Study of the Effect of Grafting Method on Surface Polarity of Tempo-Oxidized Nanocellulose Using Polycaprolactone as the Modifying Compound: Esterification versus Click-Chemistry

Abstract
:1. Introduction
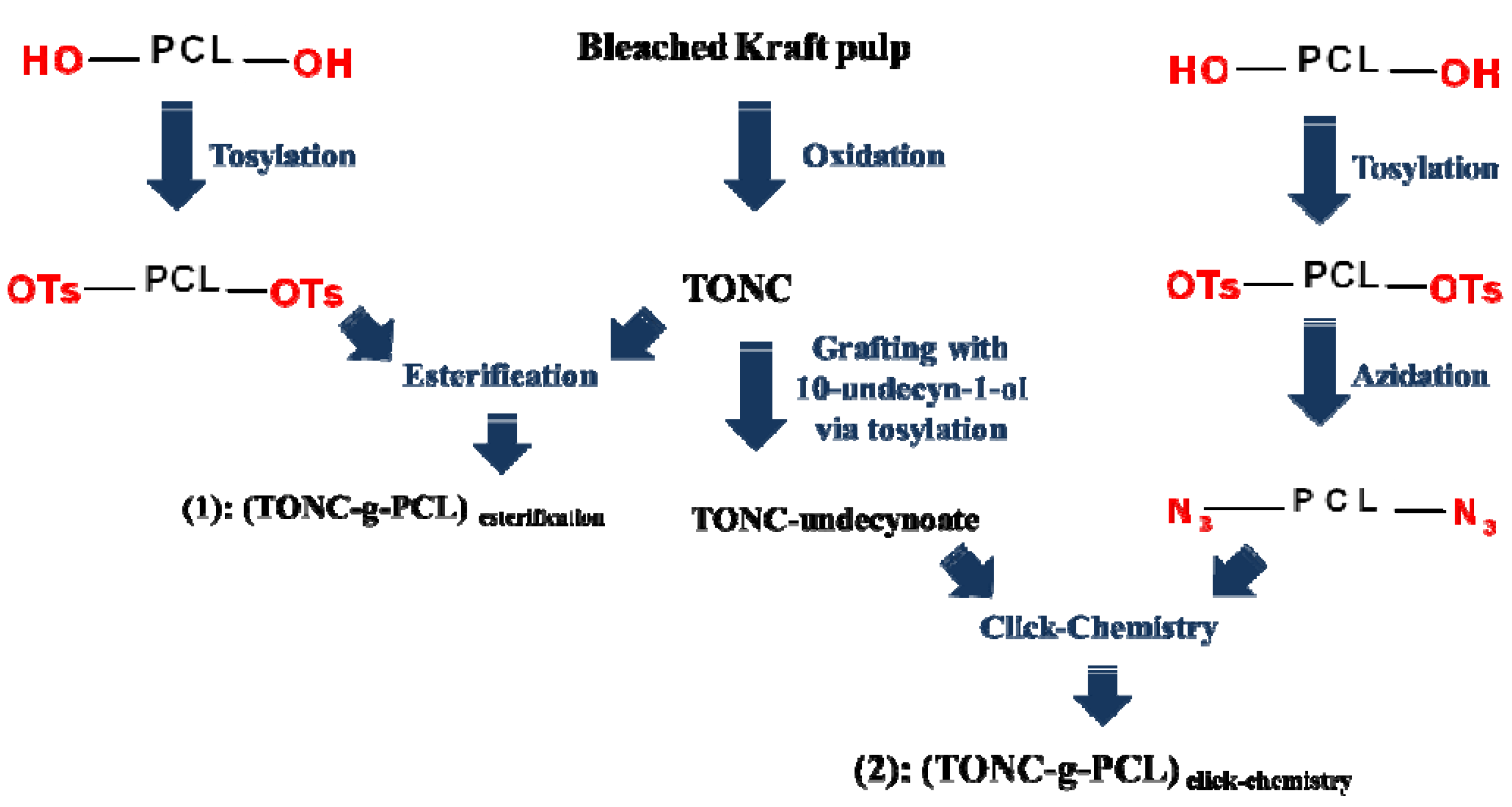
2. Results and Discussion
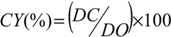
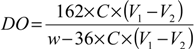
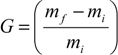
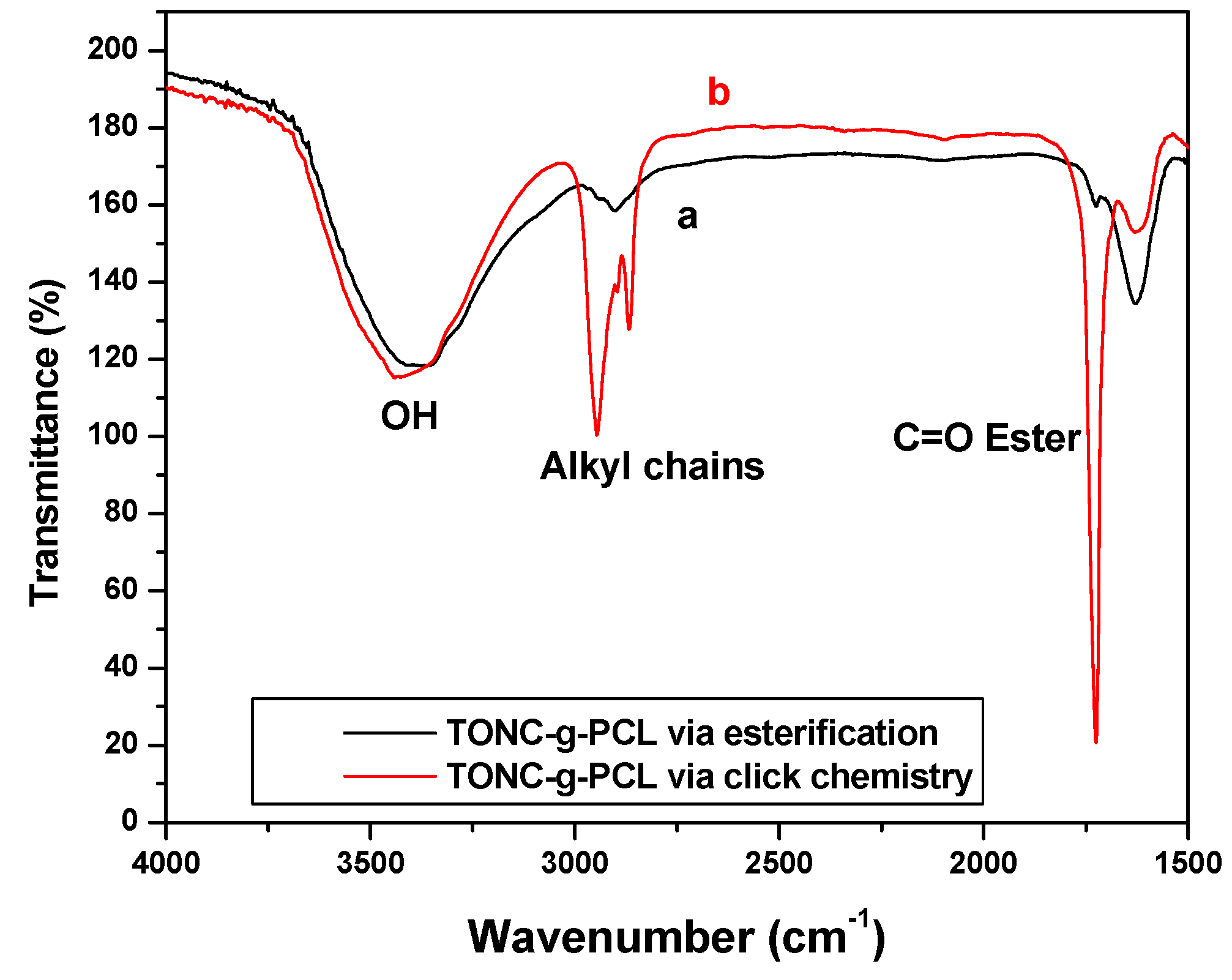
2.1. Fourier Transform Infrared Spectroscopy (FTIR) Experiments
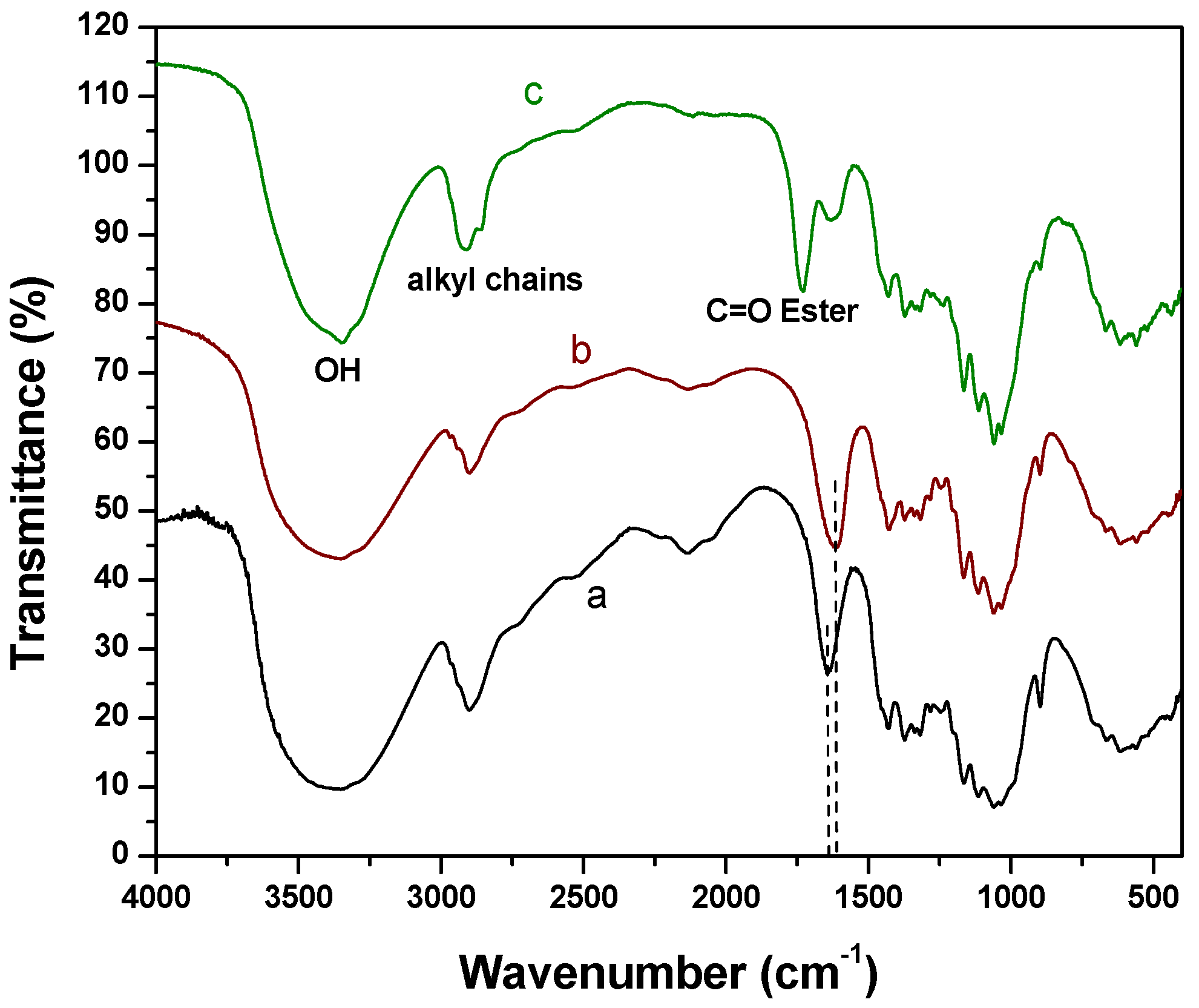
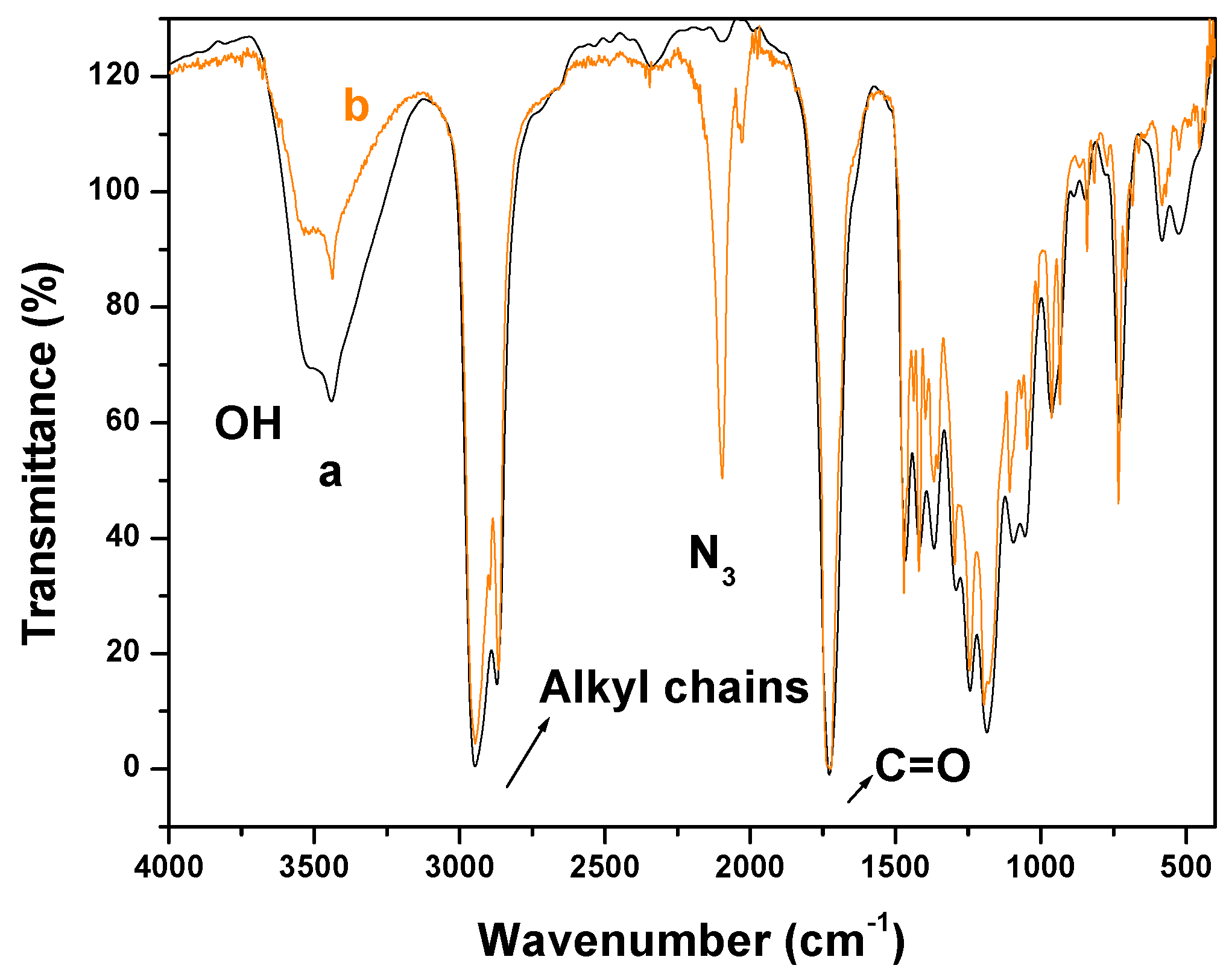
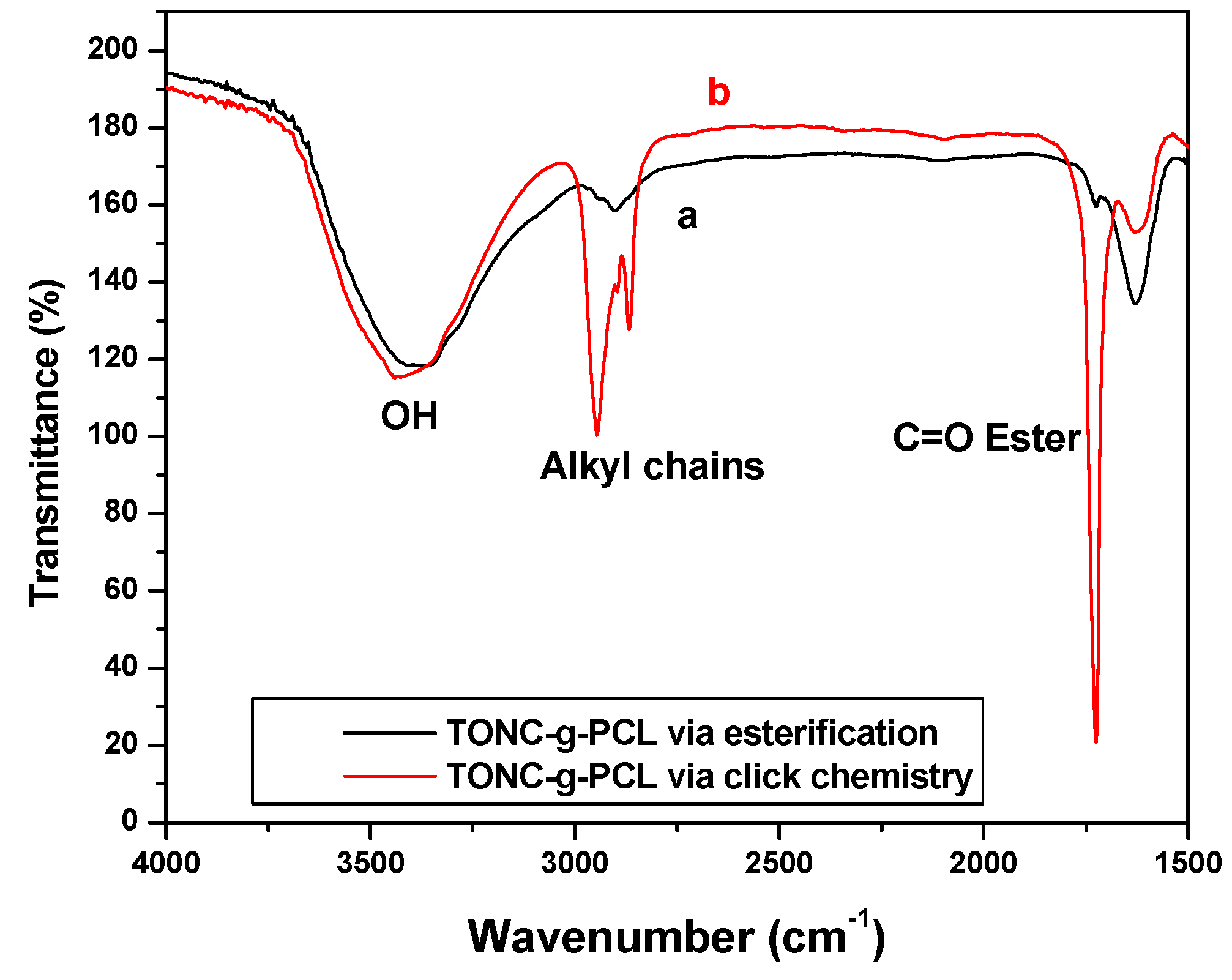
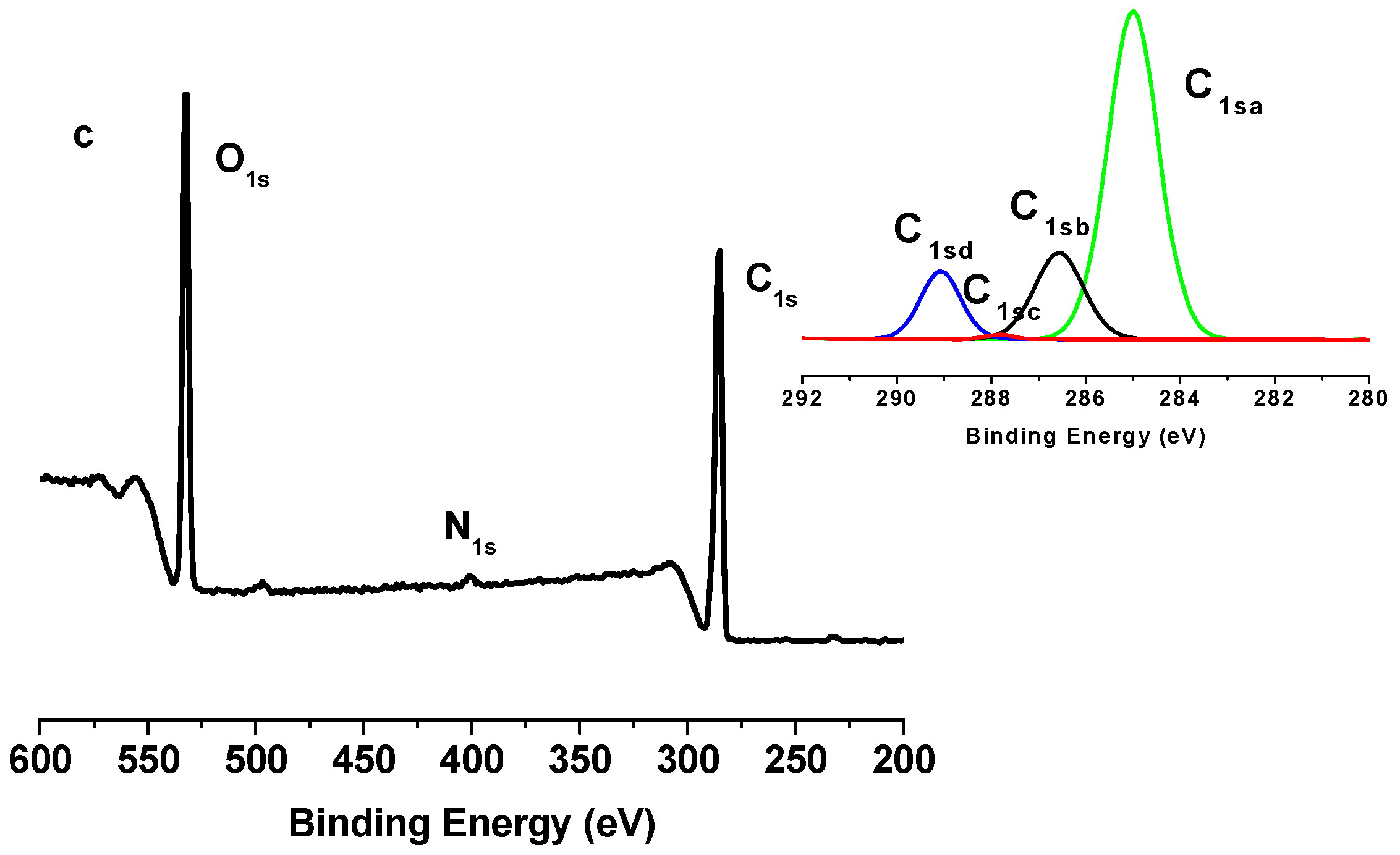
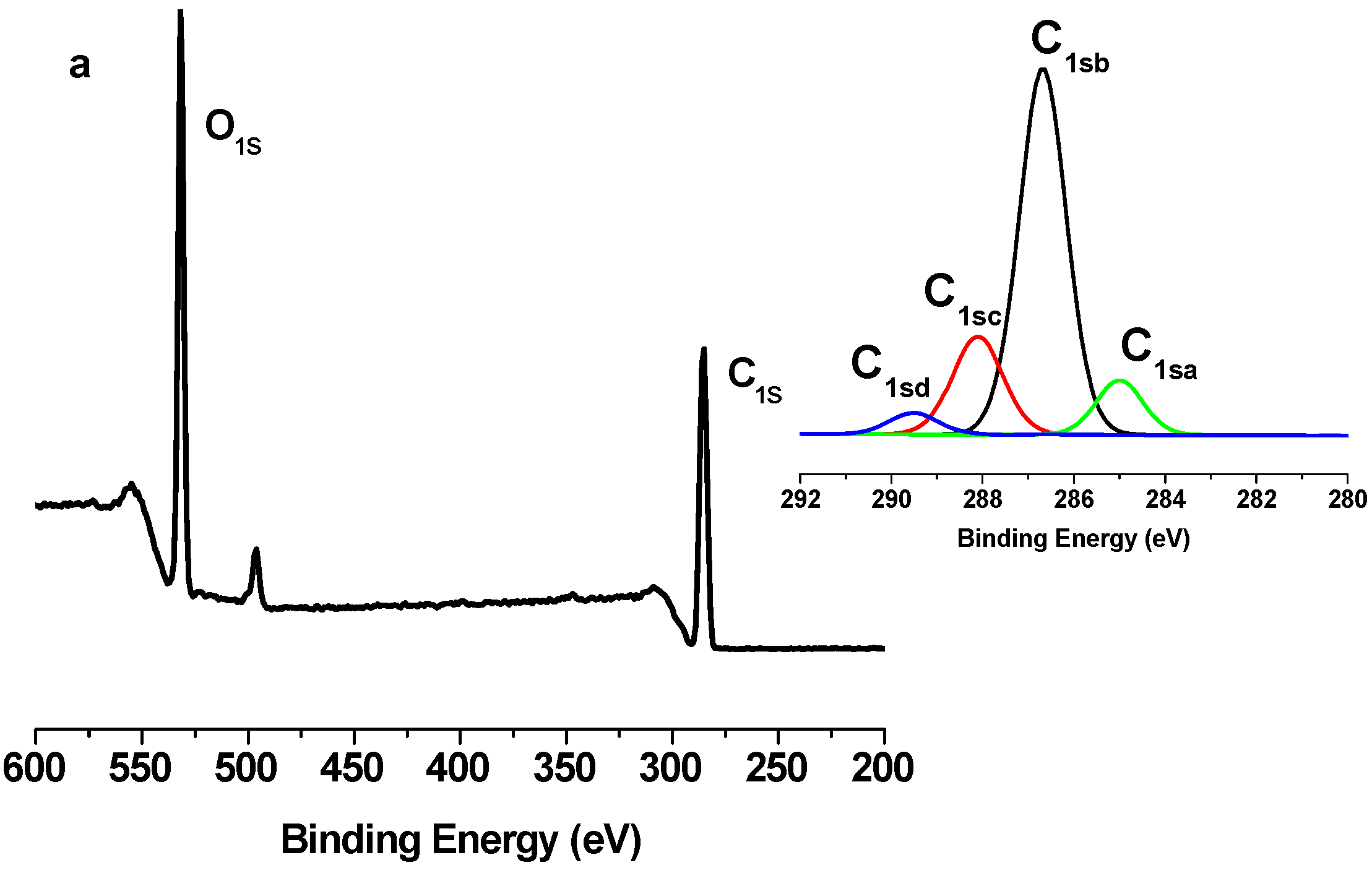
2.2. X-Ray Photoelectron Spectroscopy (XPS) Results

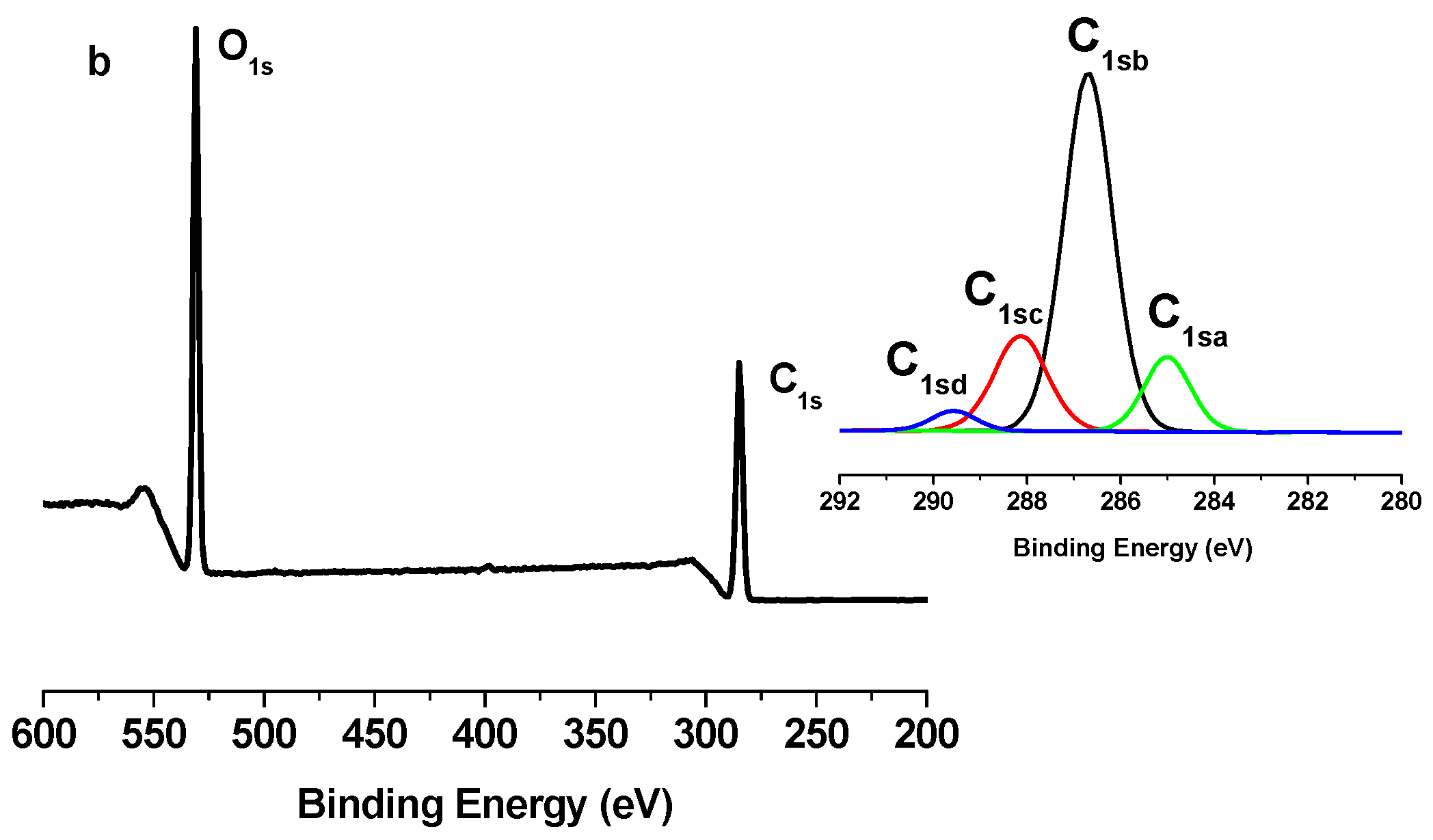

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding type | C1sa (C–C/C–H) | C1sb (C–O) | C1sc (C=O) | C1sd (O–C=O) |
|---|---|---|---|---|
| Energy (eV) | 285 | 286.5–7 | 288 | 289.2–9 |
| TONC | 9.86 | 66.91 | 19.16 | 4.07 |
| (TONC-g-PCL)ester | 12.89 | 64.96 | 18.86 | 3.29 |
| (ONC-g-PCL)click | 70.11 | 17.89 | 0.54 | 11.46 |
| Sample | Atomic content % | O/C | ||
|---|---|---|---|---|
| C | O | N | ||
| TONC | 67.11 | 32.89 | 0 | 0.49 |
| (TONC-g-PCL)ester | 68.46 | 31.55 | 0 | 0.46 |
| (ONC-g-PCL)click | 75.26 | 20.80 | 3.94 | 0.26 |
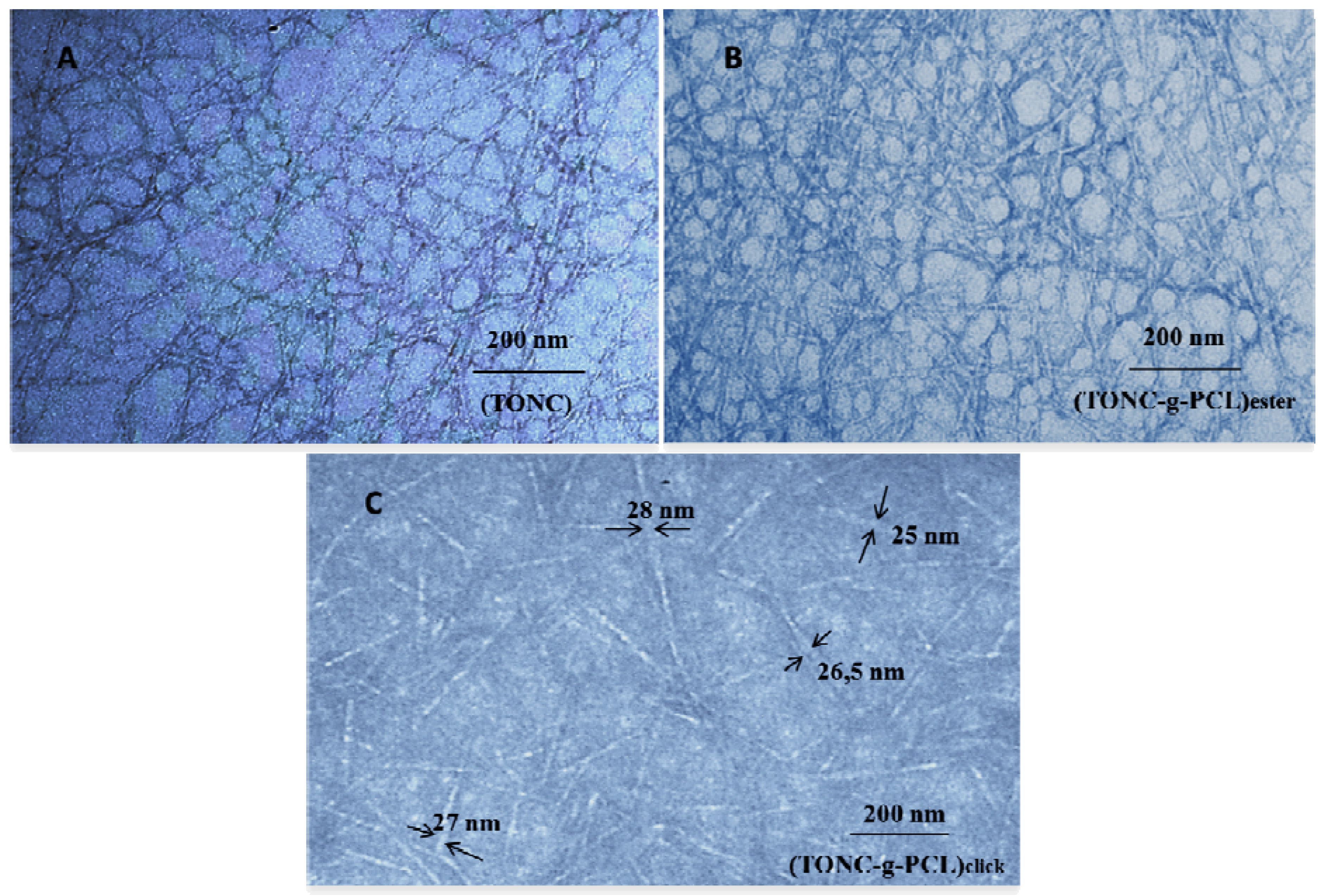
2.3. Transmission Electron Microscopy (TEM) Results
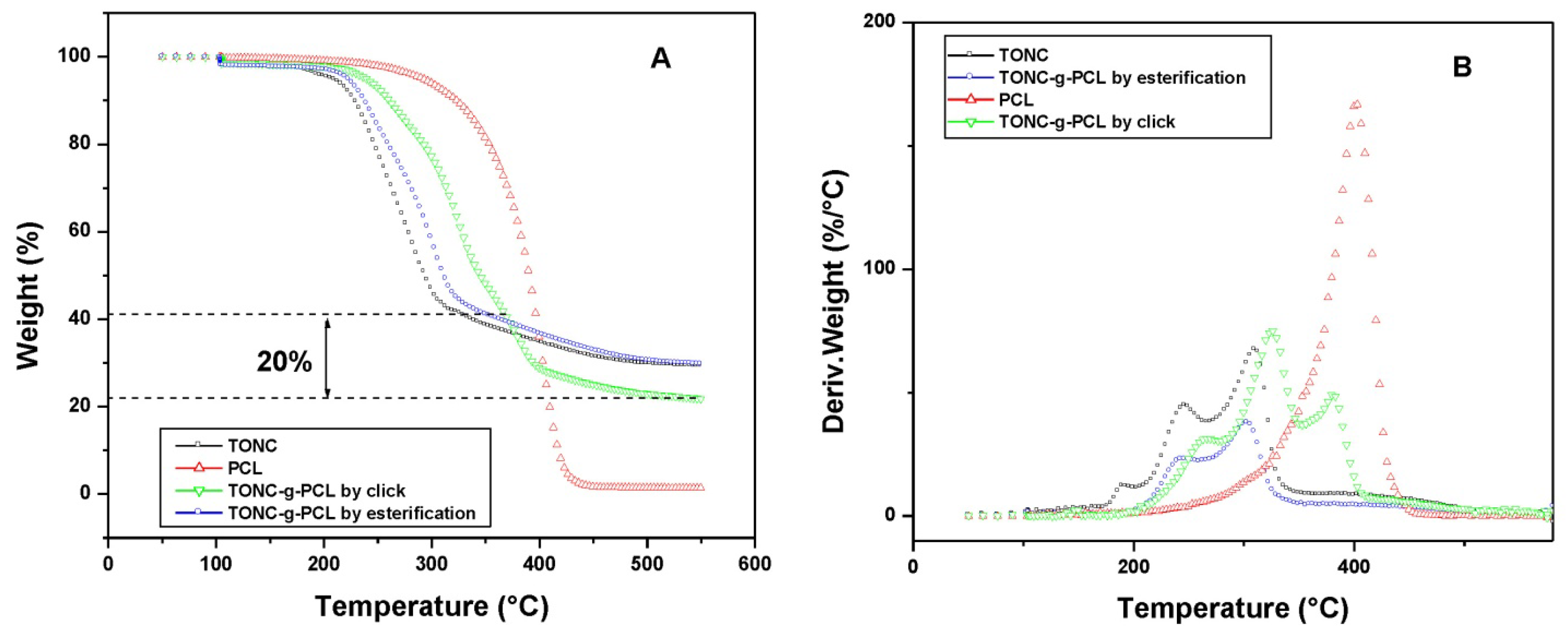
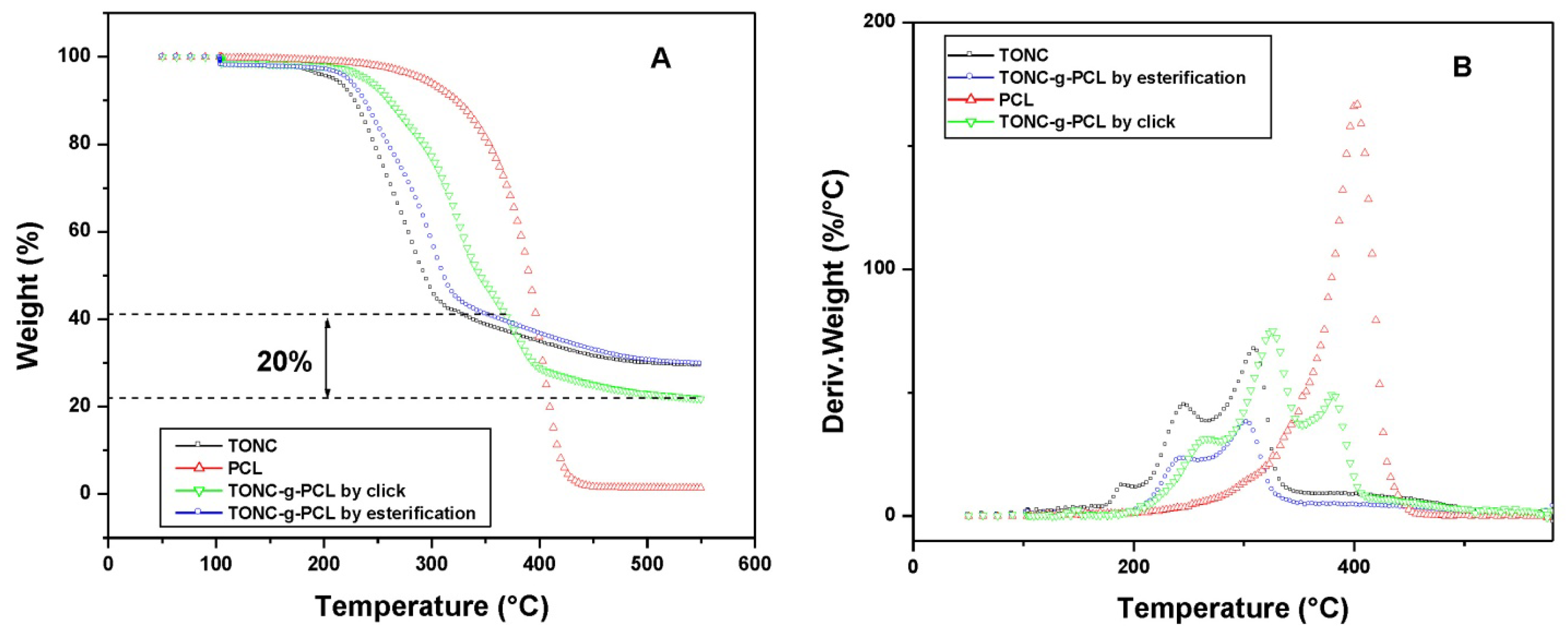
2.4. Thermogravimetry Analysis (TGA) Results
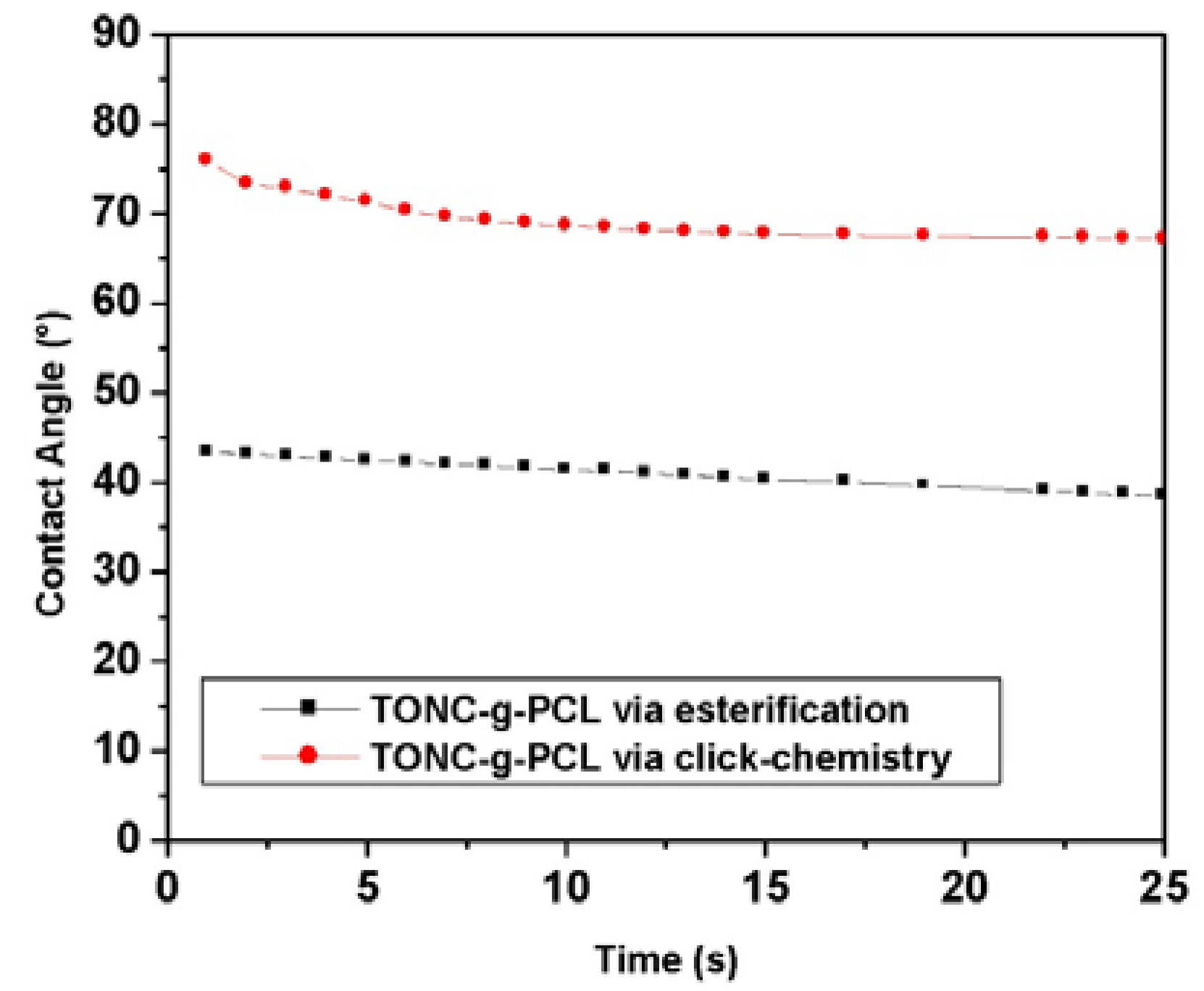
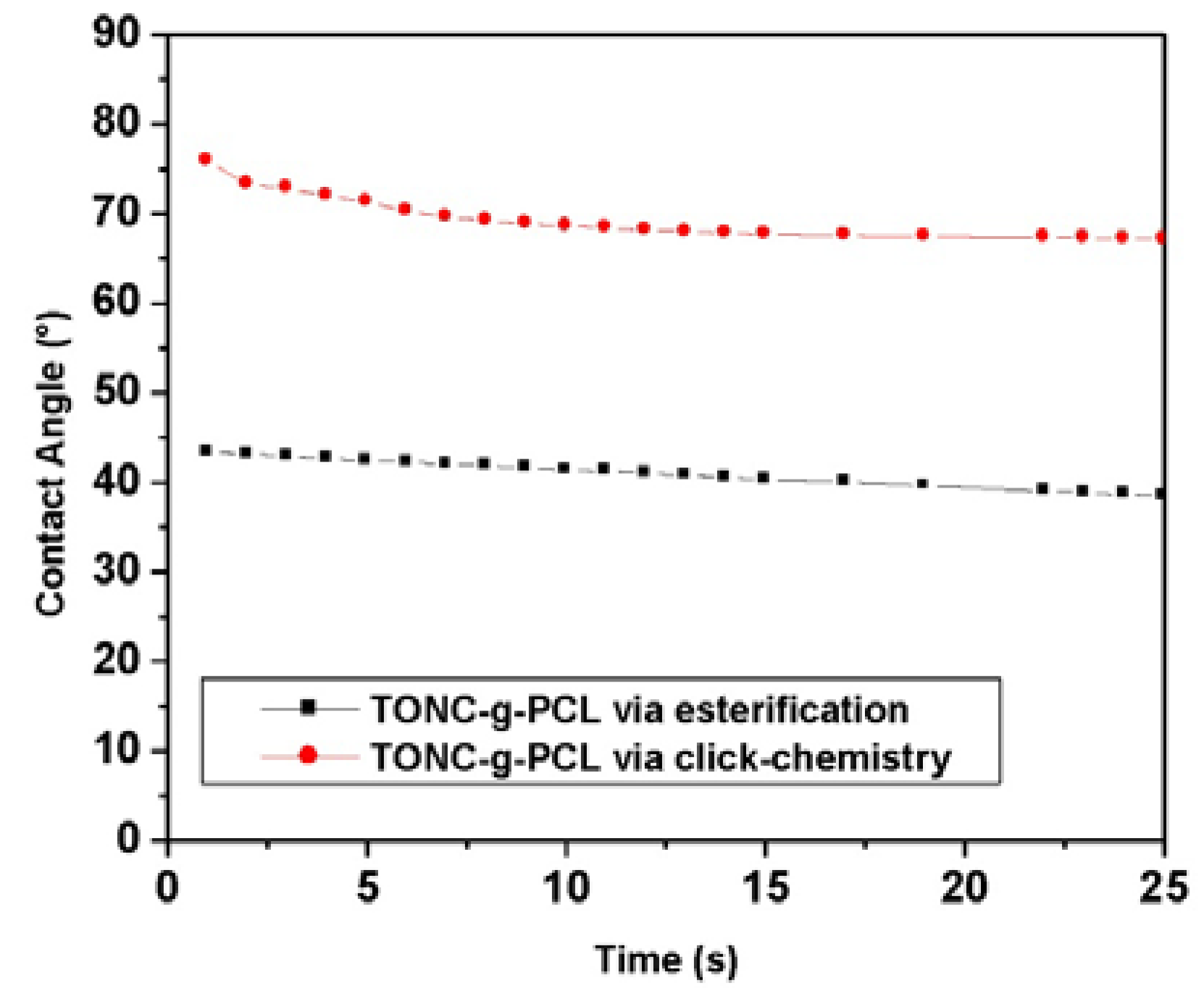
2.5. Contact Angle Results

3. Experimental Section
3.1. Materials
3.2. Preparation of Oxidized Nanocelluloses TONC by Ultrasound-TEMPO-NaBr-NaOCl-Oxidation
3.3. Measurement of Carboxyl Group Content
3.4. Production of TEMPO-Oxidized Nanocelluloses (TONC)
3.5. Synthesis of Click Precursor Bearing Alkyne Groups (TONC-Undecynoate)
3.6. Synthesis of p-toluenesulfonylpolycaprolactone (PCL-OTs)
3.7. Synthesis of Azido-polycaprolactone (PCL-N3)
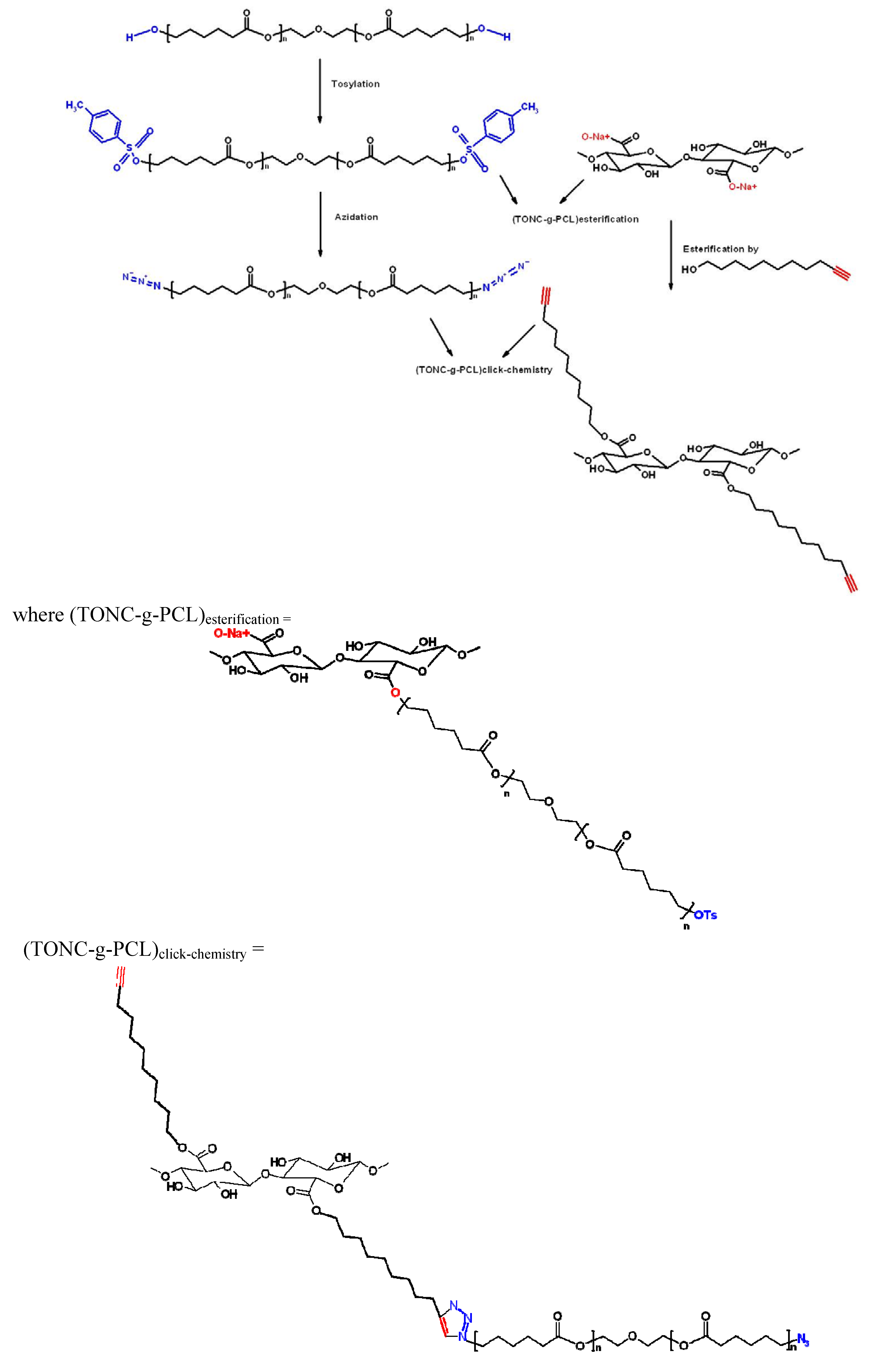
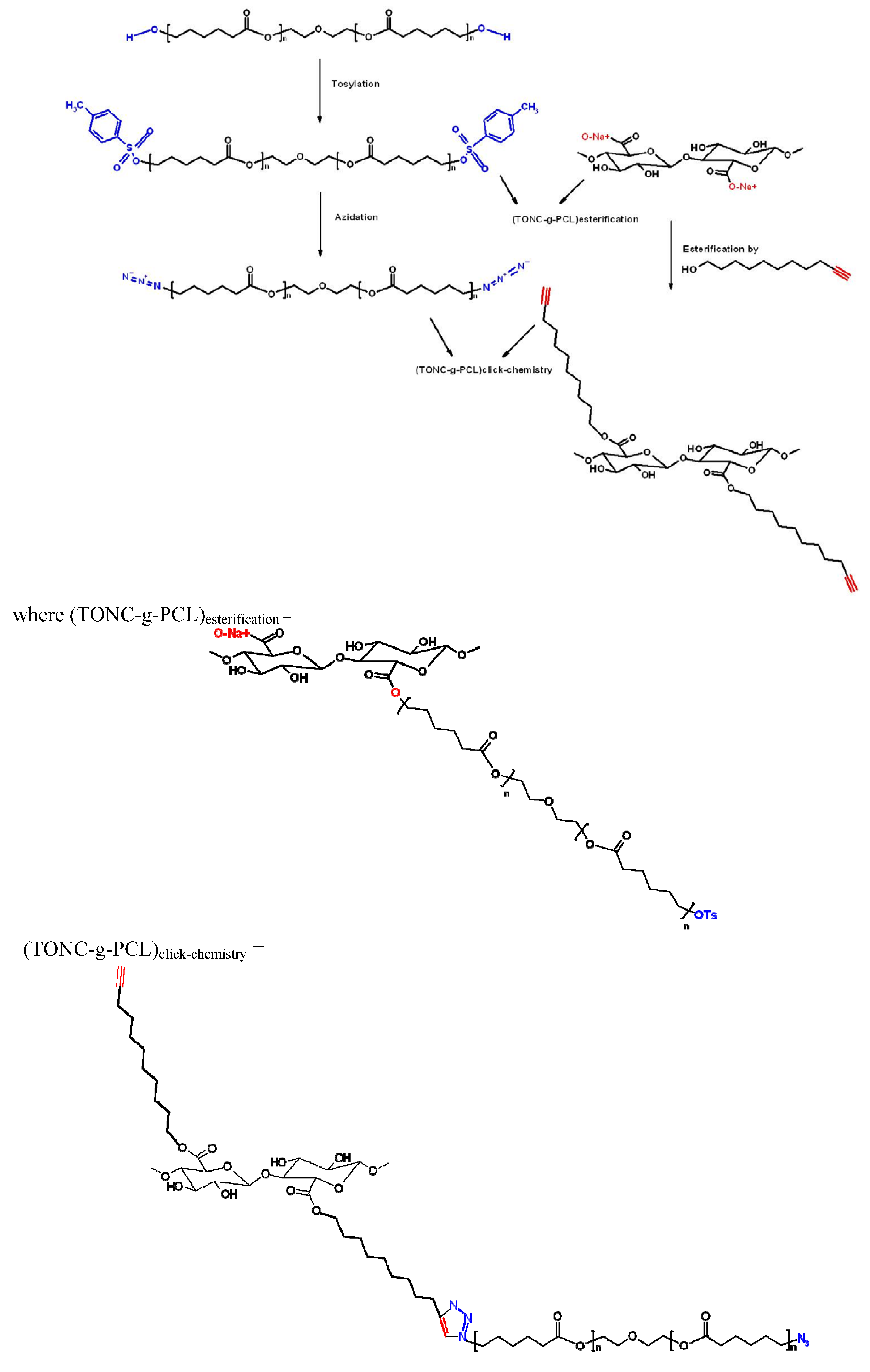
3.8. Esterification of TONC by PCL-OTs [(TONC-g-PCL)Esterification]
3.9. Grafting of PCL-N3 onto TONC-Undecynoate by Click Chemistry [(TONC-g-PCL)click-chemistry]
4. Characterization
4.1. Fourier Transform Infrared Spectrometry (FTIR)
4.2. X-Ray Photoelectron Spectroscopy (XPS)
4.3. Transmission Electron Microscopy (TEM)
4.4. Thermogravimetry Analysis (TGA)
4.5. Contact Angle (CA)
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dong, S.; Roman, M. Fluorescently labeled cellulose nanocrystals for bioimaging applications. J. Am. Chem. Soc. 2007, 129, 13810–13811. [Google Scholar] [CrossRef]
- Song, Y.B.; Zhou, J.P.; Li, Q.; Guo, Y.; Zhang, L.N. Preparation and characterization of novel quaternized cellulose nanoparticles as protein carriers. Macromol. Biosci. 2009, 9, 857–863. [Google Scholar] [CrossRef]
- Backdahl, H.; Helenius, G.; Bodin, A.; Nannmark, U.; Johansson, B.R.; Risberg, B.; Gatenholm, P. Mechanical properties of bacterial cellulose and interactions with smooth muscle cells. Biomaterials 2006, 27, 2141–2149. [Google Scholar] [CrossRef]
- Helenius, G.; Backdahl, H.; Bodin, A.; Nannmark, U.; Gatenholm, P.; Risberg, B. In vivo biocompatibility of bacterial cellulose. J Biomed. Mater. Res. A 2006, 76A, 431–438. [Google Scholar] [CrossRef]
- Svensson, A.; Nicklasson, E.; Harrah, T.; Panilaitis, B.; Kaplan, D.L.; Brittberg, M.; Gatenholm, P. Bacterial cellulose as a potential scaffold for tissue engineering of cartilage. Biomaterials 2005, 26, 419–431. [Google Scholar]
- Cai, Z.; Kim, J. Bacterial cellulose/poly(ethylene glycol) composite: Characterization and first evaluation of biocompatibility. Cellulose 2010, 17, 83–91. [Google Scholar] [CrossRef]
- Xing, Q.; Zhao, F.; Chen, S.; DeCoster, M.A.; Lvov, Y.M. Porous biocompatible three dimensional scaffolds of cellulose microfiber/gelatin composites for cell culture. Acta Biomater. 2010, 6, 2132–2139. [Google Scholar] [CrossRef]
- Lu, P.; Hsieh, Y.L. Cellulose nanocrystal-filled poly(acrylic acid) nanocomposite fibrous membranes. Nanotechnology 2009, 20. [Google Scholar] [CrossRef]
- Paralikara, S.A.; Simonsen, J.; Lombardi, J. Poly(vinyl alcohol)/cellulose nanocrystal barrier membranes. J Membr. Sci. 2008, 320, 248–258. [Google Scholar] [CrossRef]
- Dufresne, A.; Vignon, M.R. Processing and mechanical properties of natural fiber reinforced thermoplastic starch biocomposites. Macromolecules 1998, 31, 2693–2696. [Google Scholar] [CrossRef]
- Favier, V.; Chanzy, H.; Cavaille, J.Y. Polymer nanocomposites reinforced by cellulose whiskers. Macromolecules 1995, 28, 6365–6367. [Google Scholar] [CrossRef]
- Grunert, M.; Winter, W.T. Nanocomposites of cellulose acetate butyrate reinforced with cellulose nanocrystals. J. Polym. Environ. 2002, 10, 27–30. [Google Scholar]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulose whiskers versus microfibrils: Influence of the nature of the nanoparticle and its surface functionalization on the thermal and mechanical properties of nanocomposites. Biomacromolecules 2009, 10, 425–432. [Google Scholar] [CrossRef]
- Schandler, L.S.; Brinson, L.C.; Sawyer, W.G. Polymer nanocomposites: A small part of the story. JOM 2007, 59, 53–60. [Google Scholar]
- Klemm, D.; Schumann, D.; Kramer, F.; Hessler, N.; Hornung, M.; Schmauder, H.P.; Marsch, S. Nanocelluloses as innovative polymers in research and application. Adv. Polym. Sci. 2006, 205, 49–96. [Google Scholar] [CrossRef]
- Klemm, D.; Schumann, D.; Kramer, F.; Hessler, N.; Koth, D.; Sultanova, B. Nanocellulose materials—Different cellulose, different functionality. Macromol. Symp. 2009, 280, 60–71. [Google Scholar] [CrossRef]
- Battista, O.A. Microcrystal polymer science. J. Polym. Sci. 1975, 13, 625–626. [Google Scholar]
- Ranby, B.G. Fibrous macromolecular systems. Cellulose and muscle. The colloidal properties of cellulose micelles. Discuss. Faraday Soc. 1951, 11, 158–164. [Google Scholar] [CrossRef]
- Nakagaito, A.N.; Yano, H. Novel high-strength biocomposites based on microfibrillated cellulose having nano-order-unit web-like network structure. Appl. Phys. A 2005, 80, 155–159. [Google Scholar] [CrossRef]
- Baiardo, M.; Frisoni, G.; Scandola, M.; Licciardello, A. Surface chemical modification of natural cellulose fibers. J. Appl. Polym. Sci. 2002, 83, 38–45. [Google Scholar] [CrossRef]
- Dong, X.M.; Revol, J.F.; Gray, D.G. Effect of microcrystallite preparation conditions on the formation of colloid crystals of cellulose. Cellulose 1998, 5, 19–32. [Google Scholar] [CrossRef]
- Nakagaito, A.N.; Yano, H. The effect of morphological changes from pulp fiber towards nano-scale fibrillated cellulose on the mechanical properties of high-strength plant fiber based composites. Appl. Phys. A 2004, 78, 547–552. [Google Scholar] [CrossRef]
- Andresen, M.; Johansson, L.S.; Tanem, B.S.; Stenius, P. Properties and characterization of hydrophobized microfibrillated cellulose. Cellulose 2006, 13, 665–677. [Google Scholar] [CrossRef]
- Gousse, C.; Chanzy, H.; Excoffier, G.; Soubeyrand, L.; Fleury, E. Stable suspensions of partially silylated cellulose whiskers dispersed in organic solvents. Polymer 2002, 43, 2645–2651. [Google Scholar] [CrossRef]
- Yuan, H.H.; Nishiyama, Y.; Wada, M.; Kuga, S. Surface acylation of cellulose whiskers by drying aqueous emulsion. Biomacromolecules 2006, 7, 696–700. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S. Steric stabilization of a cellulose microcrystal suspension by poly(ethylene glycol) grafting. Langmuir 2001, 17, 21–27. [Google Scholar] [CrossRef]
- Ahmed Said Azizi, S.; Lonnberg, H.; Fogelstrom, L.; Berglund, L.; Malmstrom, E.; Anders, H. Surface grafting of microfibrillated cellulose with poly(caprolactone)—Synthesis and characterization. Eur. Polym. J. 2008, 44, 2991–2997. [Google Scholar] [CrossRef]
- Heux, L.; Chauve, G.; Bonini, C. Nonflocculatingand chiral-nematic self-ordering of cellulose microcrystals suspensions in nonpolar solvents. Langmuir 2000, 16, 8210–8212. [Google Scholar] [CrossRef]
- Zhou, Q.; Brumer, H.; Teeri, T.T. Self-organization of cellulose nanocrystals adsorbed with xyloglucan oligosaccharide-poly(ethylene-glycol)-polystyrene triblock copolymer. Macromolecules 2009, 42, 5430–5432. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, Y.; Putaux, J.L.; Vignon, M.; Isogai, A. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 2006, 7, 1687–1691. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Du Prez, F.E. “Clicking” polymers or just efficient linking: What is the difference? Angew. Chem. Int. Ed. 2011, 50, 60–62. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.-L.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(epsilon-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 41, 5002–5010. [Google Scholar]
- Krouit, M.; Bras, J. Cellulose surface grafting with polycaprolactone by heterogeneous click-chemistry. Eur. Polym. J. 2008, 44, 4074–4081. [Google Scholar] [CrossRef]
- Julien, O.; Krouit, M.; Bras, P.; Thielemans, W.; Belgacem, M.N. Surface modification of cellulose by PCL grafts. Acta Mater. 2010, 58, 792–801. [Google Scholar] [CrossRef]
- Benkaddour, A.; Jradi, K.; Daneault, C.; Sylvain, R. Grafting of polycaprolactone on oxidized nanocelluloses by click chemistry. Nanomaterials 2013, 3, 141–157. [Google Scholar] [CrossRef]
- Varma, A.J.; Chavan, V.B. Thermal properties of oxidized cellulose. Cellulose 1995, 2, 41–49. [Google Scholar]
- Lerdkanchanaporn, S.; Dollimore, D.; Alexander, K.S. A simultaneous TG-DTA study of the degradation in nitrogen of cellulose to carbon, alone and in the presence of other pharmaceutical excipients. Thermochim. Acta 1998, 324, 25–32. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Okita, Y.; Isogai, A. Thermal stabilization of TEMPO-oxidized cellulose. Polym. Degrad. Stab. 2010, 95, 1502–1508. [Google Scholar] [CrossRef]
- Biresaw, G.; Carriere, C.J.J. Correlation between mechanical adhesion and interfacial properties of starch/biodegradable polyester blends. Polym. Sci. B 2001, 39, 920–930. [Google Scholar] [CrossRef]
- Habibi, Y.; Dufresne, A. Highly filled bionanocomposites from functionalized polysaccharide nanocrystals. Biomacromolecules 2008, 9, 1974–1980. [Google Scholar] [CrossRef]
- Mishra, S.P.; Thirree, J.; Manent, A.S.; Chabot, B.; Daneault, C. Ultrasound-catalyzed TEMPO-mediated oxydation of native cellulose for the production of nanocellulose: Effect of process variables. BioResources 2011, 6, 121–143. [Google Scholar]
- Saito, T.; Isogai, A. Ion-exchange behavior of carboxylate groups in fibrous cellulose oxidized by the TEMPO mediated system. Carbohydr. Polym. 2005, 61, 183–190. [Google Scholar] [CrossRef]
- Benoit, D.; Grimaldi, S.; Robin, S.; Finet, J.P.; Tordo, P.; Gnanou, Y. Kinetics and mechaism of controlled free-radical polymerization of styrene and n-butyl acrylate in the presence of an acyclic phosphonylated nitroxide. J. Am. Chem. Soc. 2000, 122, 5929–5939. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Davis, T.P.; Fane, A.G. Honeycomb structured porous films prepared from carbohydrate based polymers synthesised via the RAFT process. J. Mater. Chem. 2003, 13, 2090–2097. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Benkaddour, A.; Jradi, K.; Robert, S.; Daneault, C. Study of the Effect of Grafting Method on Surface Polarity of Tempo-Oxidized Nanocellulose Using Polycaprolactone as the Modifying Compound: Esterification versus Click-Chemistry. Nanomaterials 2013, 3, 638-654. https://doi.org/10.3390/nano3040638
Benkaddour A, Jradi K, Robert S, Daneault C. Study of the Effect of Grafting Method on Surface Polarity of Tempo-Oxidized Nanocellulose Using Polycaprolactone as the Modifying Compound: Esterification versus Click-Chemistry. Nanomaterials. 2013; 3(4):638-654. https://doi.org/10.3390/nano3040638
Chicago/Turabian StyleBenkaddour, Abdelhaq, Khalil Jradi, Sylvain Robert, and Claude Daneault. 2013. "Study of the Effect of Grafting Method on Surface Polarity of Tempo-Oxidized Nanocellulose Using Polycaprolactone as the Modifying Compound: Esterification versus Click-Chemistry" Nanomaterials 3, no. 4: 638-654. https://doi.org/10.3390/nano3040638
APA StyleBenkaddour, A., Jradi, K., Robert, S., & Daneault, C. (2013). Study of the Effect of Grafting Method on Surface Polarity of Tempo-Oxidized Nanocellulose Using Polycaprolactone as the Modifying Compound: Esterification versus Click-Chemistry. Nanomaterials, 3(4), 638-654. https://doi.org/10.3390/nano3040638