
The Photocatalytic Conversion of Carbon Dioxide to Fuels Using Titanium Dioxide Nanosheets/Graphene Oxide Heterostructure as Photocatalyst
 and
and 
Abstract
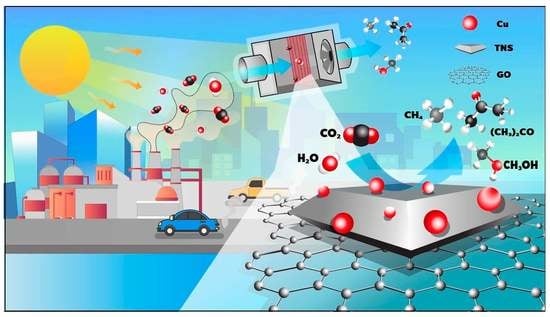
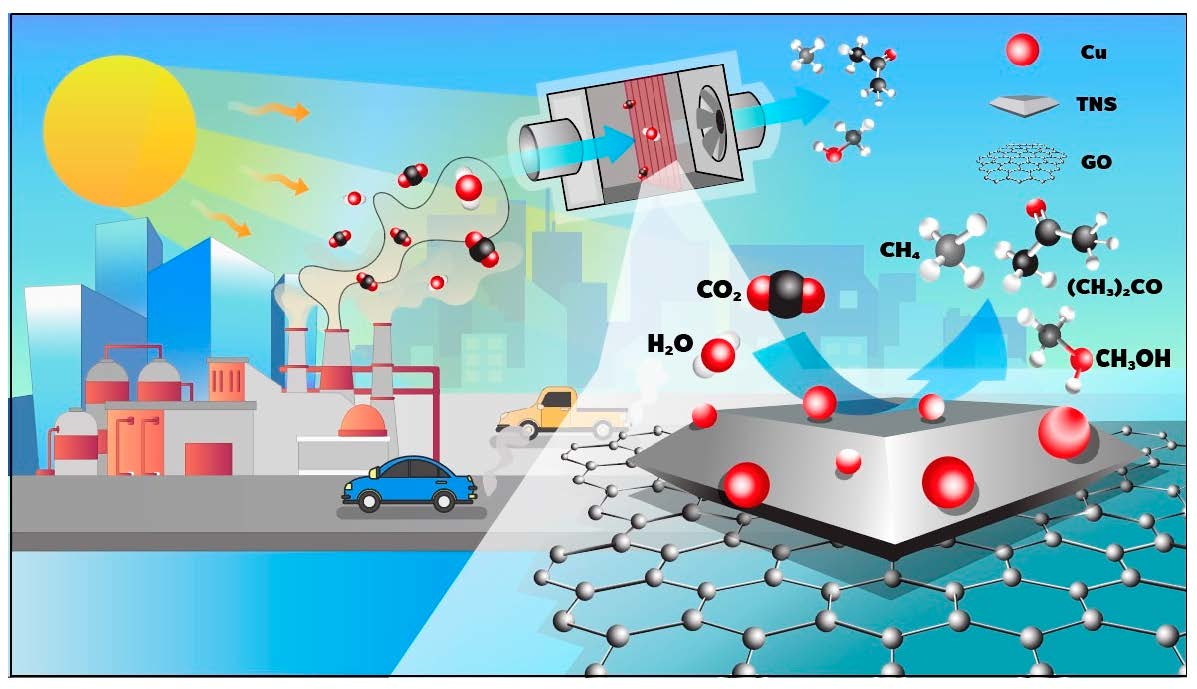
:
1. Introduction
2. Materials and Methods
2.1. GO Synthesis
2.2. Synthesis of TiO2 Nanosheet (TNS) and TiO2 Nanoparticle (TNP)
2.3. Synthesis of the TNS/GO Composites
2.4. CO2 Photoreduction
2.5. Sample Characterizations
3. Results and Discussion
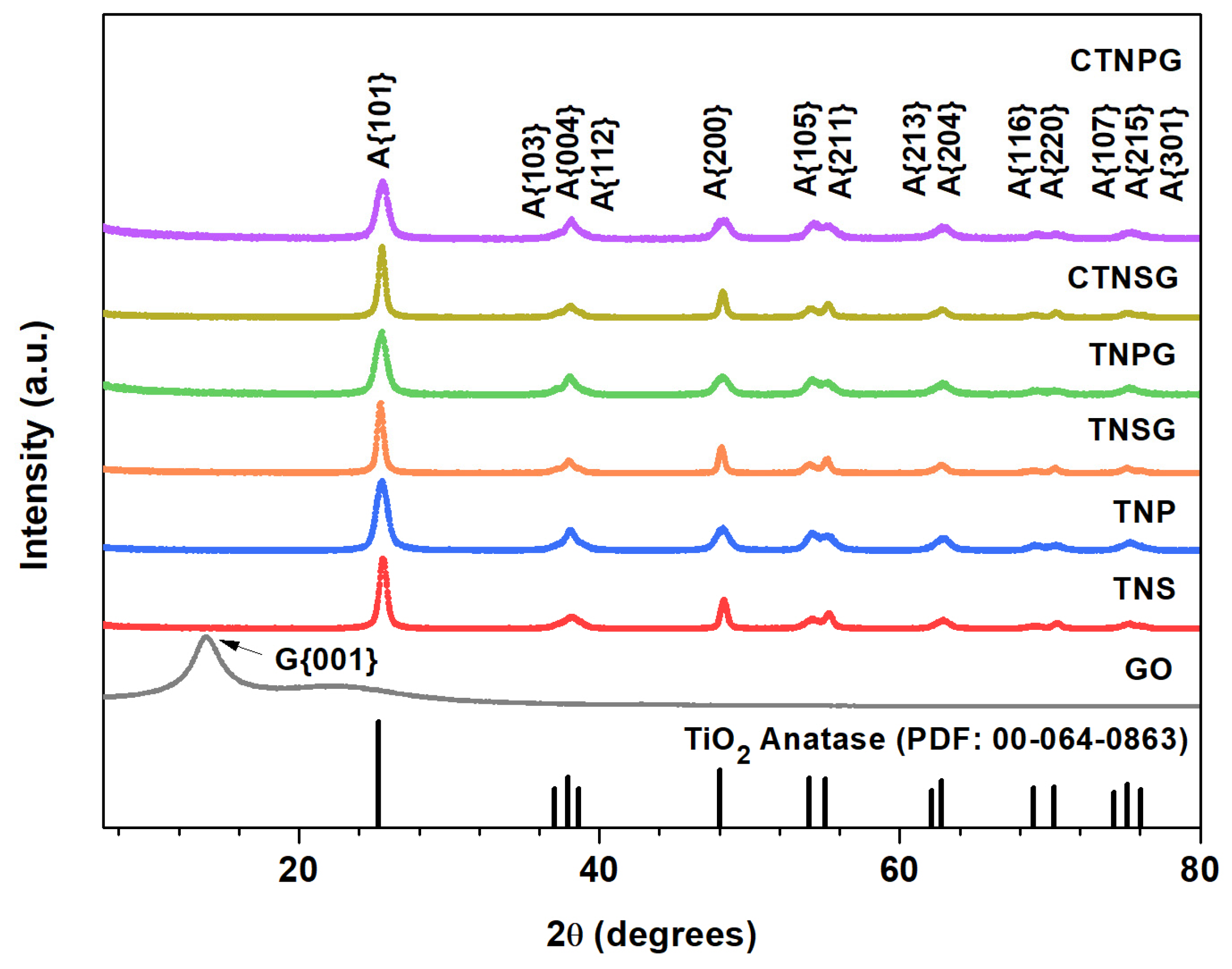
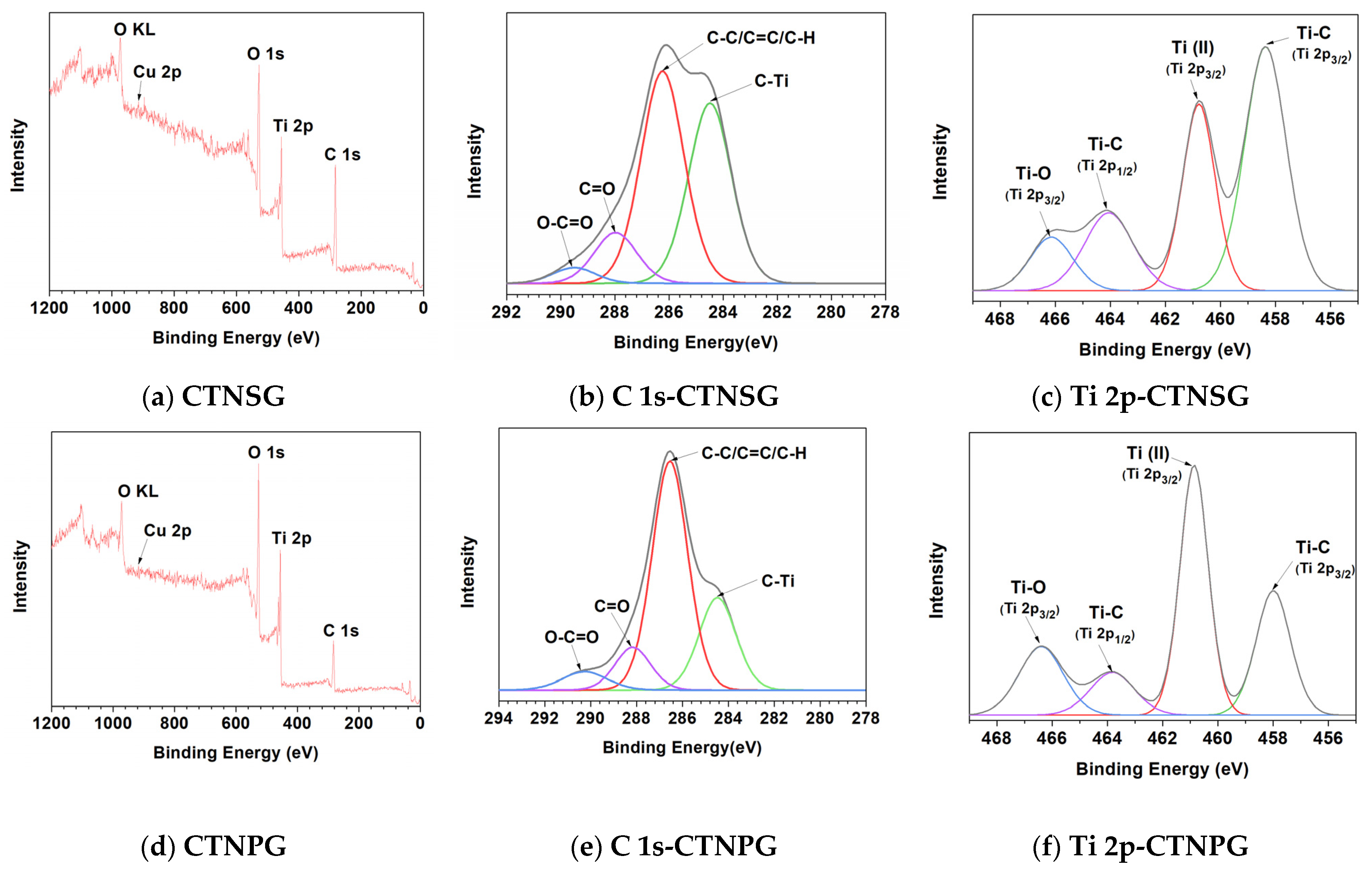
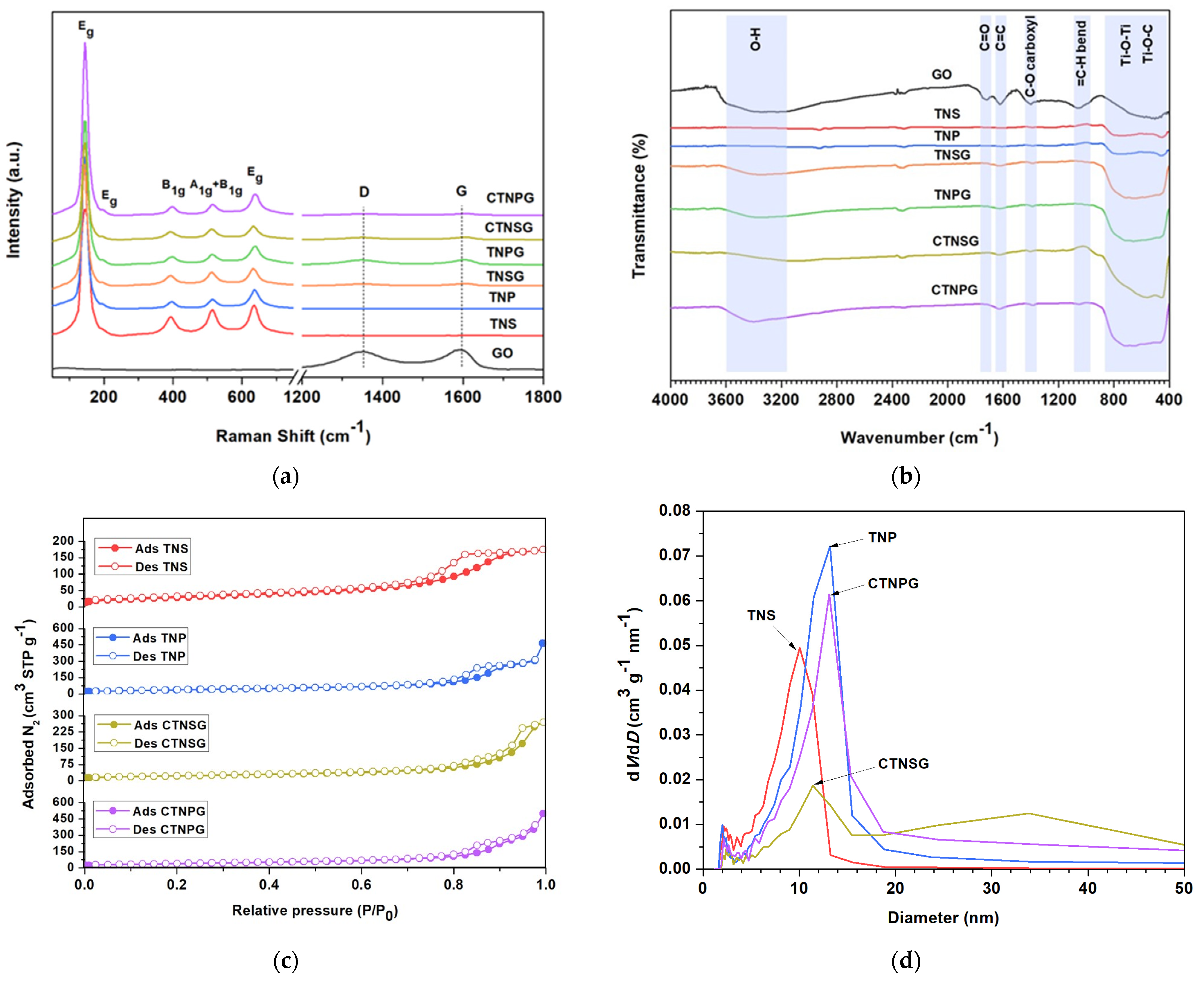
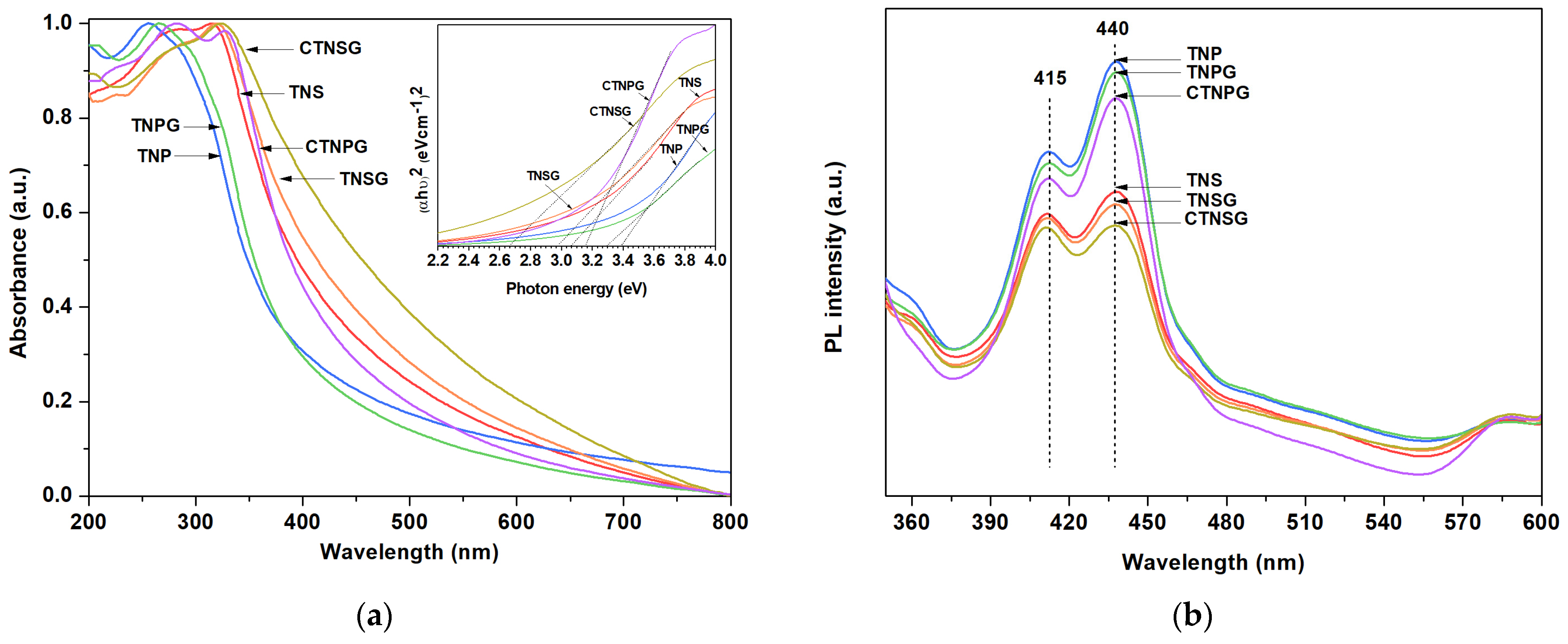
3.1. Material Characterizations
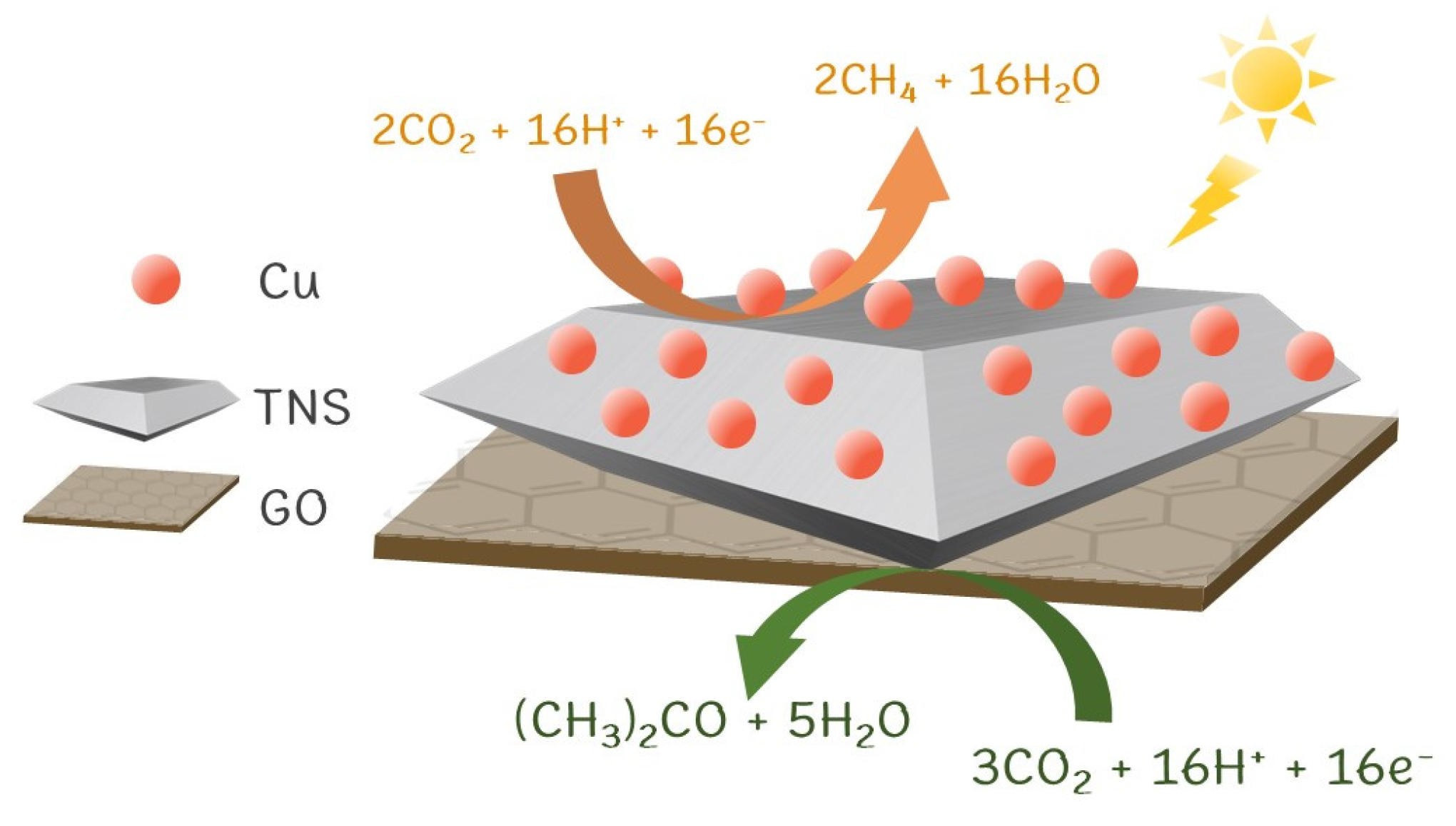
3.2. CO2 Photoreduction
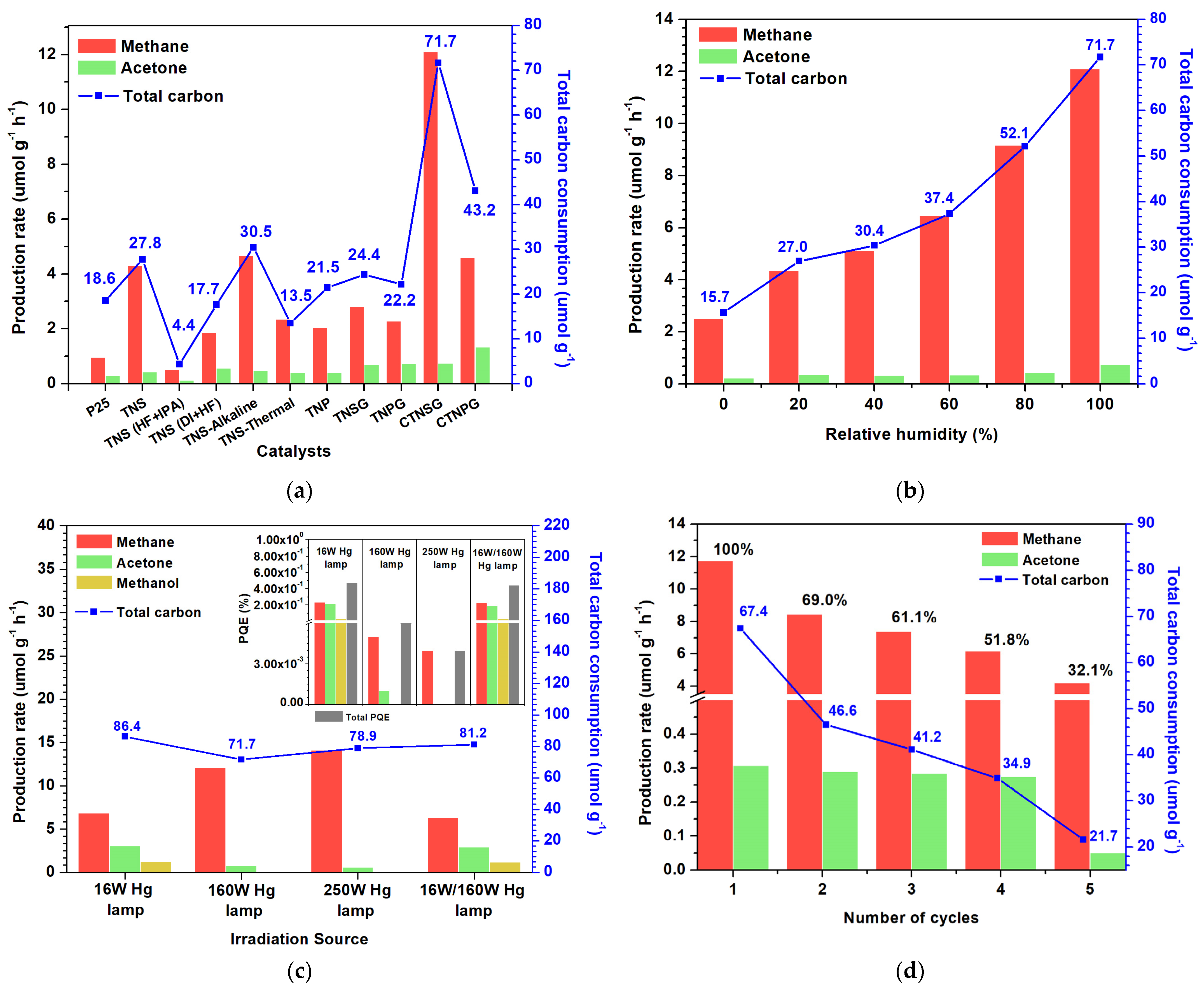
3.2.1. Photoactivity of Photocatalyst
3.2.2. Effect of Humidity
3.2.3. Effect of the Light Source
3.2.4. Reusability and Stability of Photocatalyst
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nogueira, A.E.; Oliveira, J.A.; da Silva, G.T.; Ribeiro, C. Insights into the role of CuO in the CO2 photoreduction process. Sci. Rep. 2019, 9, 1316. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.P.; Nguyen, D.L.T.; Nguyen, V.-H.; Le, T.-H.; Vo, D.-V.N.; Trinh, Q.T.; Bae, S.-R.; Chae, S.Y.; Kim, S.Y.; Le, Q.V. Recent advances in TiO2-based photocatalysts for reduction of CO2 to fuels. Nanomaterials 2020, 10, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglyuvatyy, E. Climate Change and International Response. In Climate Change, Forests and Federalism; Springer: Singapore, 2022; pp. 1–12. [Google Scholar]
- Salvi, B.L.; Jindal, S. Recent developments and challenges ahead in carbon capture and sequestration technologies. SN Appl. Sci. 2019, 1, 885. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhu, D.; Jin, Z.; Tian, H.; Zhou, B.; Jiang, P.; Meng, Q.; Wu, X.; Xu, H.; Hu, T. Carbon capture and storage for long-term and safe sealing with constrained natural CO2 analogs. Renew. Sustain. Energy Rev. 2023, 171, 113000. [Google Scholar] [CrossRef]
- Xin, C.; Hu, M.; Wang, K.; Wang, X. Significant enhancement of photocatalytic reduction of CO2 with H2O over ZnO by the formation of basic zinc carbonate. Langmuir 2017, 33, 6667–6676. [Google Scholar] [CrossRef]
- Li, P.; Hu, H.; Luo, G.; Zhu, S.; Guo, L.; Qu, P.; Shen, Q.; He, T. Crystal facet-dependent CO2 photoreduction over porous ZnO nanocatalysts. ACS Appl. Mater. Interfaces 2020, 12, 56039–56048. [Google Scholar] [CrossRef]
- Wang, J.; Xia, T.; Wang, L.; Zheng, X.; Qi, Z.; Gao, C.; Zhu, J.; Li, Z.; Xu, H.; Xiong, Y. Enabling visible-light-driven selective CO2 reduction by doping quantum dots: Trapping electrons and suppressing H2 evolution. Angew. Chem. Int. Ed. 2018, 57, 16447–16451. [Google Scholar] [CrossRef]
- Almomani, F.; Bhosale, R.; Khraisheh, M.; Kumar, A.; Tawalbeh, M. Photocatalytic conversion of CO2 and H2O to useful fuels by nanostructured composite catalysis. Appl. Surf. Sci. 2019, 483, 363–372. [Google Scholar] [CrossRef]
- Lertthanaphol, N.; Pienutsa, N.; Chusri, K.; Sornsuchat, T.; Chanthara, P.; Seeharaj, P.; Kim-Lohsoontorn, P.; Srinives, S. One-Step Hydrothermal Synthesis of Precious Metal-Doped Titanium Dioxide–Graphene Oxide Composites for Photocatalytic Conversion of CO2 to Ethanol. ACS Omega 2021, 6, 35769–35779. [Google Scholar] [CrossRef]
- Larimi, A.; Rahimi, M.; Khorasheh, F. Carbonaceous supports decorated with Pt–TiO2 nanoparticles using electrostatic self-assembly method as a highly visible-light active photocatalyst for CO2 photoreduction. Renew. Energy 2020, 145, 1862–1869. [Google Scholar] [CrossRef]
- Lertthanaphol, N.; Prawiset, N.; Soontornapaluk, P.; Kitjanukit, N.; Neamsung, W.; Pienutsa, N.; Chusri, K.; Sornsuchat, T.; Chanthara, P.; Phadungbut, P. Soft template-assisted copper-doped sodium dititanate nanosheet/graphene oxide heterostructure for photoreduction of carbon dioxide to liquid fuels. RSC Adv. 2022, 12, 24362–24373. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-L.; Ong, W.-J.; Chai, S.-P.; Goh, B.T.; Mohamed, A.R. Visible-light-active oxygen-rich TiO2 decorated 2D graphene oxide with enhanced photocatalytic activity toward carbon dioxide reduction. Appl. Catal. B Environ. 2015, 179, 160–170. [Google Scholar] [CrossRef]
- Tan, L.-L.; Ong, W.-J.; Chai, S.-P.; Mohamed, A.R. Photocatalytic reduction of CO2 with H2O over graphene oxide-supported oxygen-rich TiO2 hybrid photocatalyst under visible light irradiation: Process and kinetic studies. Chem. Eng. J. 2017, 308, 248–255. [Google Scholar] [CrossRef]
- Olivo, A.; Ghedini, E.; Signoretto, M.; Compagnoni, M.; Rossetti, I. Liquid vs. Gas Phase CO2 photoreduction process: Which is the effect of the reaction medium? Energies 2017, 10, 1394. [Google Scholar] [CrossRef] [Green Version]
- Lettieri, S.; Pavone, M.; Fioravanti, A.; Santamaria Amato, L.; Maddalena, P. Charge carrier processes and optical properties in TiO2 and TiO2-based heterojunction photocatalysts: A Review. Materials 2021, 14, 1645. [Google Scholar] [CrossRef]
- Wang, X.; Yan, W.; Li, H.; Wang, Z.-J. Nitrogen-Doped Ni/TiO2 Catalyst with Enhanced Activity and Improved Selectivity for CO2 Methanation. Energy Fuels 2022, 36, 11647–11653. [Google Scholar] [CrossRef]
- Mekprasart, W.; Pavasupree, S.; Jayasankar, C.; Ravuri, B.R.; Wattanawikkam, C.; Pecharapa, W. Characterization, X-ray Absorption Spectroscopic Analysis and Photocatalytic Activity of Co/Zn Co-Doped TiO2 Nanoparticles Synthesized by One-Step Sonochemical Process. Crystals 2021, 11, 1254. [Google Scholar] [CrossRef]
- Zhang, Z.; Zuo, F.; Feng, P. Hard template synthesis of crystalline mesoporous anatase TiO2 for photocatalytic hydrogen evolution. J. Mater. Chem. 2010, 20, 2206–2212. [Google Scholar] [CrossRef]
- Han, X.; Kuang, Q.; Jin, M.; Xie, Z.; Zheng, L. Synthesis of titania nanosheets with a high percentage of exposed (001) facets and related photocatalytic properties. J. Am. Chem. Soc. 2009, 131, 3152–3153. [Google Scholar] [CrossRef]
- Yang, H.G.; Liu, G.; Qiao, S.Z.; Sun, C.H.; Jin, Y.G.; Smith, S.C.; Zou, J.; Cheng, H.M.; Lu, G.Q. Solvothermal synthesis and photoreactivity of anatase TiO2 nanosheets with dominant {001} facets. J. Am. Chem. Soc. 2009, 131, 4078–4083. [Google Scholar] [CrossRef]
- Sun, R.; Jiang, X.; Zhang, M.; Ma, Y.; Jiang, X.; Liu, Z.; Wang, Y.; Yang, J.; Xie, M.; Han, W. Dual quantum dots decorated TiO2 nanorod arrays for efficient CO2 reduction. J. Catal. 2019, 378, 192–200. [Google Scholar] [CrossRef]
- Alkanad, K.; Hezam, A.; Al-Zaqri, N.; Bajiri, M.A.; Alnaggar, G.; Drmosh, Q.A.; Almukhlifi, H.A.; Neratur Krishnappagowda, L. One-Step Hydrothermal Synthesis of Anatase TiO2 Nanotubes for Efficient Photocatalytic CO2 Reduction. ACS Omega 2022, 7, 38686–38699. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Hsu, Y.-H. Effects of reaction temperature on the photocatalytic activity of TiO2 with Pd and Cu cocatalysts. Catalysts 2021, 11, 966. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, H.; Guo, L.-H.; Zhao, L. Two-dimensional interface engineering of a titania–graphene nanosheet composite for improved photocatalytic activity. ACS Appl. Mater. Interfaces 2013, 5, 13035–13041. [Google Scholar] [CrossRef]
- Xiong, Z.; Luo, Y.; Zhao, Y.; Zhang, J.; Zheng, C.; Wu, J.C. Synthesis, characterization and enhanced photocatalytic CO2 reduction activity of graphene supported TiO2 nanocrystals with coexposed {001} and {101} facets. Phys. Chem. Chem. Phys. 2016, 18, 13186–13195. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Low, J.; Xiao, W.; Zhou, P.; Jaroniec, M. Enhanced photocatalytic CO2-reduction activity of anatase TiO2 by coexposed {001} and {101} facets. J. Am. Chem. Soc. 2014, 136, 8839–8842. [Google Scholar] [CrossRef]
- Ruan, L.; Wang, X.; Wang, T.; Ren, Z.; Chen, Y.; Zhao, R.; Zhou, D.; Fu, G.; Li, S.; Gao, L. Surface defect-controlled growth and high photocatalytic H2 production efficiency of anatase TiO2 nanosheets. ACS Appl. Mater. Interfaces 2019, 11, 37256–37262. [Google Scholar] [CrossRef]
- Yasin, G.; Arif, M.; Shakeel, M.; Dun, Y.; Zuo, Y.; Khan, W.Q.; Tang, Y.; Khan, A.; Nadeem, M. Exploring the nickel-graphene nanocomposite coatings for superior corrosion resistance: Manipulating the effect of deposition current density on its morphology, mechanical properties, and erosion-corrosion performance. Adv. Eng. Mater. 2018, 20, 1701166. [Google Scholar] [CrossRef]
- Reddy, M.J.K.; Ryu, S.H.; Shanmugharaj, A. Synthesis of SnO2 pillared carbon using long chain alkylamine grafted graphene oxide: An efficient anode material for lithium ion batteries. Nanoscale 2016, 8, 471–482. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, W.; Yu, L. Photocatalytic decomposition of gaseous HCHO over Ag modified TiO2 nanosheets at ambient temperature. Nanomaterials 2019, 9, 338. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Fang, J.; Li, D.; Chen, J.; Zhang, X.; Shao, Y.; He, Y. A facile synthesis of CdSe quantum dots-decorated anatase TiO2 with exposed {0 0 1} facets and its superior photocatalytic activity. Appl. Catal. B Environ. 2016, 181, 838–847. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Wang, F.; Liu, X.; Ren, C. Fabrication of two lanthanides co-doped Bi2MoO6 photocatalyst: Selection, design and mechanism of Ln1/Ln2 redox couple for enhancing photocatalytic activity. Appl. Catal. B Environ. 2017, 217, 378–387. [Google Scholar] [CrossRef]
- Wang, W.-S.; Wang, D.-H.; Qu, W.-G.; Lu, L.-Q.; Xu, A.-W. Large ultrathin anatase TiO2 nanosheets with exposed {001} facets on graphene for enhanced visible light photocatalytic activity. J. Phys. Chem. C 2012, 116, 19893–19901. [Google Scholar] [CrossRef]
- Yan, J.; Wu, G.; Guan, N.; Li, L.; Li, Z.; Cao, X. Understanding the effect of surface/bulk defects on the photocatalytic activity of TiO2: Anatase versus rutile. Phys. Chem. Chem. Phys. 2013, 15, 10978–10988. [Google Scholar] [CrossRef]
- Seriani, N.; Pinilla, C.; Crespo, Y. Presence of gap states at Cu/TiO2 anatase surfaces: Consequences for the photocatalytic activity. J. Phys. Chem. C 2015, 119, 6696–6702. [Google Scholar] [CrossRef]
- Chen, B.-R.; Nguyen, V.-H.; Wu, J.C.; Martin, R.; Kočí, K. Production of renewable fuels by the photohydrogenation of CO2: Effect of the Cu species loaded onto TiO2 photocatalysts. Phys. Chem. Chem. Phys. 2016, 18, 4942–4951. [Google Scholar] [CrossRef]
- Azam, M.U.; Tahir, M.; Umer, M.; Jaffar, M.M.; Nawawi, M. Engineering approach to enhance photocatalytic water splitting for dynamic H2 production using La2O3/TiO2 nanocatalyst in a monolith photoreactor. Appl. Surf. Sci. 2019, 484, 1089–1101. [Google Scholar] [CrossRef]
- Rodríguez, V.; Camarillo, R.; Martínez, F.; Jiménez, C.; Rincón, J. High-pressure synthesis of rGO/TiO2 and rGO/TiO2/Cu catalysts for efficient CO2 reduction under solar light. J. Supercrit. Fluids 2021, 174, 105265. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, Y.; Wu, X.; Zheng, H.; Zhao, Z.; Liu, J.; Li, J. Graphene-wrapped Pt/TiO2 photocatalysts with enhanced photogenerated charges separation and reactant adsorption for high selective photoreduction of CO2 to CH4. Appl. Catal. B Environ. 2018, 226, 360–372. [Google Scholar] [CrossRef]
- Yang, Z.; Yin, T.; Zhang, F.; Wu, W.; Lin, M.; Dong, Z.; Zhang, J. Investigation on dispersion properties of CO2 and ester solvent mixtures using in situ FTIR spectroscopy. RSC Adv. 2020, 10, 18192–18199. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Condition | Production Rate (µmol h−1gcat−1) | Reference | ||
|---|---|---|---|---|---|
| Methane | Methanol | Acetone | |||
| Copper−TiO2 nanosheet/GO (CTNSG) | -10 mgcat -160 Watts mercury -5 h reaction time | 12.09 | 0 | 0.74 | This work |
| Copper−TiO2 nanosheet/GO (CTNSG) | -10 mgcat -16 Watts mercury -5 h reaction time | 6.84 | 1.23 | 3.07 | This work |
| TiO2 nanotubes | -50 mgcat -300 Watts Xe -5 h reaction time | 47.00 | - | - | [23] |
| Copper−TiO2/rGO | -50 mgcat -450 Watts Xe -3 h reaction time | 2.20 | 0 | 0 | [39] |
| Pt−TiO2/rGO Pt−TiO2/MWCNT Pt−TiO2/SWCNT | -100 mgcat -15 Watts energy-saving daylight lamp (Philips) -12 h reaction time | 0.50 1.90 0.70 | 0 | 0 | [11] |
| Pt−TiO2 nanosheet/rGO | -300 Watts Xe -8 h reaction time | 4.70 | 0 | 0 | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karawek, A.; Kittipoom, K.; Tansuthepverawongse, L.; Kitjanukit, N.; Neamsung, W.; Lertthanaphol, N.; Chanthara, P.; Ratchahat, S.; Phadungbut, P.; Kim-Lohsoontorn, P.; et al. The Photocatalytic Conversion of Carbon Dioxide to Fuels Using Titanium Dioxide Nanosheets/Graphene Oxide Heterostructure as Photocatalyst. Nanomaterials 2023, 13, 320. https://doi.org/10.3390/nano13020320
Karawek A, Kittipoom K, Tansuthepverawongse L, Kitjanukit N, Neamsung W, Lertthanaphol N, Chanthara P, Ratchahat S, Phadungbut P, Kim-Lohsoontorn P, et al. The Photocatalytic Conversion of Carbon Dioxide to Fuels Using Titanium Dioxide Nanosheets/Graphene Oxide Heterostructure as Photocatalyst. Nanomaterials. 2023; 13(2):320. https://doi.org/10.3390/nano13020320
Chicago/Turabian StyleKarawek, Apisit, Kittipad Kittipoom, Labhassiree Tansuthepverawongse, Nutkamol Kitjanukit, Wannisa Neamsung, Napat Lertthanaphol, Prowpatchara Chanthara, Sakhon Ratchahat, Poomiwat Phadungbut, Pattaraporn Kim-Lohsoontorn, and et al. 2023. "The Photocatalytic Conversion of Carbon Dioxide to Fuels Using Titanium Dioxide Nanosheets/Graphene Oxide Heterostructure as Photocatalyst" Nanomaterials 13, no. 2: 320. https://doi.org/10.3390/nano13020320