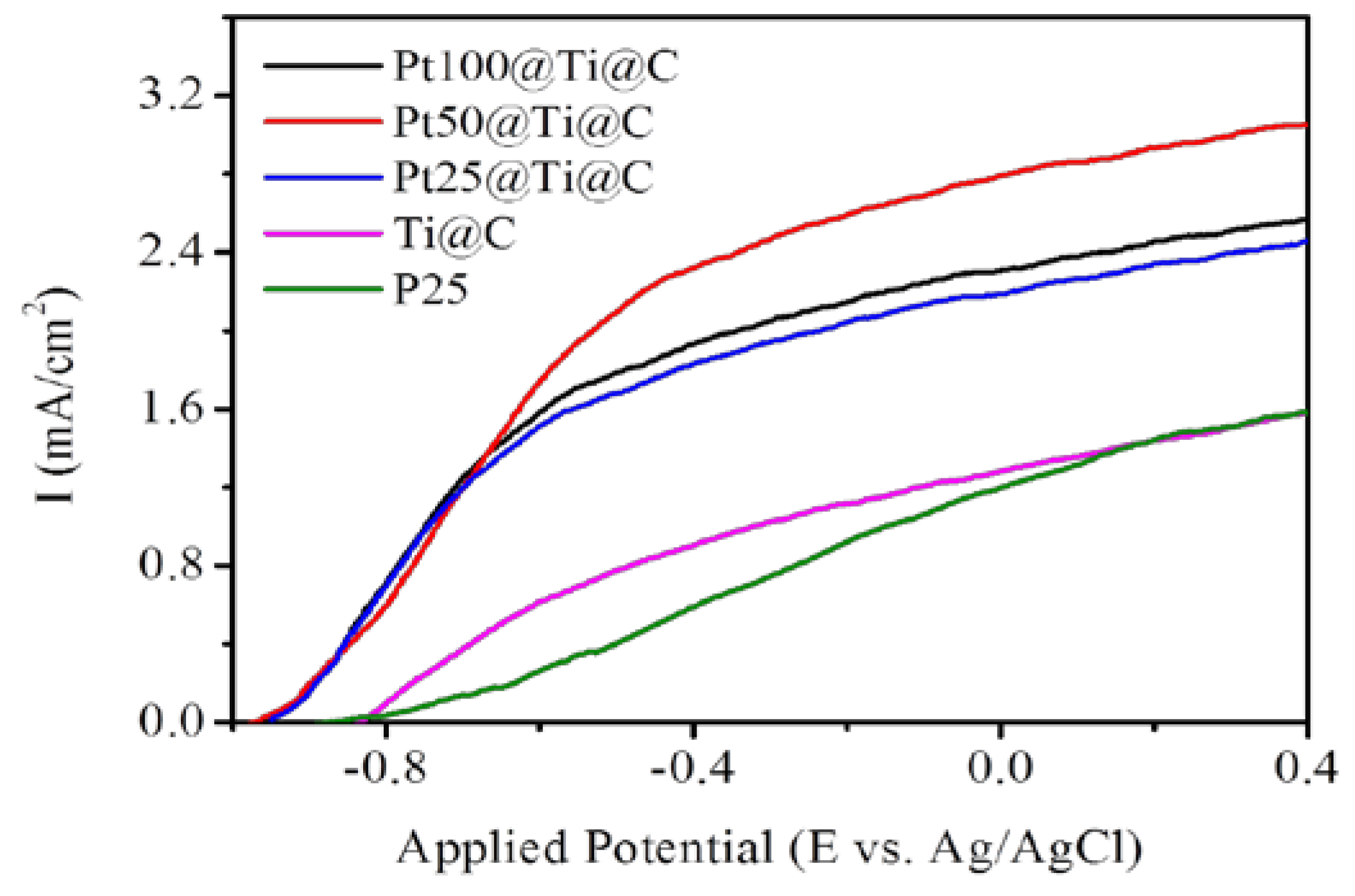
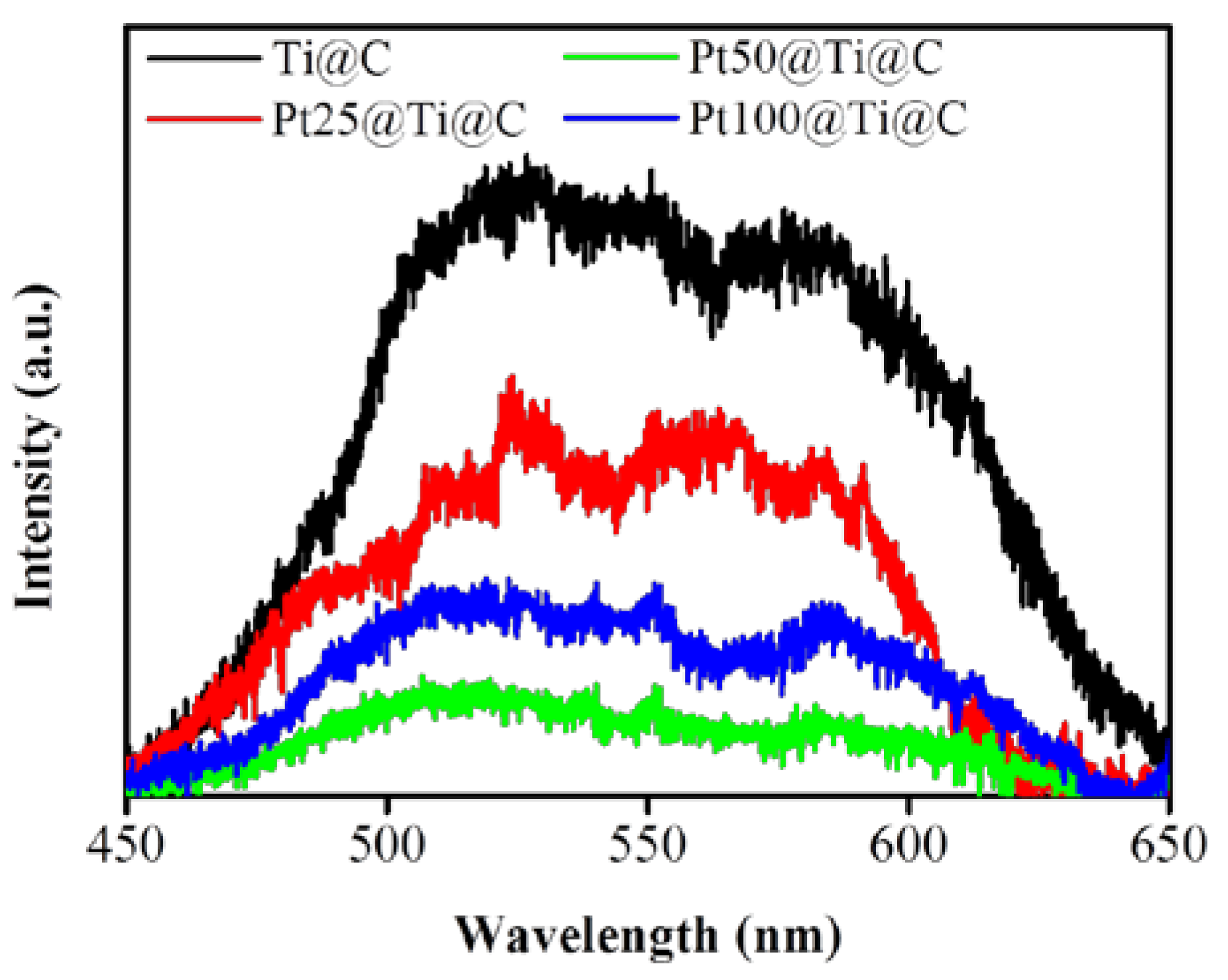
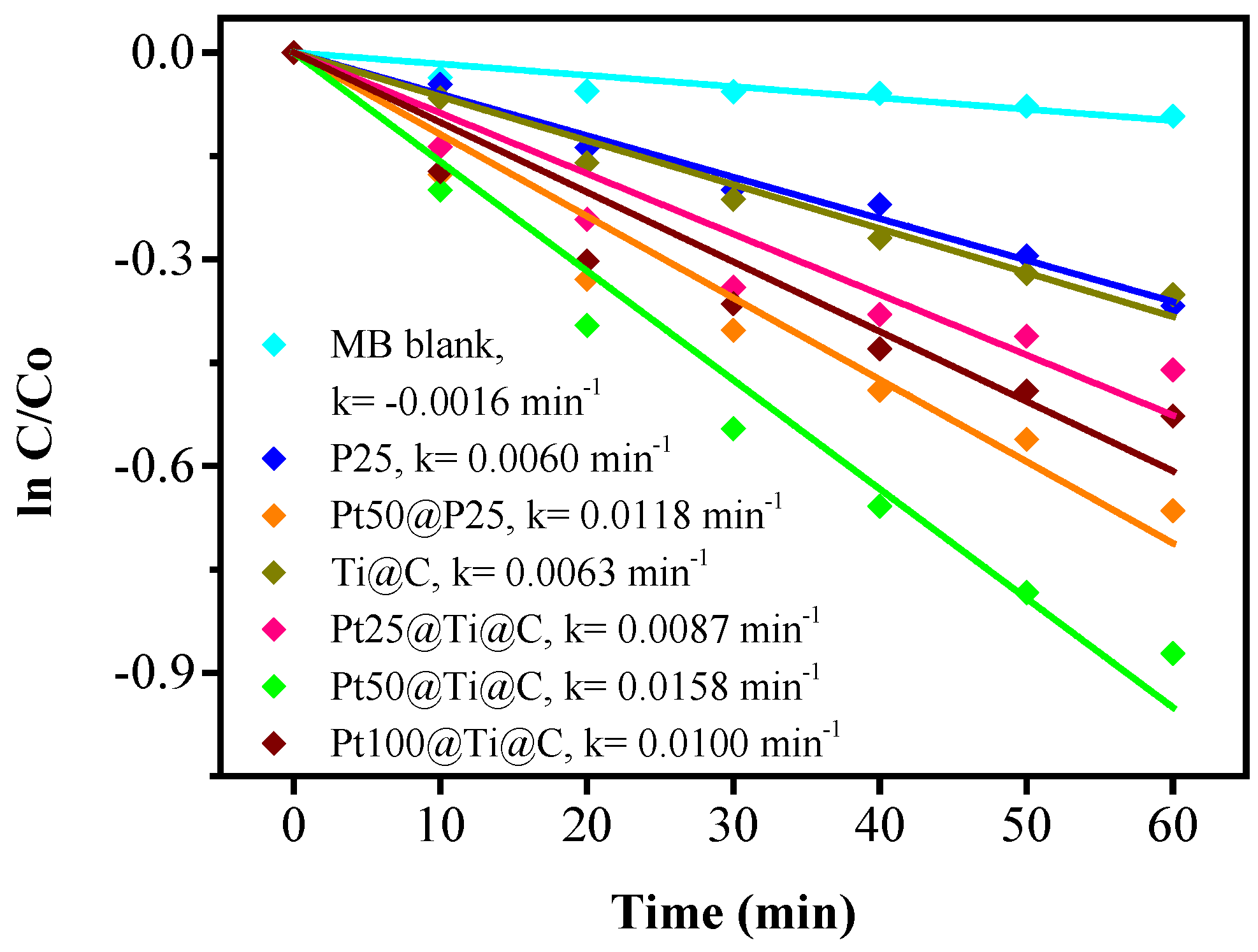
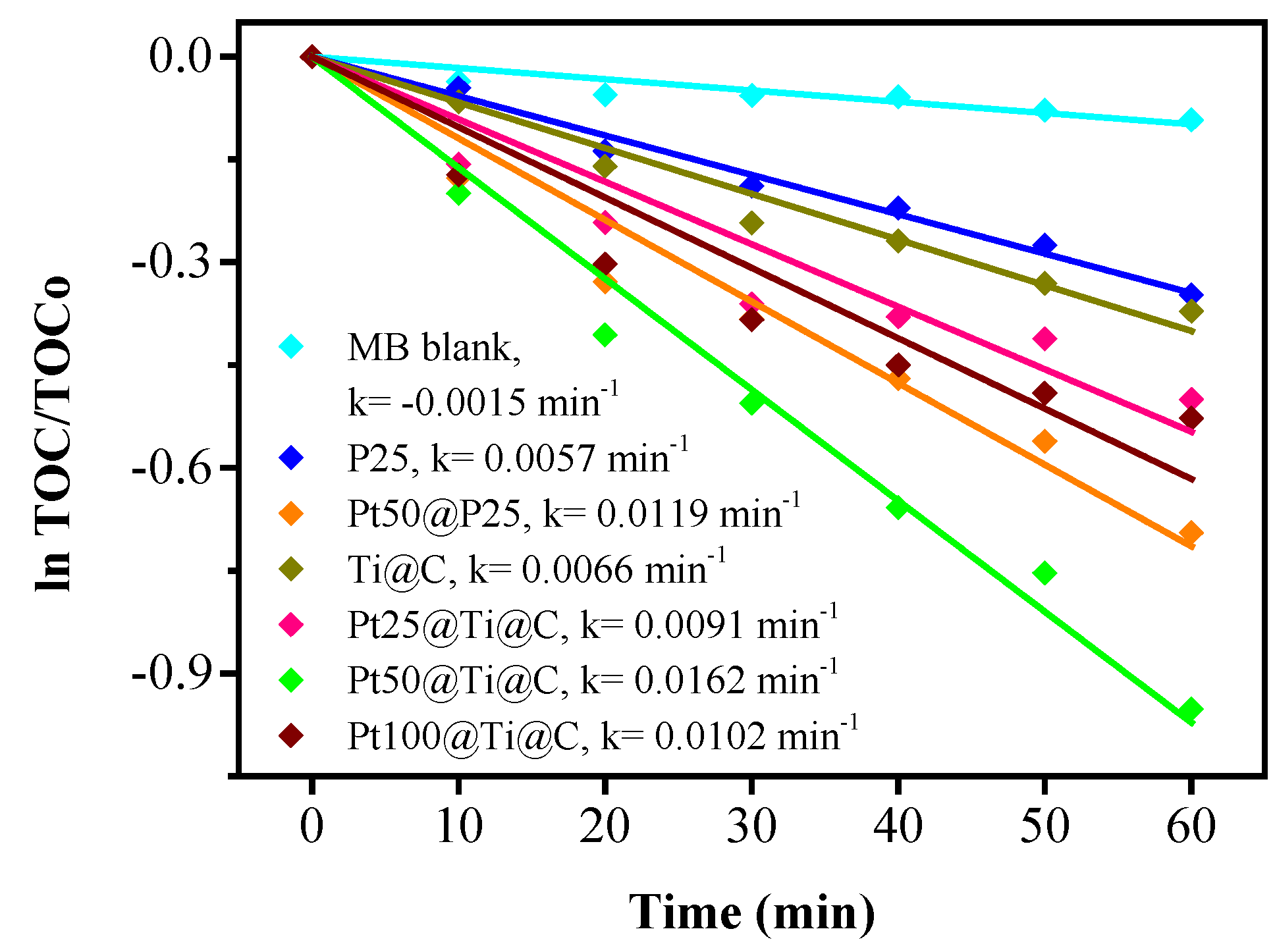
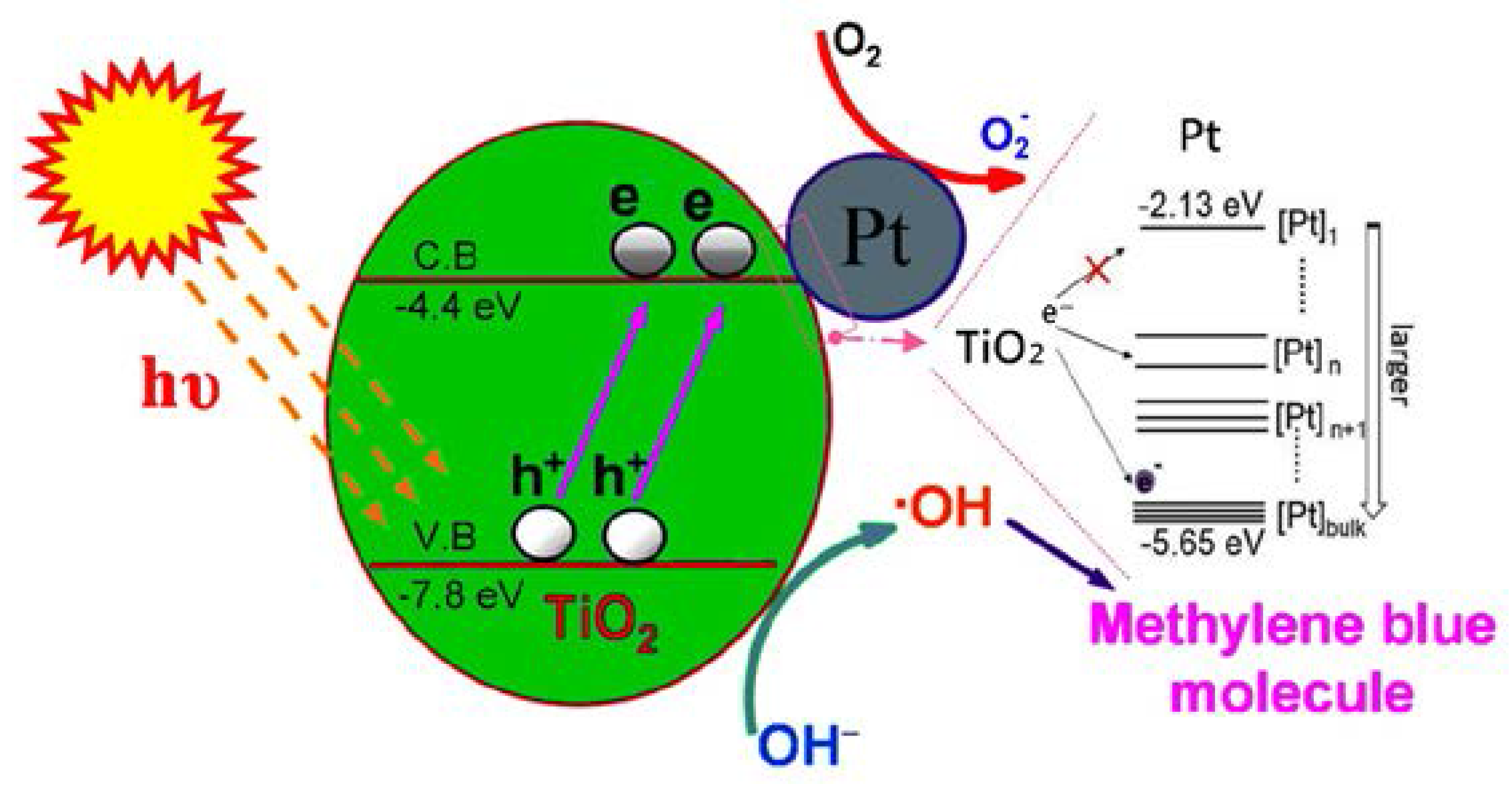
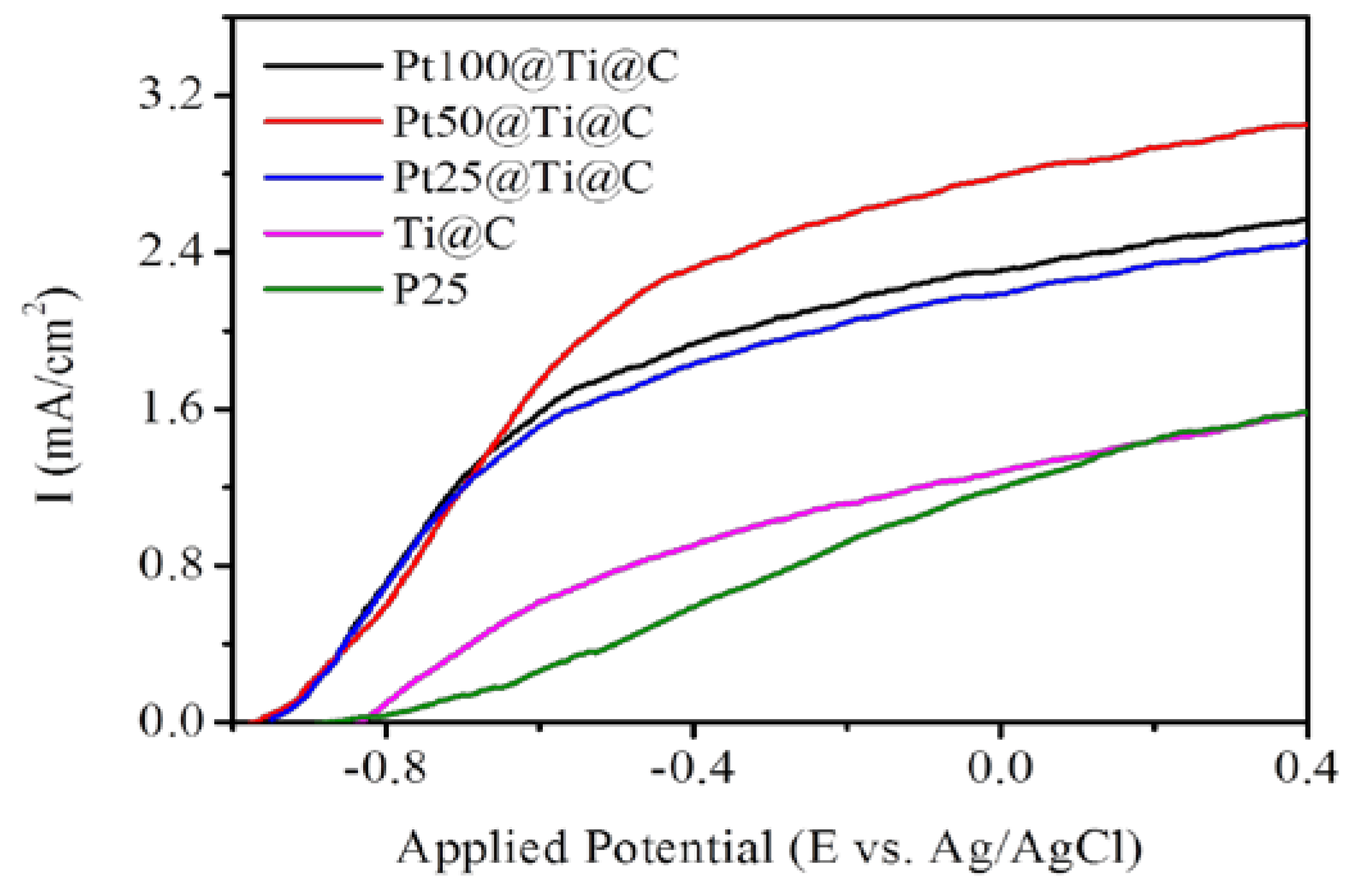
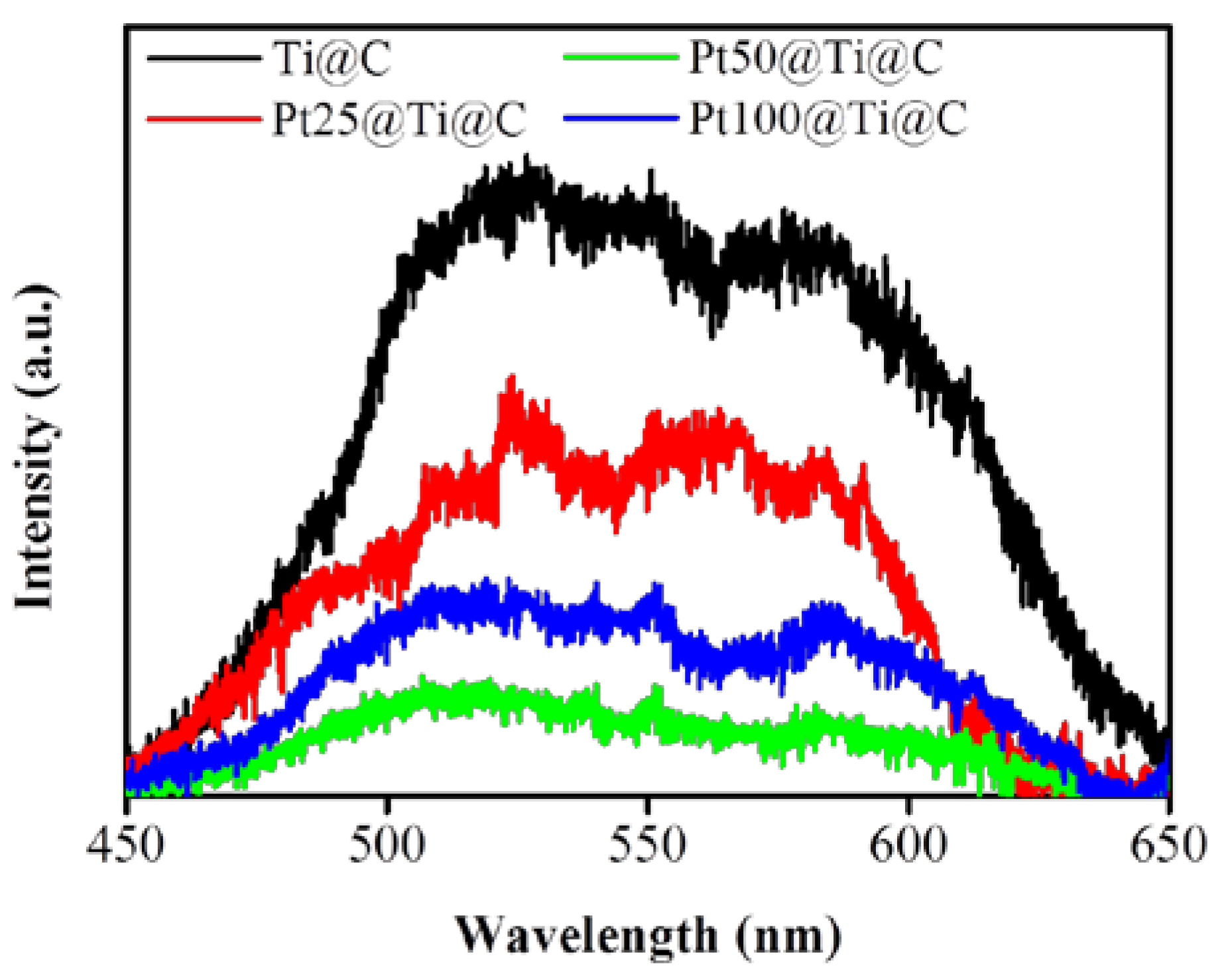
3.4. Photocatalytic Efficiency of Pt@TiO2@CNTs Structure
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Serpone, N.; Lawless, D.; Khairutdinov, R.; Pelizzetti, E. Subnanosecond relaxation dynamics in TiO2 colloidal sols (particle sizes RP = 1.0-13.4 nm). Relevance to heterogeneous photocatalysis. J. Phys. Chem. 1995, 99, 16655–16661. [Google Scholar] [CrossRef]
- Wang, C.C.; Zhang, Z.B.; Ying, J.Y. Photocatalytic decomposition of halogenated organics over nanocrystalline titania. Nanostruct. Mater. 1997, 9, 583–586. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Wang, C.C.; Zakaria, R.; Ying, J.Y. Role of particle size in nanocrystalline TiO2-based photocatalysts. J. Phys. Chem. B 1998, 102, 10871–10878. [Google Scholar] [CrossRef]
- Parra, S.; Stanca, S.E.; Guasaquillo, I.; Thampi, K.R. Photocatalytic degradation of atrazine using suspended and supported TiO2. Appl. Catal. B Environ. 2004, 51, 107–116. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Gao, X.P.; Lan, Y.; Song, D.Y.; Xi, Y.X.; Zhao, J.C. Photocatalytic degradation of atrazine using suspended and supported TiO2. J. Am. Chem. Soc. 2004, 126, 8380–8381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Banfield, J.F. Understanding polymorphic phase transformation behavior during growth of nanocrystalline aggregates: Insights from TiO2. J. Phys. Chem. B 2000, 104, 3481–3487. [Google Scholar] [CrossRef]
- Britto, P.J.; Santhanam, K.S.V.; Rubio, A.; Alonso, J.A.; Ajayan, P.M. Improved charge transfer at carbon nanotube electrodes. Adv. Mater. 1999, 11, 154–157. [Google Scholar] [CrossRef]
- Che, J.W.; Cagin, T.; Goddard, W.A. Thermal conductivity of carbon nanotubes. Nanotechnology 2000, 11, 65–69. [Google Scholar] [CrossRef]
- Collins, P.C.; Arnold, M.S.; Avouris, P. Engineering carbon nanotubes and nanotube circuits using electrical breakdown. Science 2001, 292, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Orlanducci, S.; Sessa, V.; Terranova, M.L.; Battiston, G.A.; Battiston, S.; Gerbasi, R. Nanocrystalline TiO2 on single walled carbon nanotube arrays: Towards the assembly of organized C/TiO2 nanosystems. Carbon 2006, 44, 2839–2843. [Google Scholar] [CrossRef]
- Li, C.; Yuan, J.; Han, B.; Jiang, L.; Shangguan, W. TiO2 nanotubes incorporated with CdS for photocatalytic hydrogen production from splitting water under visible light irradiation. Int. J. Hydrog. Energy 2010, 35, 7073–7079. [Google Scholar] [CrossRef]
- Li, F.B.; Li, X.Z. The enhancement of photodegradation efficiency using Pt-TiO2 catalyst. Chemosphere 2002, 48, 1103–1111. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen-production from a water-ethanol solution: A compasison activities of Au-TiO2 and Pt-TiO2. J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Gomes, H.T.; Machado, B.F.; Silva, A.M.T.; Dražić, G.; Faria, J.L. Photodeposition of Pt nanoparticles on TiO2–carbon xerogel composites. Mater. Lett. 2011, 65, 966–969. [Google Scholar] [CrossRef]
- Chen, M.-L.; Zhang, F.-J.; Oh, W.-C. Preparation and Catalytic Properties of Pt/CNT/TiO2 Composite. J. Korean Ceram. Soc. 2010, 47, 269–275. [Google Scholar] [CrossRef]
- Bedolla-Valdez, Z.I.; Verde-Gómez, Y.; Valenzuela-Muñiz, A.M.; Gochi-Ponce, Y.; Oropeza-Guzmán, M.T.; Berhault, G.; Alonso-Núñez, G. Sonochemical synthesis and characterization of Pt/CNT, Pt/TiO2, and Pt/CNT/TiO2 electrocatalysts for methanol electro-oxidation. Electrochim. Acta 2015, 186, 76–84. [Google Scholar] [CrossRef]
- Marichy, C.; Pinna, N. Carbon-nanostructures coated/decorated by atomic layer deposition: Growth and applications. Coordin. Chem. Rev. 2013, 257, 3232–3253. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.C.; Kei, C.C.; Hsueh, Y.C.; Perng, T.P. Atomic layer deposition of platinum nanoparticles on carbon nanotubes for application in proton-exchange membrane fuel cells. Small 2009, 5, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.C.; Wang, C.C.; Kei, C.C.; Lin, Y.H.; Liu, C.; Perng, T.P. Fabrication of catalyst by atomic layer deposition for high specific power density proton exchange membrane fuel cells. J. Catal. 2012, 294, 63–68. [Google Scholar] [CrossRef]
- Hsueh, Y.C.; Wang, C.C.; Liu, C.; Kei, C.C.; Perng, T.P. Deposition of platinum on oxygen plasma treated carbon nanotubes by atomic layer deposition. Nanotechnology 2012, 23, 405603. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Wang, C.C.; Liao, S.Y.; Gan, J.Y.; Perng, T.P. CNT/TiO2 core-shell structures prepared by atomic layer deposition and characterization of their photocatalytic properties. Thin Solid Films 2015, 616, 151–159. [Google Scholar] [CrossRef]
- Zhang, J.K.; Chen, C.Q.; Chen, S.; Hu, Q.M.; Gao, Z.; Li, Y.Q.; Qin, Y. Highly dispersed Pt nanoparticles supported on carbon nanotubes produced by atomic layer deposition for hydrogen generation from hydrolysis of ammonia borane. Catal. Sci. Technol. 2017, 7, 322–329. [Google Scholar] [CrossRef]
- Lin, C.J.; Yu, Y.H.; Liou, Y.H. Free-standing TiO2 nanotube array films sensitized with CdS as highly active solar light-driven photocatalysts. Appl. Catal. B Environ. 2009, 93, 119–125. [Google Scholar] [CrossRef]
- El-Hendawy, A.N.A. Influence of HNO3 oxidation on the structure and adsorptive properties of corncob-based activated carbon. Carbon 2003, 41, 713–722. [Google Scholar] [CrossRef]
- Pradhan, B.K.; Sandle, N.K. Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 1999, 37, 1323–1332. [Google Scholar] [CrossRef]
- Aarik, J.; Aidla, A.; Mandar, H.; Uustare, T. Atomic layer deposition of titanium dioxide from TiCl4 and H2O: Investigation of growth mechanism. Appl. Surf. Sci. 2001, 172, 148–158. [Google Scholar] [CrossRef]
- Puurunen, R.L. Surface chemistry of atomic layer deposition: A case study for the trimethylaluminum/water process. J. Appl. Phys. 2005, 97, 121301. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Guerra-Nunez, C.; Utke, I.; Michler, J.; Rossell, M.D.; Erni, R. Morphology and crystallinity control of ultrathin TiO2 layers deposited on carbon nanotubes by temperature-step atomic layer deposition. J. Phys. Chem. C 2015, 119, 3379–3387. [Google Scholar] [CrossRef]
- Kočí, K.; Obalová, L.; Matějová, L.; Plachá, D.; Lacný, Z.; Jirkovský, J.; Šolcová, O. Effect of TiO2 particle size on the photocatalytic reduction of CO2. Appl. Catal. B Environ. 2009, 89, 494–502. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Barka, N.; Qourzal, S.; Assabbane, A.; Nounah, A.; Ait-Ichou, Y. Factors influencing the photocatalytic degradation of Rhodamine B by TiO2-coated non-woven paper. J. Photochem. Photobiol. A 2008, 195, 346–351. [Google Scholar] [CrossRef]
- Wang, C.C.; Hsueh, Y.C.; Su, C.Y.; Kei, C.C.; Perng, T.P. Deposition of uniform Pt nanoparticles with controllable size on TiO2-based nanowires by atomic layer deposition and their photocatalytic properties. Nanotechnology 2015, 26, 254002. [Google Scholar] [CrossRef] [PubMed]
- Rosseler, O.; Shankar, M.V.; Du, M.K.-L.; Schmidlin, L.; Keller, N.; Keller, V. Solar light photocatalytic hydrogen production from water over Pt and Au/TiO2 (anatase/rutile) photocatalysts: Influence of noble metal and porogen promotion. J. Catal. 2010, 269, 179–190. [Google Scholar] [CrossRef]
- Kiwi, J.; Gratzel, M. Optimization of conditions for photochemical water cleavage-aqueous Pt/TiO2 (anatase) dispersions under ultraviolet-light. J. Phys. Chem. 1984, 88, 1302–1307. [Google Scholar] [CrossRef]
- Sadeghi, M.; Liu, W.; Zhang, T.G.; Stavropoulos, P.; Levy, B. Role of photoinduced charge carrier separation distance in heterogeneous photocatalysis: Oxidative degradation of CH3OH vapor in contact with Pt/TiO2 and cofumed TiO2-Fe2O3. J. Phys. Chem. 1996, 100, 19466–19474. [Google Scholar] [CrossRef]
- Wang, W.N.; An, W.J.; Ramalingam, B.; Mukherjee, S.; Niedzwiedzki, D.M.; Gangopadhyay, S.; Biswas, P. Size and structure matter: enhanced CO2 photoreduction efficiency by size-resolved ultrafine Pt nanoparticles on TiO2 single crystals. J. Am. Chem. Soc. 2012, 134, 11276–11281. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Kei, C.C.; Perng, T.P. Fabrication of TiO2 nanotubes by atomic layer deposition and their photocatalytic and photoelectrochemical applications. Nanotechnology 2011, 22, 365702. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Li, F.B. Study of Au/Au3+-TiO2 photocatalysts toward visible photooxidation for water and wastewater treatment. Environ. Sci. Technol. 2001, 35, 2381–2387. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Li, F.B.; Yang, C.L.; Ge, W.K. Photocatalytic activity of WOx-TiO2 under visible light irradiation. J. Photochem. Photobiol. A 2001, 141, 209–217. [Google Scholar] [CrossRef]
- Yu, J.C.; Yu, J.G.; Ho, W.K.; Jiang, Z.T.; Zhang, L.Z. Effects of F-doping on the photocatalytic activity and microstructures of nanocrystalline TiO2 powders. Chem. Mater. 2002, 14, 3808–3816. [Google Scholar] [CrossRef]
- Isaifan, R.J.; Ntais, S.; Couillard, M.; Baranova, E.A. Size-dependent activity of Pt/yttria-stabilized zirconia catalyst for ethylene and carbon monoxide oxidation in oxygen-free gas environment. J. Catal. 2015, 324, 32–40. [Google Scholar] [CrossRef]
- Ciftci, A.; Ligthart, D.; Hensen, E.J.M. Influence of Pt particle size and Re addition by catalytic reduction on aqueous phase reforming of glycerol for carbon-supported Pt(Re) catalysts. Appl. Catal. B Environ. 2015, 174, 126–135. [Google Scholar] [CrossRef]
- Feng, X.J.; Sloppy, J.D.; LaTemp, T.J.; Paulose, M.; Komarneni, S.; Bao, N.Z.; Grimes, C.A. Synthesis and deposition of ultrafine Pt nanoparticles within high aspect ratio TiO2 nanotube arrays: Application to the photocatalytic reduction of carbon dioxide. J. Mater. Chem. 2011, 21, 13429–13433. [Google Scholar] [CrossRef]
- Yoo, J.; Altomare, M.; Mokhtar, M.; Alshehri, A.; Al-Thabaiti, S.A.; Mazare, A.; Schmuki, P. Photocatalytic H2 generation using dewetted Pt-decorated TiO2 nanotubes: Optimized dewetting and oxide crystallization by a multiple annealing process. J. Phy. Chem. C 2016, 120, 15884–15892. [Google Scholar] [CrossRef]
- Kim, G.J.; Kwon, D.W.; Hong, S.C. Effect of Pt particle size and valence state on the performance of Pt/TiO2 catalysts for CO oxidation at room temperature. J. Phy. Chem. C 2016, 120, 17996–18004. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Size Distribution (nm) | Optimized Size (nm) | Preparation Method a | Test Reagent | Platinum Precursor | Ref. |
|---|---|---|---|---|---|
| 1.9–6.7 | 1.9 | Hydrothermal | CO, C2H4 | PtCl4 | [42] |
| 1.2–4.2 | 2 | Impregnation | Glycerol | H2PtCl6·6H2O | [43] |
| 1.8–4.9 | 1.75 | MW-solvothermal | CO2 | H2PtCl6 | [44] |
| 1–15 | 2 | sputter | EtOH/H2O | Pt target | [45] |
| 2.2–16.7 | 2.2 | H2 reduction | CO2 | N.A | [46] |
| 1.47–3.9 | 2.41 | PEALD | MB | MeCpPtMe3 | This study |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, S.-Y.; Yang, Y.-C.; Huang, S.-H.; Gan, J.-Y. Synthesis of Pt@TiO2@CNTs Hierarchical Structure Catalyst by Atomic Layer Deposition and Their Photocatalytic and Photoelectrochemical Activity. Nanomaterials 2017, 7, 97. https://doi.org/10.3390/nano7050097
Liao S-Y, Yang Y-C, Huang S-H, Gan J-Y. Synthesis of Pt@TiO2@CNTs Hierarchical Structure Catalyst by Atomic Layer Deposition and Their Photocatalytic and Photoelectrochemical Activity. Nanomaterials. 2017; 7(5):97. https://doi.org/10.3390/nano7050097
Chicago/Turabian StyleLiao, Shih-Yun, Ya-Chu Yang, Sheng-Hsin Huang, and Jon-Yiew Gan. 2017. "Synthesis of Pt@TiO2@CNTs Hierarchical Structure Catalyst by Atomic Layer Deposition and Their Photocatalytic and Photoelectrochemical Activity" Nanomaterials 7, no. 5: 97. https://doi.org/10.3390/nano7050097