
Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
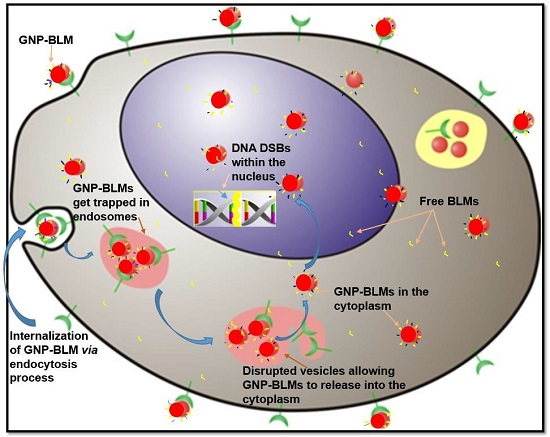
1. Introduction
- 1
- Will the efficacy of the drug be compromised by conjugating them onto the GNP surface?
- 2
- The action of the drug is through binding onto DNA. Does this mean NPs can also reach the nucleus?
- 3
- Will drug conjugated GNPs (GNP-BLM) be more effective than the BLM alone (free drug)?
2. Results
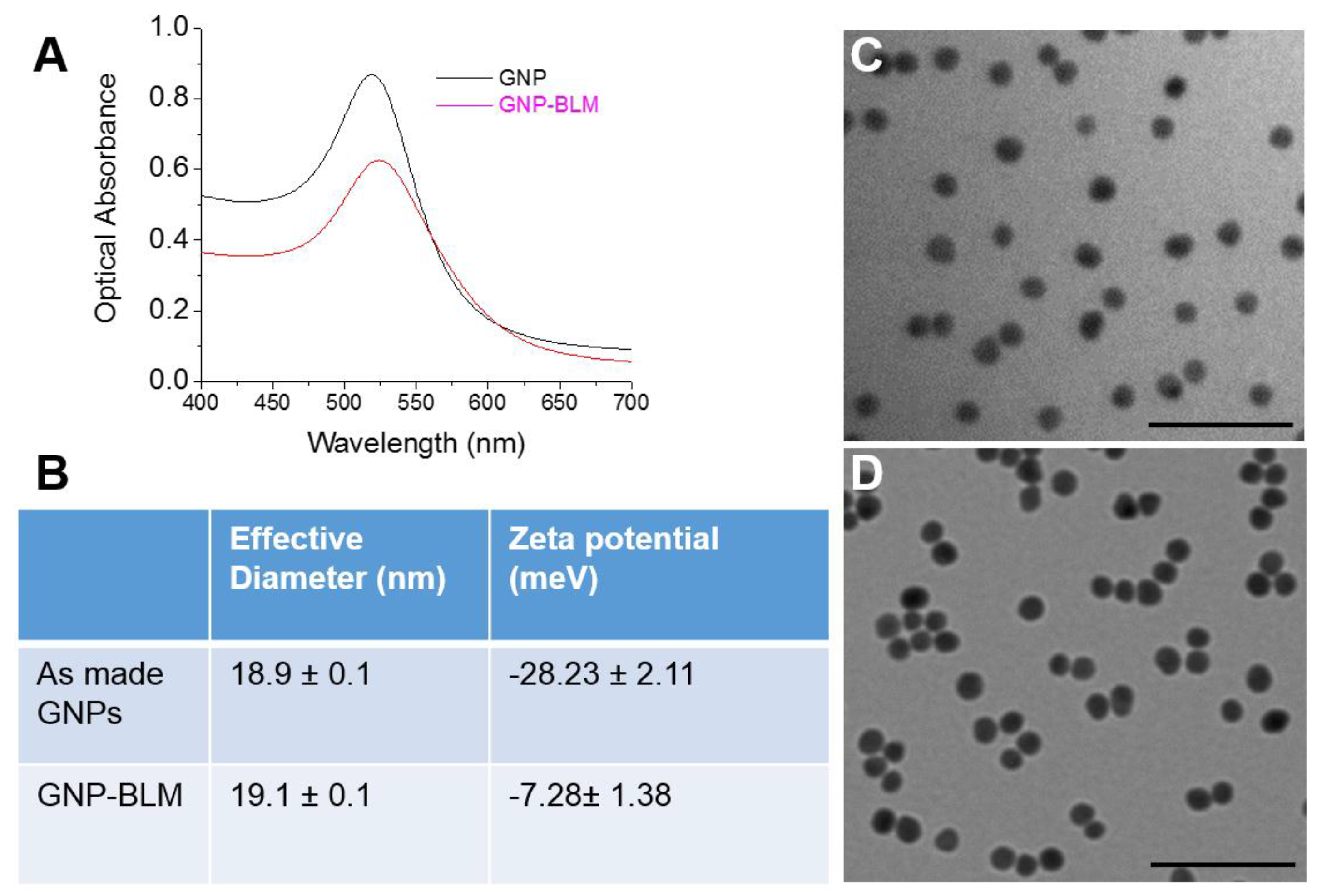
2.1. Characterization of Nanoparticles
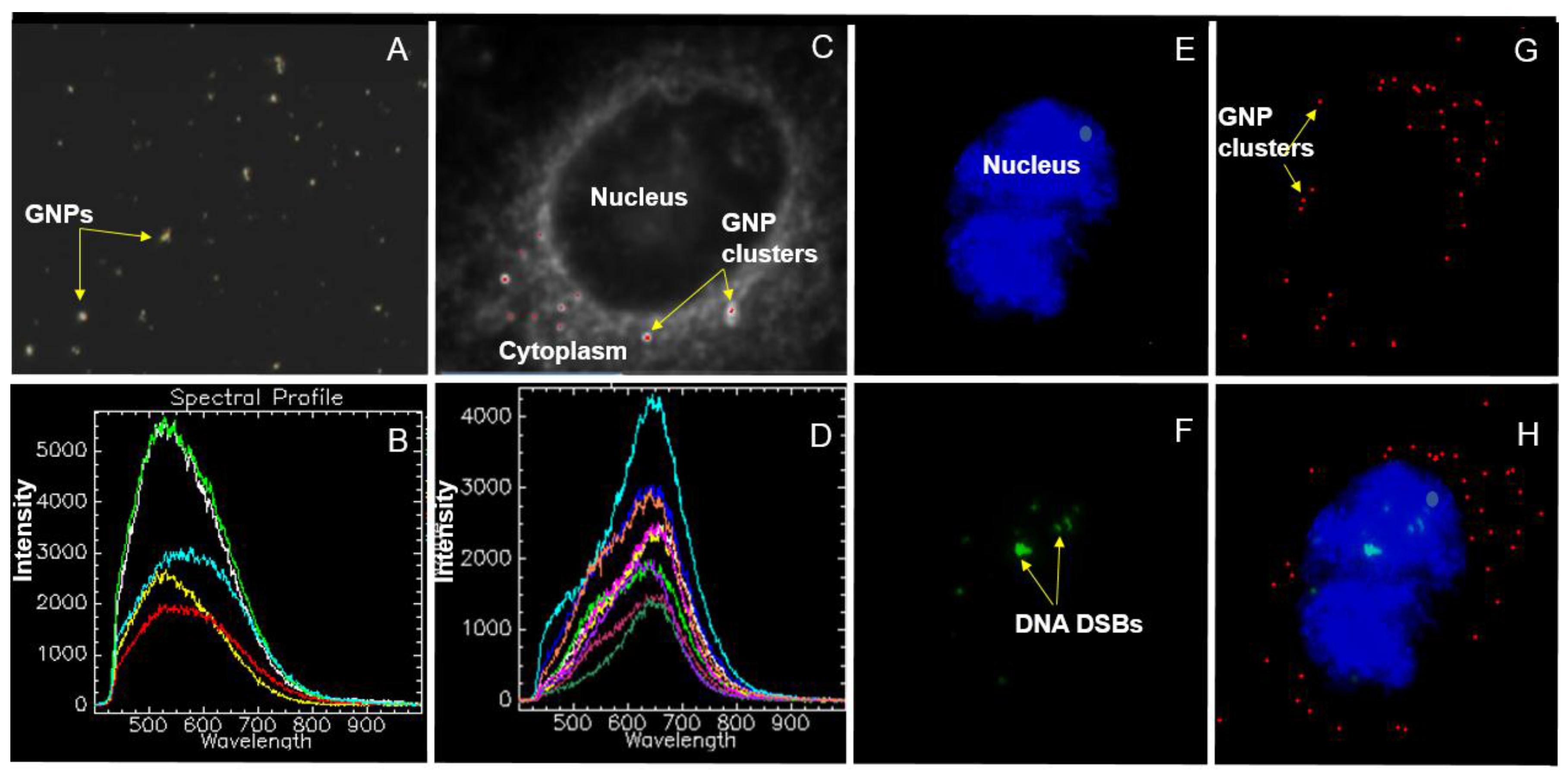
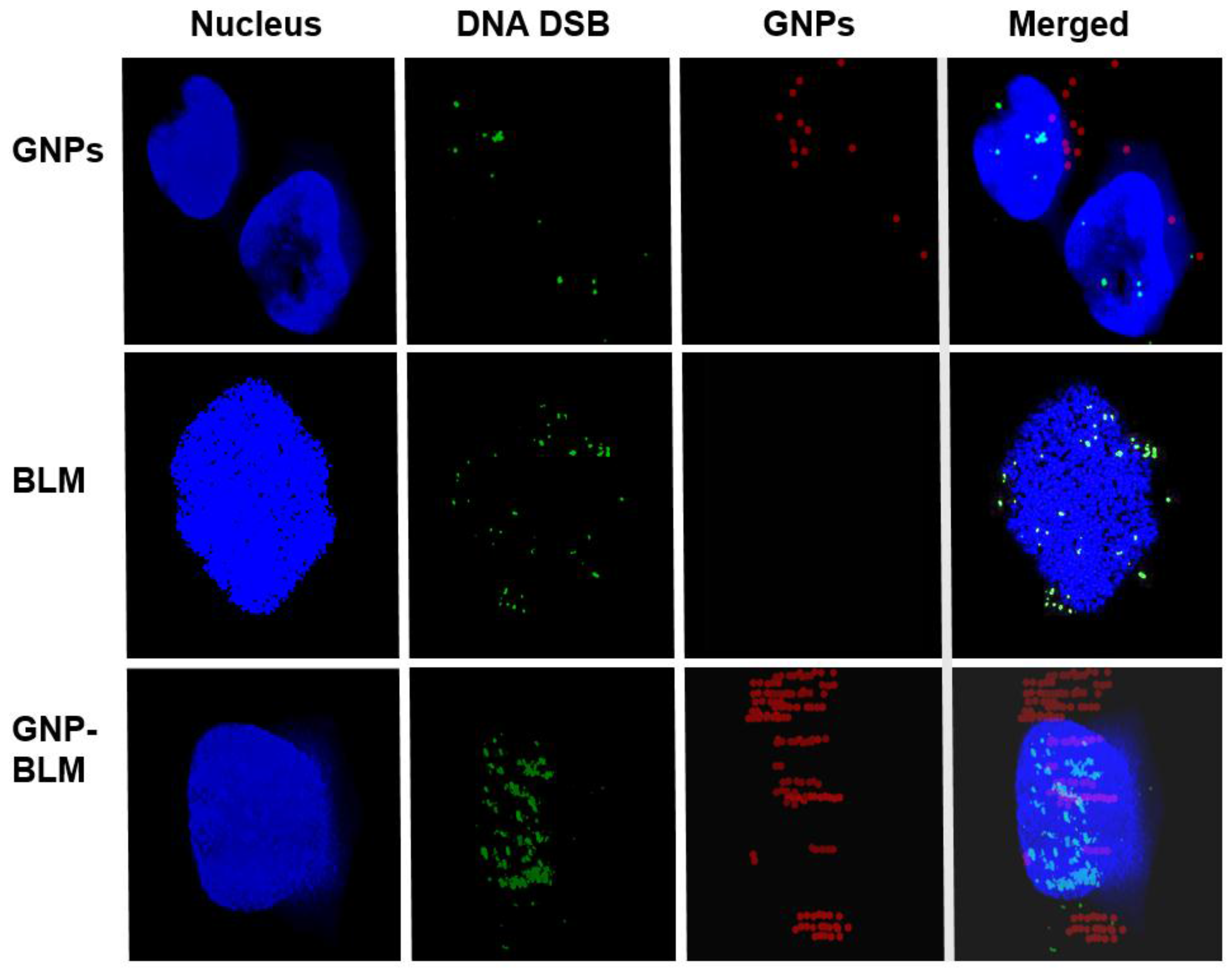
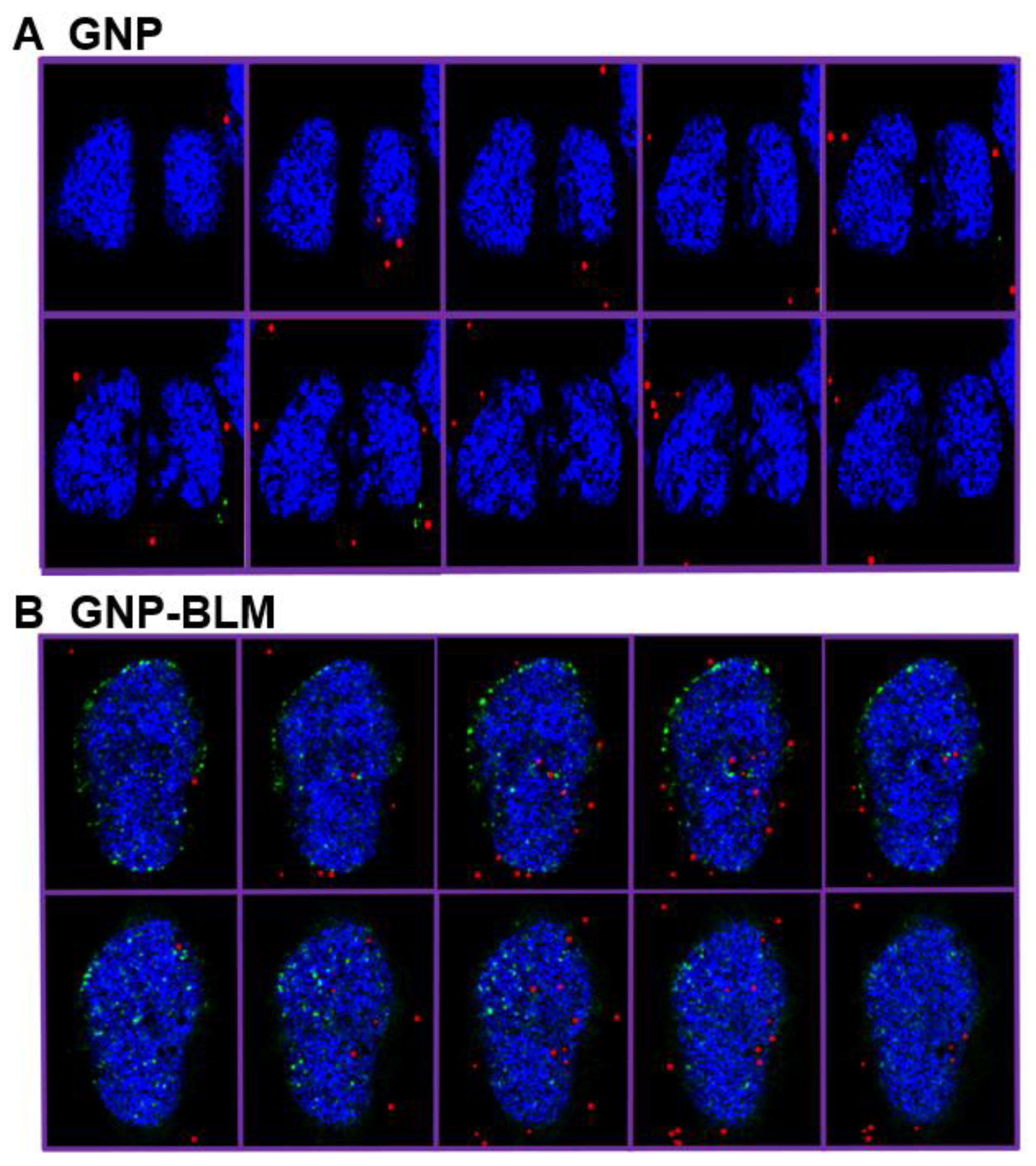
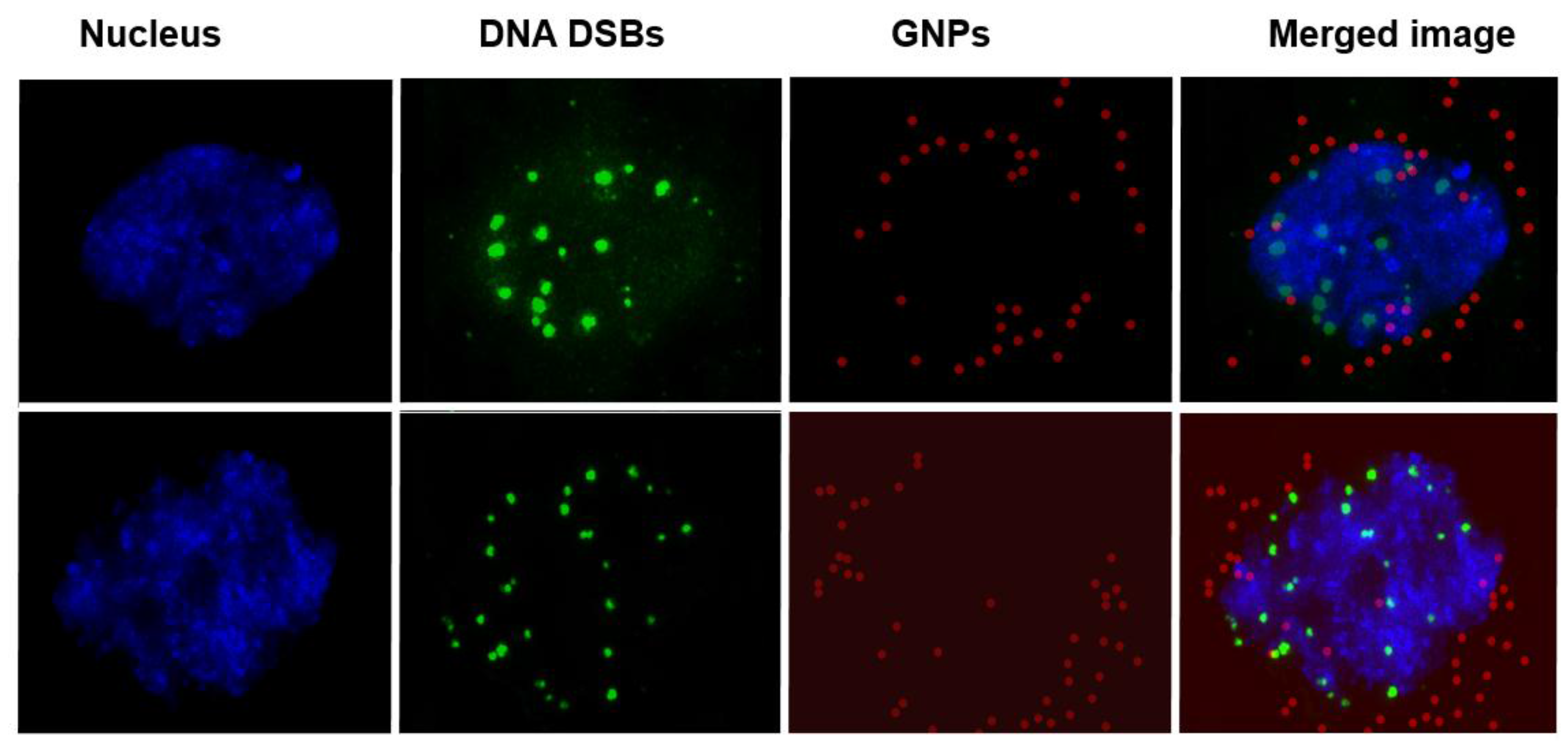
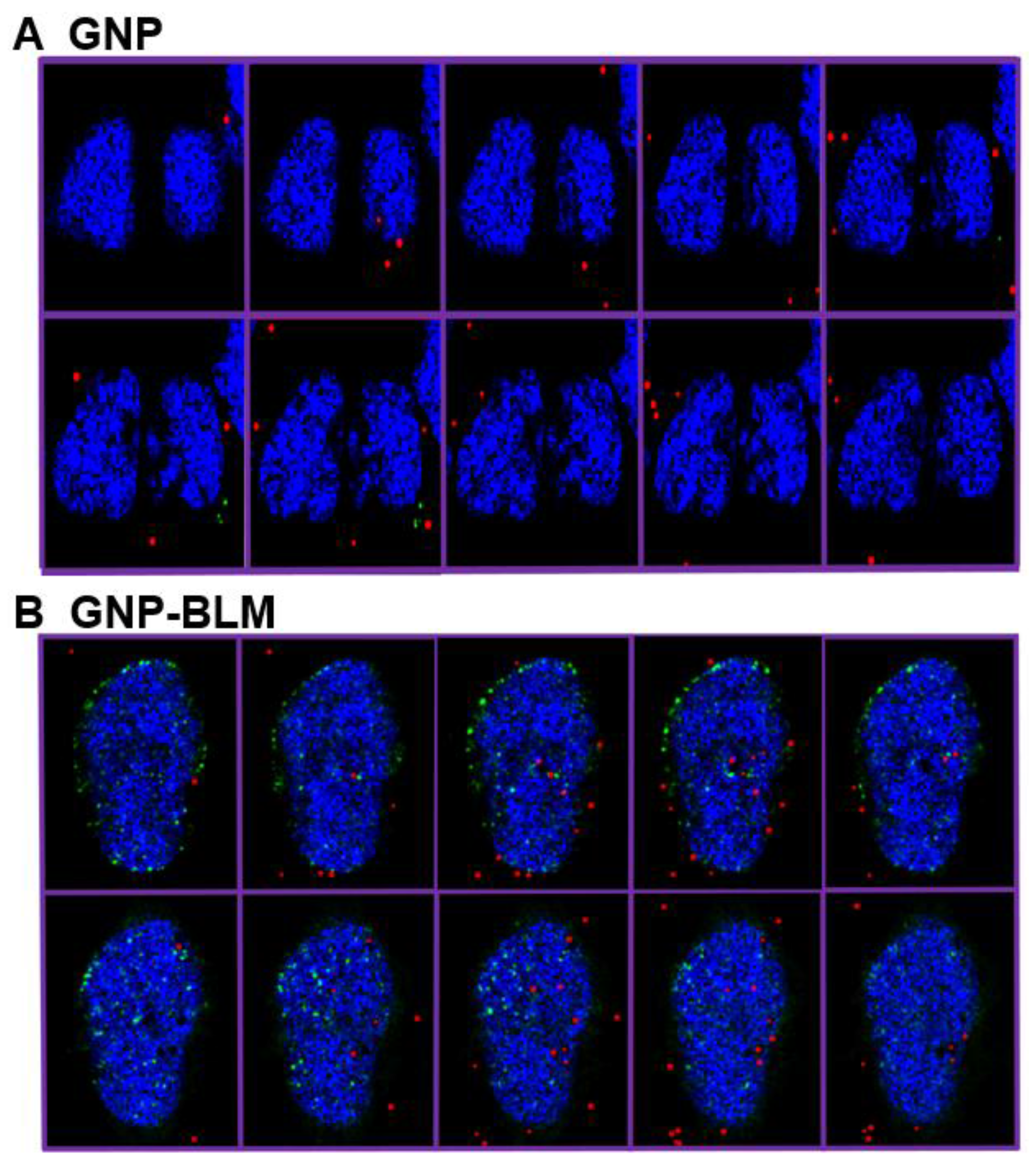
2.2. Mapping of NP Distribution Using Hyperspectral Imaging Technique
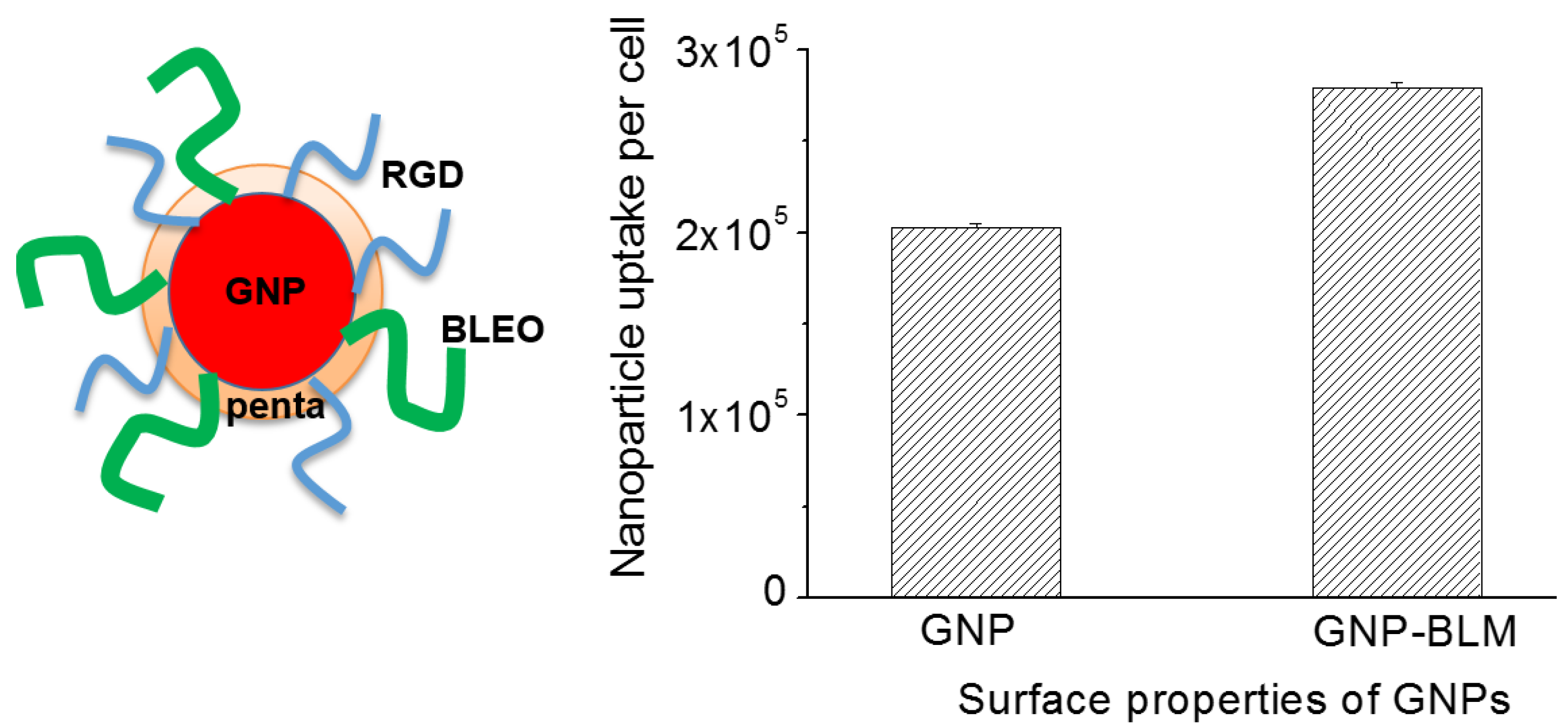
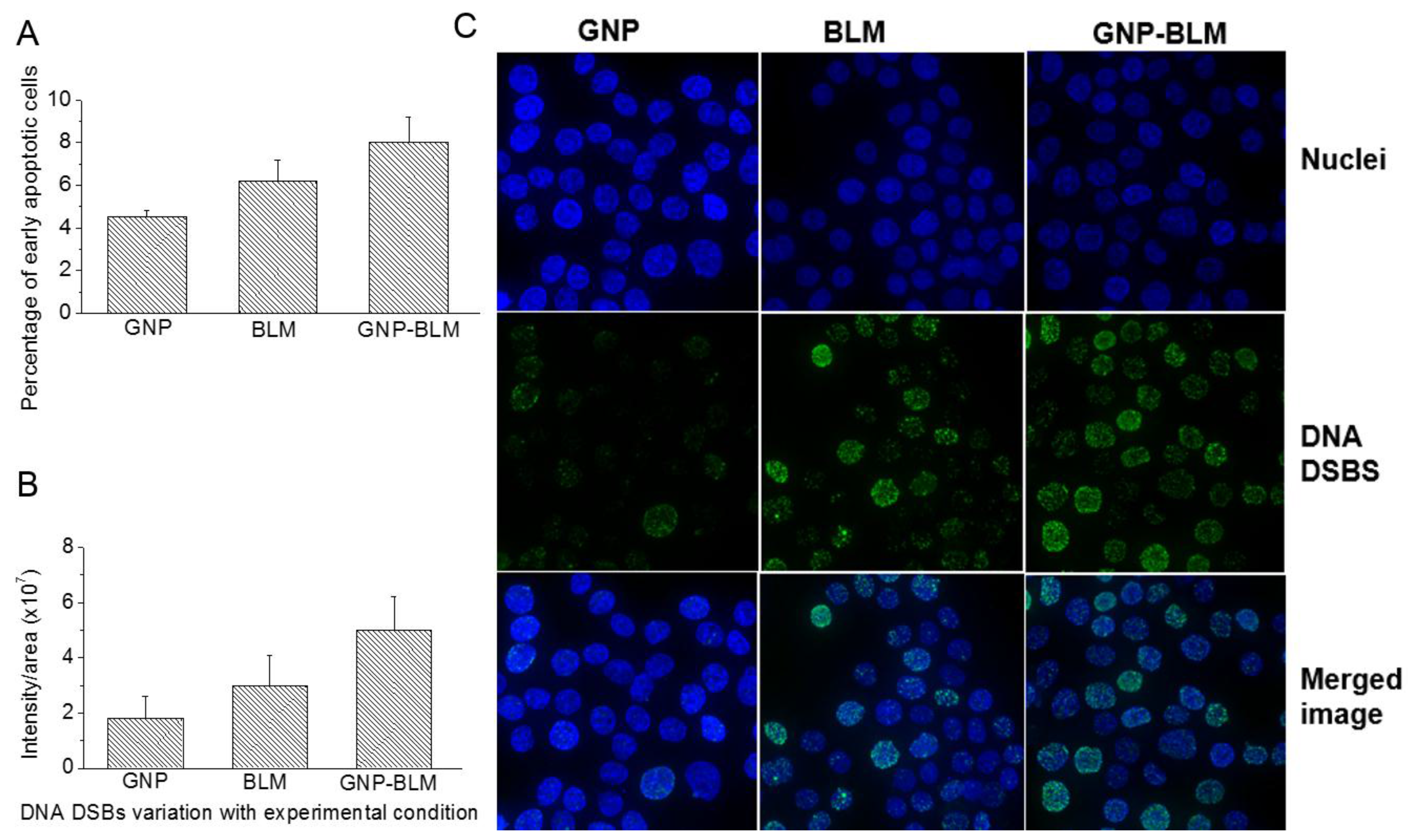
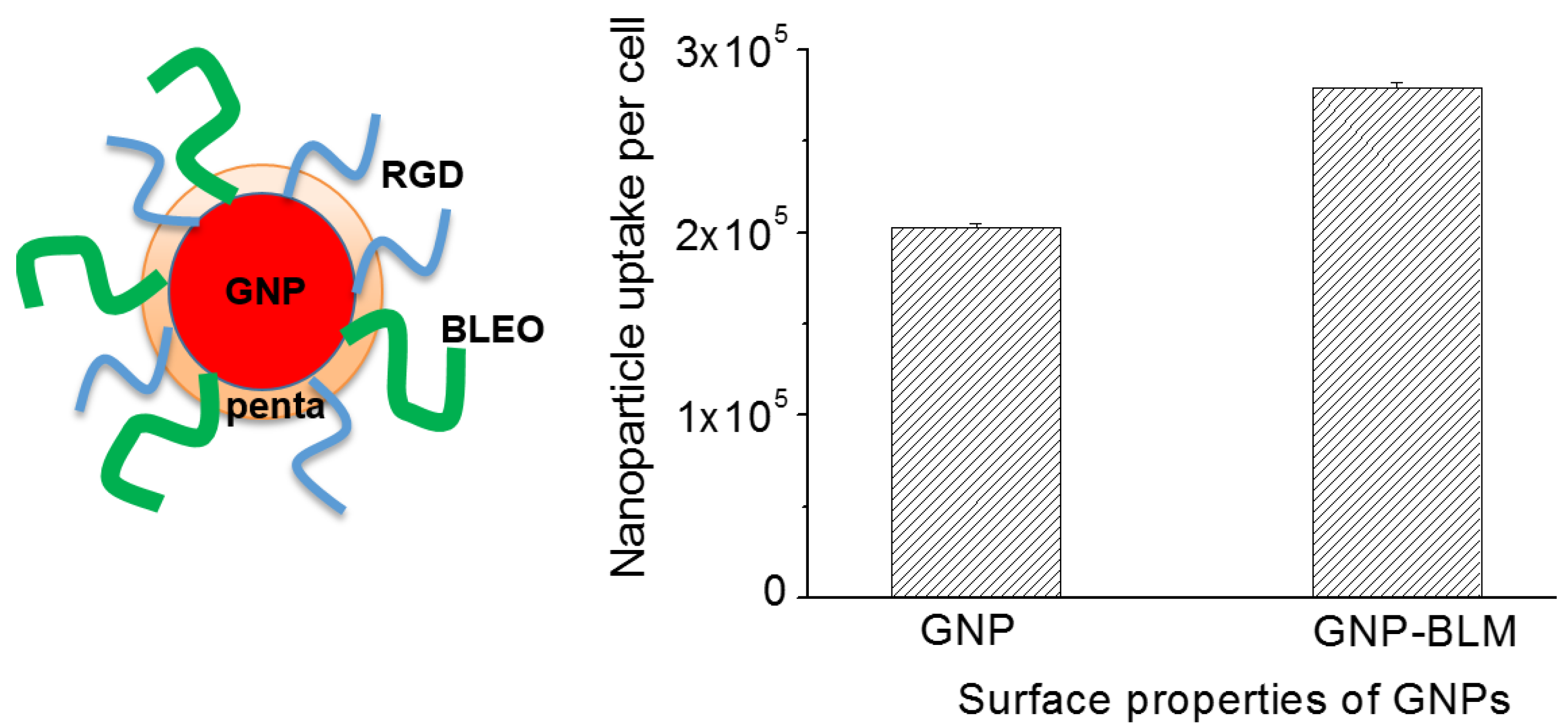
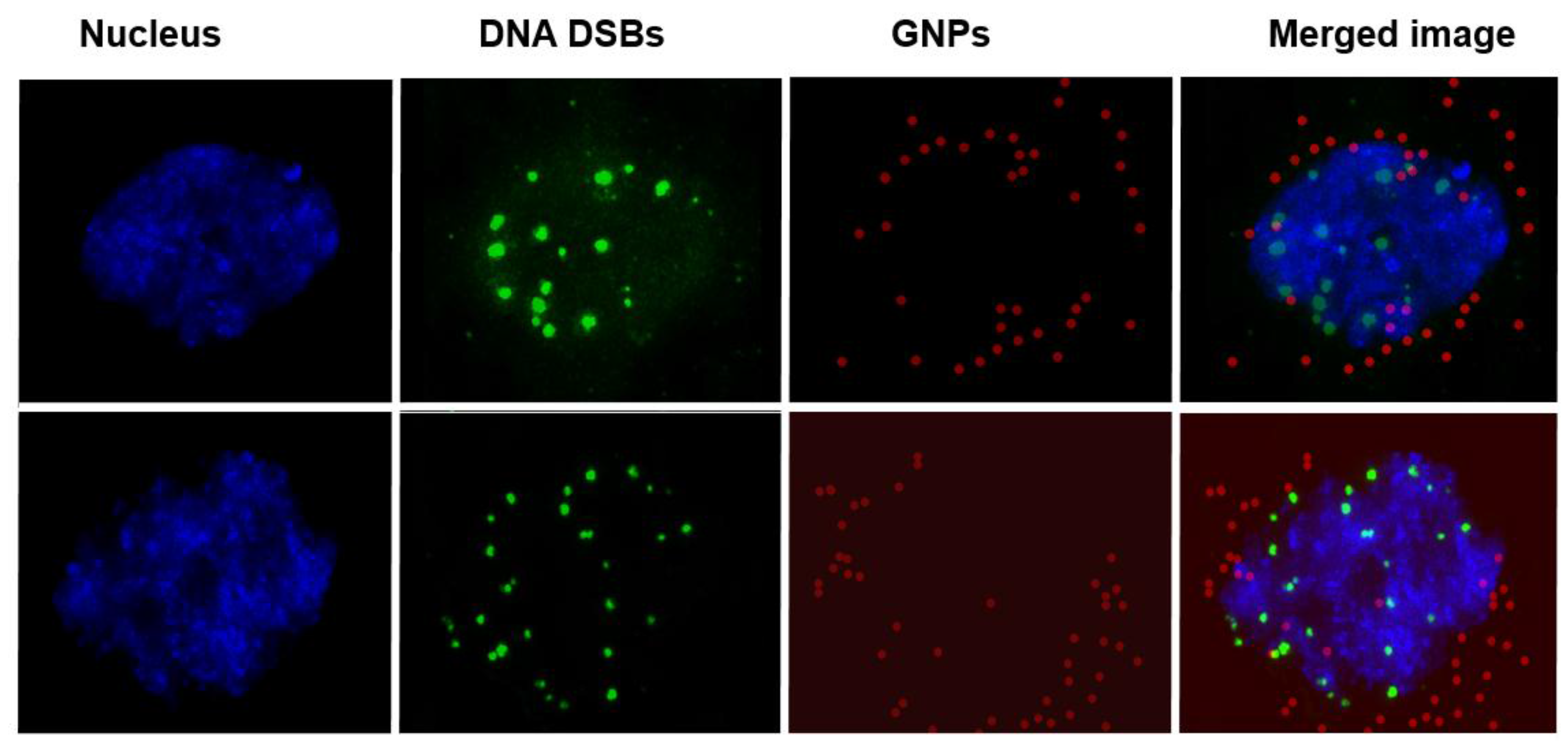
2.3. Cellular Uptake of BLM Conjugated GNPs
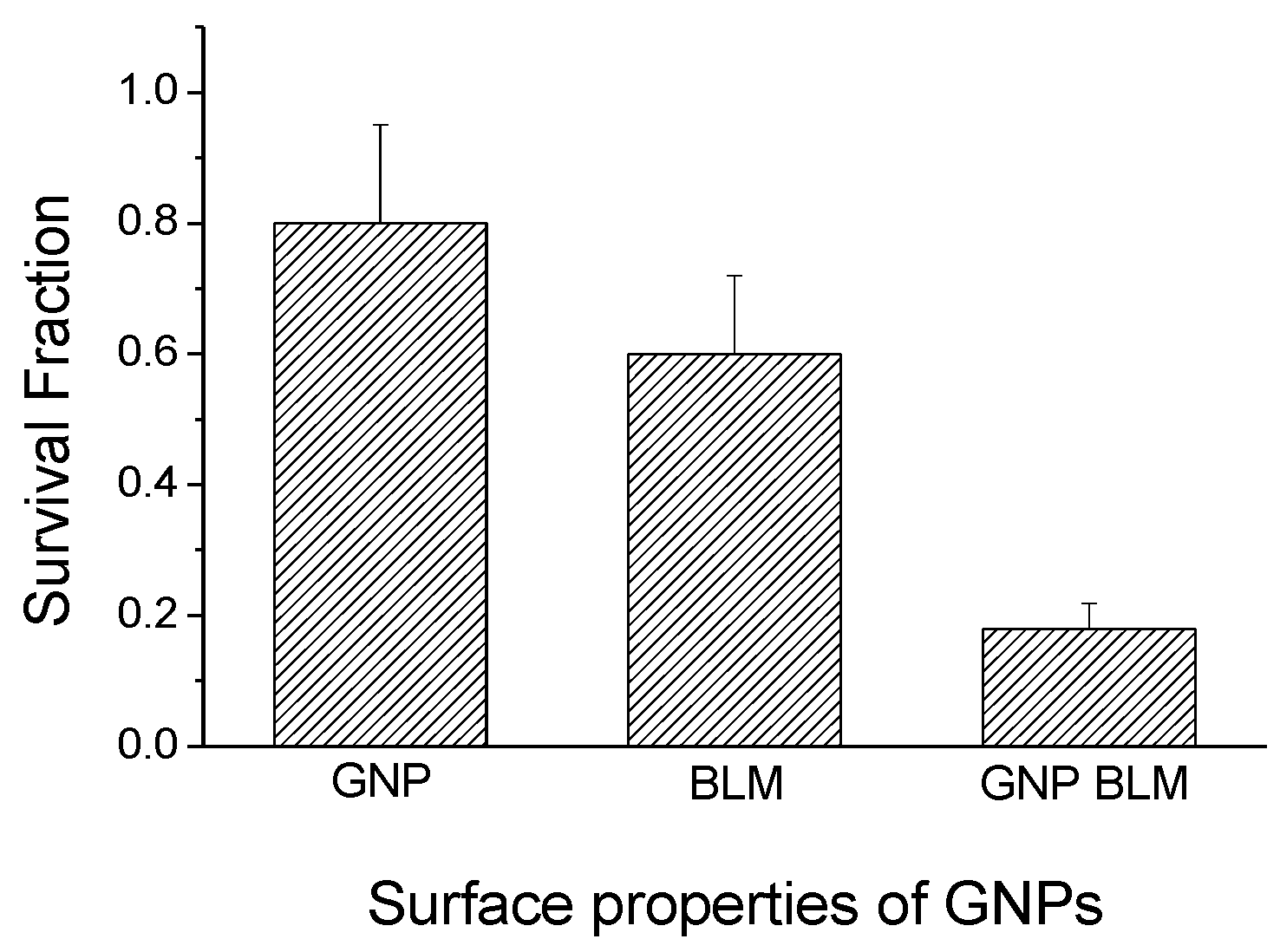
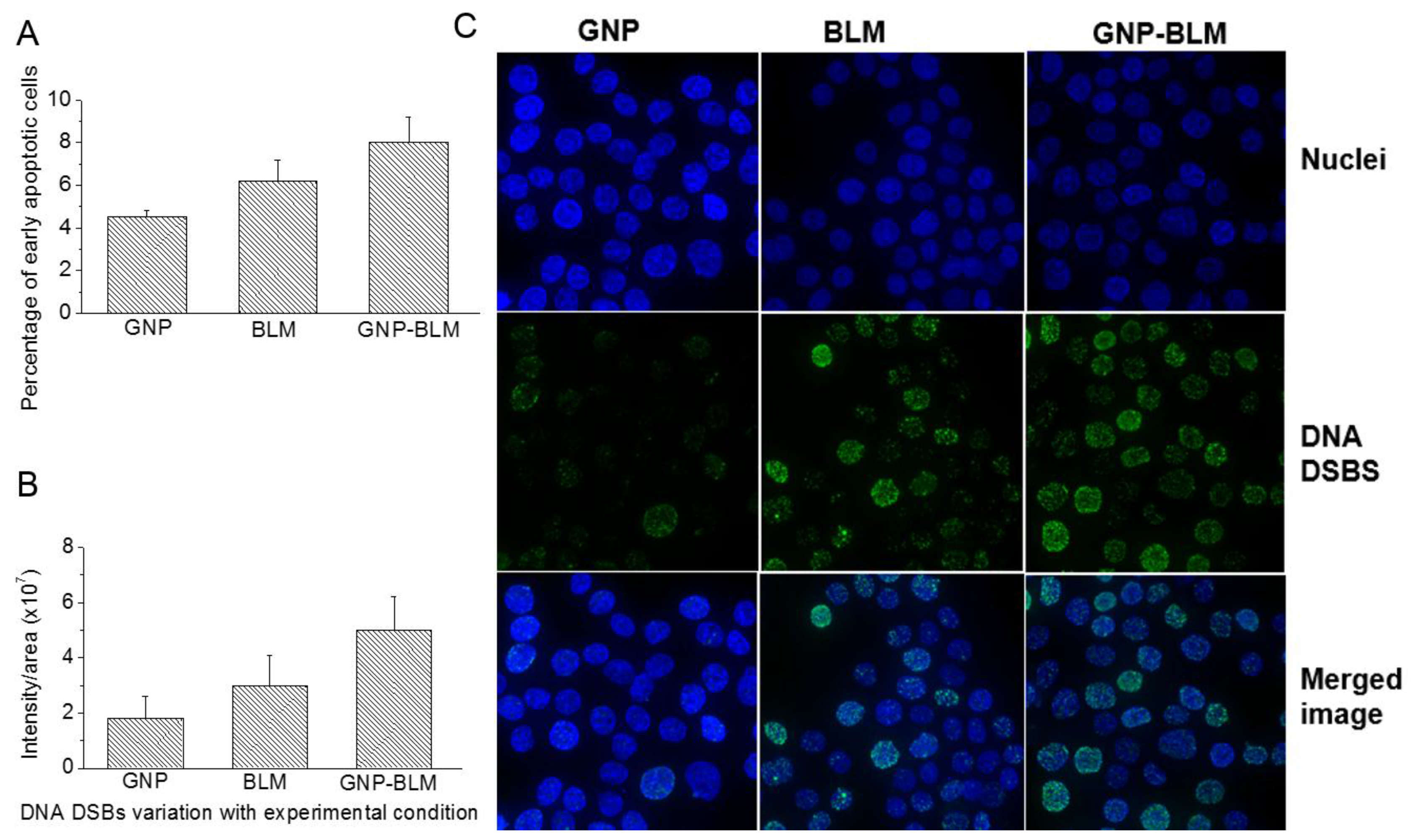
2.4. Therapeutic Efficacy Due to GNP-BLM vs BLM
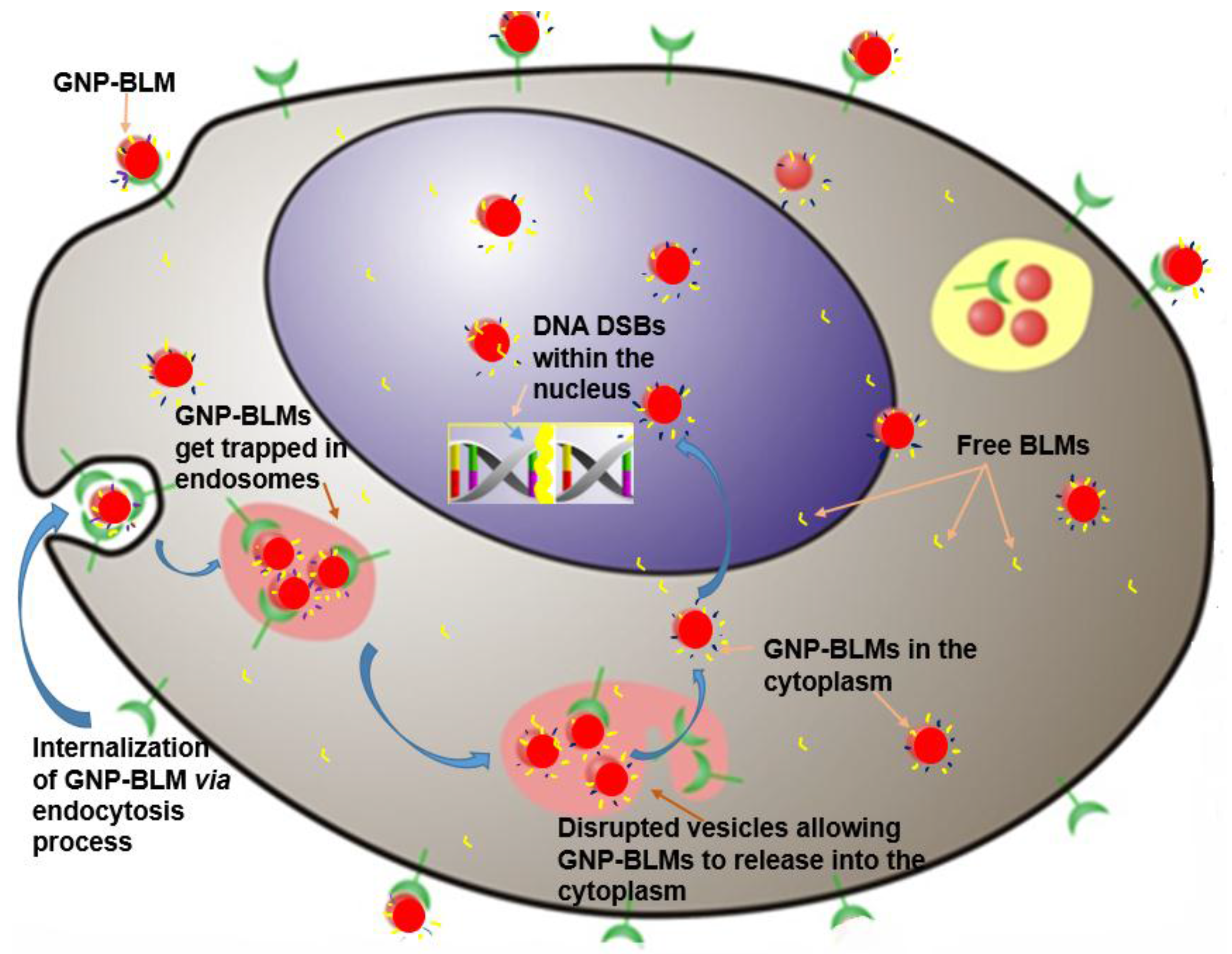
3. Discussion
- (1)
- A RGD peptide on the GNP-BLM complex allows for the release of these GNP-BLMs from these vesicles into the cytoplasm [34]. This will provide more time for the drug to be released from NPs into the cytoplasm.
- (2)
- BLM is known to reach the nucleus and bind to DNA causing DNA DSBs. If a RGD peptide assists the transport of the GNP-BLM complex to the cytoplasm, it is also possible that we can even transport some GNPs into the nucleus with the help of BLM (if they are still available on the GNP surface). This would benefit GNP mediated combined therapeutic strategies of chemotherapy and radiation therapy in the near future.
4. Experimental Section
4.1. Synthesis of Colloidal Gold Nanoparticles (GNPs)
4.2. Drug Conjugation
4.3. In vitro Experiments with the Drug
4.4. Quantification of Nanoparticle Uptake
4.5. Clonogenic Assay
4.6. Immunofluorescence Assay
4.7. Apoptotic Assay
4.8. Hyperspectral Imaging
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jain, R.K. Transport of molecules, particles, and cells in solid tumors. Annu. Rev. Biomed. Eng. 1999, 1, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Georgelin, T.; Bombard, S.; Siaugue, J.-M.; Cabuil, V. Nanoparticle-mediated delivery of bleomycin. Angew. Chem. Int. Ed. 2010, 49, 8897–8901. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Gullotti, E.; Yeo, Y. Extracellularly Activated Nanocarriers: A New Paradigm of Tumor Targeted Drug Delivery. Mol. Pharm. 2009, 6, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Babu, S.; Heckert, E.; Dowding, J.; Hirst, S.M.; Inerbaev, T.M.; Self, W.T.; Reilly, C.M.; Masunov, A.E.; Rahman, T.S.; et al. Protonated nanoparticle surface governing ligand tethering and cellular targeting. ACS Nano 2009, 3, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Emerich, D.F.; Thanos, C.G. Targeted nanoparticle-based drug delivery and diagnosis. J. Drug Target. 2007, 15, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Giersig, M.; Welte, T.; Pison, U. Nanoparticle-based diagnosis and therapy. Curr. Drug Targets 2006, 7, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Nanoparticle Therapeutics: An Emerging Treatment Modality for Cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.; Kim, C.; Rotello, V.M. Gold nanoparticle platforms as drug and biomacromolecule delivery systems. J. Controll. Release 2010, 148, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Lavan, D.A.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Homayoni, H.; Menon, J.U.; Nguyen, K.T. Chitosan-based nanoparticles for drug delivery. Rev. Nanosci. Nanotechnol. 2014, 3, 133–148. [Google Scholar] [CrossRef]
- Bindhani, B.K.; Parida, U.K.; Biswal, S.K.; Panigrahi, A.K.; Nayak, P.L. Gold nanoparticles and their biomedical applications. Rev. Nanosci. Nanotechnol. 2013, 2, 247–260. [Google Scholar] [CrossRef]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Mwamuka, J.; Gole, A.; Murphy, C.J.; Wyatt, M.D. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small 2005, 1, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Male, K.B.; Lachance, B.; Hrapovic, S.; Sunahara, G.; Luong, J.H.T. Assessment of cytotoxicity of quantum dots and gold nanoparticles using cell-based impedance spectroscopy. Anal. Chem. 2008, 80, 5487–5493. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.M.; Vig, K.; Dennis, V.A.; Singh, S.R. Functionalized gold nanoparticles and their biomedical applications. Nanomaterials 2011, 1, 31–63. [Google Scholar] [CrossRef]
- Brown, S.D.; Nativo, P.; Smith, J.-A.; Stirling, D.; Edwards, P.R.; Venugopal, B.; Flint, D.J.; Plumb, J.A.; Graham, D.; Wheate, N.J. Gold nanoparticles for the improved anticancer drug delivery of the active component of oxaliplatin. J. Am. Chem. Soc. 2010, 132, 4678–4684. [Google Scholar] [CrossRef] [PubMed]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Yohan, D.; Charmainne, C.; Lu, X.; Chithrani, B.D. Size dependent gold nanoparticle interaction at nano-micro interface using both monolayer and multilayer (tissue-like) cell models. Nano-Micro Lett. 2016, 8, 44–53. [Google Scholar] [CrossRef]
- Gao, H.; Shi, W.; Freund, L.B.; Freund, L.B. Mechanics of receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 2005, 102. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Heller, D.A.; Strano, M.S.; Sharma, R.; Strano, M.S. Size-Dependent Cellular Uptake and Expulsion of Single-Walled Carbon Nanotubes: Single Particle Tracking and a Generic Uptake Model for Nanoparticles. ACS Nano 2009, 3, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Wang, J.; Fan, X.; Gao, H. Size and shape effects on diffusion and absorption of colloidal particles near a partially absorbing sphere: Implications for uptake of nanoparticles in animal cells. Phys. Rev. E 2008, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lee, C.W.; Chiou, A.; Wei, P.K. Size-dependent endocytosis of gold nanoparticles studied by three-dimensional mapping of plasmonic scattering images. J. Nanobiotechnol. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Smythe, E.; Warren, G. The mechanism of Receptor-mediated endocytosis. Eur. J. Biochem. 1991, 202, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Lykotrafitis, G.; Bao, G.; Suresh, S. Size-dependent endocytosis of nanoparticles. Adv. Mater. 2009, 21, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Ramanujan, S.; Pluen, A.; McKee, T.D.; Brown, E.B.; Boucher, Y.; Jain, R.K. Diffusion and Convection in Collagen Gels: Implications for Transport in the Tumor Interstitium. Biophys. J. 2002, 83, 1650–1660. [Google Scholar] [CrossRef]
- Grantab, R.; Sivananthan, S.; Tannock, I.F. The penetration of anticancer drugs through tumor tissue as a function of cellular adhesion and packing density of tumor cells. Cancer Res. 2006, 66, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar] [PubMed]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Alexandrakis, G.; Brown, E.B.; Tong, R.T.; McKee, T.D.; Campbell, R.B.; Boucher, Y.; Jain, R.K. Two-photon fluorescence correlation microscopy reveals the two-phase nature of transport in tumors. Nat. Med. 2004, 10, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Uertz, J.; Yohan, D.; Chithrani, B.D. Peptide modified gold nanoparticles for improved cellular uptake, nuclear transport, and intracellular retention. Nanoscale 2014, 6, 12026–12033. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Neshatian, M.; van Prooijen, M.; Chithrani, B.D. Cancer Nanotechnology: Enhanced Therapeutic Response Using Peptide-Modified Gold Nanoparticles. J. Nanosci. Nanotechnol. 2014, 14, 4813–4819. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, Y.; Morishima, H.; Saito, S.; Takita, T.; Umezawa, H.; Kobayashi, S.; Otsuka, M.; Narita, M.; Ohno, M. Synthesis of the pyrimidine moiety of bleomycin. J. Am. Chem. Soc. 1980, 102, 6630–6631. [Google Scholar] [CrossRef]
- Hecht, S.M. The chemistry of activated bleomycin. Acc. Chem. Res. 1986, 19, 383–391. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell. Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Haiss, W.; Thanh, N.T.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV-Vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Chequer, N.; González, J.; Cordova, T. Alternative metodology for gold nanoparticles diameter characterization using PCA technique and UV-Vis spectrophotometry. Nanosci. Nanotechnol. 2012, 2, 184–189. [Google Scholar] [CrossRef]
- Saulis, G.; Saulė, R. Size of the pores created by an electric pulse: Microsecond vs. millisecond pulses. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 3032–3039. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Bristow, R.G. Discordance between phosphorylation and recruitment of 53BP1 in response to DNA double-strand breaks. Cell. Cycle 2012, 11, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Joh, D.Y.; Sun, L.; Stangl, M.; Al Zaki, A.; Murty, S.; Santoiemma, P.P.; Davis, J.J.; Baumann, B.C.; Alonso-Basanta, M.; Bhang, D.; et al. Selective targeting of brain tumors with gold nanoparticle-induced radiosensitization. PLOS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Shah, N.H.; Malick, A.W.; Rhodes, C.T. Controlled drug delivery by biodegradable poly(ester) devices: Different preparative approaches. Drug Dev. Indust. Pharm. 1998, 24, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Kohler, N.; Sun, C.; Fichtenholtz, A.; Gunn, J.; Fang, C.; Zhang, M. Methotrexate-immobilized poly(ethylene glycol) magnetic nanoparticles for MR imaging and drug delivery. Small 2006, 2, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Medarova, Z.; Pham, W.; Farrar, C.; Petkova, V.; Moore, A. In vivo imaging of siRNA delivery and silencing in tumors. Nat. Med. 2007, 13, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Cheng, R.; Deng, C.; Zhong, Z. Intracellular drug release nanosystems. Mater. Today 2012, 15, 436–442. [Google Scholar] [CrossRef]
- Jain, S.; Coulter, J.A.; Hounsell, A.R.; Butterworth, K.T.; McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Dickson, G.R.; Prise, K.M.; Currell, F.J. Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Povirk, L.F.; Wubker, W.; Kohnlein, W.; Hutchinson, F. DNA double-strand breaks and alkali-labile bonds produced by bleomycin. Nucleic Acids Res. 1977, 4, 3573–3579. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, K.D.; Lewis, M.K.; Long, E.C.; Georgiadis, M.M. Crystal structure of DNA-bound Co(III)·bleomycin B2: Insights on intercalation and minor groove binding. PNAS 2008, 105, 5052–5056. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.M. Bleomycin: New Perspectives on the Mechanism of Action. J. Nat. Prod. 2000, 63, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Jelveh, S.; Chithrani, D.B. Gold nanostructures as a platform for combinational therapy in future cancer therapeutics. Cancers 2011, 3, 1081–1110. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Uertz, J.; Chithrani, D.B. Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy. Nanomaterials 2016, 6, 48. https://doi.org/10.3390/nano6030048
Yang C, Uertz J, Chithrani DB. Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy. Nanomaterials. 2016; 6(3):48. https://doi.org/10.3390/nano6030048
Chicago/Turabian StyleYang, Celina, Jamie Uertz, and Devika B. Chithrani. 2016. "Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy" Nanomaterials 6, no. 3: 48. https://doi.org/10.3390/nano6030048
APA StyleYang, C., Uertz, J., & Chithrani, D. B. (2016). Colloidal Gold-Mediated Delivery of Bleomycin for Improved Outcome in Chemotherapy. Nanomaterials, 6(3), 48. https://doi.org/10.3390/nano6030048